
Abstract
Gait disability is a major health care problem worldwide. Powered exoskeletons have recently emerged as devices that can enable users with gait disabilities to ambulate in an upright posture, and potentially bring other clinical benefits. In 2014, the US Food and Drug Administration approved marketing of the ReWalk™ Personal Exoskeleton as a class II medical device with special controls. Since then, Indego™ and Ekso™ have also received regulatory approval. With similar trends worldwide, this industry is likely to grow rapidly. On the other hand, the regulatory science of powered exoskeletons is still developing. The type and extent of probable risks of these devices are yet to be understood, and industry standards are yet to be developed. To address this gap, Manufacturer and User Facility Device Experience, Clinicaltrials.gov, and PubMed databases were searched for reports of adverse events and inclusion and exclusion criteria involving the use of lower limb powered exoskeletons. Current inclusion and exclusion criteria, which can determine probable risks, were found to be diverse. Reported adverse events and identified risks of current devices are also wide-ranging. In light of these findings, current regulations, standards, and regulatory procedures for medical device applications in the USA, Europe, and Japan were also compared. There is a need to raise awareness of probable risks associated with the use of powered exoskeletons and to develop adequate countermeasures, standards, and regulations for these human-machine systems. With appropriate risk mitigation strategies, adequate standards, comprehensive reporting of adverse events, and regulatory oversight, powered exoskeletons may one day allow individuals with gait disabilities to safely and independently ambulate.
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Introduction
The Centers for Disease Control and Prevention reported that 17.1 million American adults are unable to or find it very difficult to walk a quarter mile.Citation1 Gait impairment significantly contributes to long-term disability and ambulatory dysfunction in daily living.Citation2 It is reported that in the USA, there are about 800,000 stroke incidents annuallyCitation3 and more than a quarter of a million people currently living with a spinal cord injury (SCI),Citation4 both of which are common causes of gait impairment.
Physical rehabilitation continues to remain a mainstay for individuals to regain functional independence after a stroke. However, rising health care costs tend to limit access to rehabilitation clinicians and facilities, thereby acting as a bottleneck to functional recovery.Citation5 More recently, body weight-supported robot-assisted treadmill training has been shown to lead to comparable or improved functional outcomes for stroke surviors.Citation6,Citation7 Similarly, manually assisted or robot-assisted locomotor training can result in beneficial effects, even for subjects with complete SCI.Citation8 However, these robotic devices remain largely restricted to clinical or research settings due to their size and cost, and they are less amenable to training with other functional tasks such as over-ground walking, stair climbing, etc. As a result, a number of newer assistive and therapeutic, wearable lower limb robotic devices, namely powered exoskeletons, have been developed.Citation9–Citation15
Several studies have systematically reviewed lower limb powered exoskeletons in a clinical context. A recent scoping review reported that clinical trials of powered robotic exoskeletons for post-stroke rehabilitation were free of serious adverse events.Citation16 Other studies have focused on assessing the usefulness, safety, or effectiveness of powered exoskeletons as assistive devices for individuals with SCI.Citation17,Citation18 One review concluded that exoskeletons are safe to use in real-world settings and known to yield health benefits.Citation18 However, this study was later challenged as it included duplicate subjects and studies, rendering its conclusions questionable.Citation19
Overall, the protocols, outcomes, inclusion/exclusion criteria, and metrics in published clinical exoskeleton studies with individuals with SCI were found to vary greatly across studies.Citation20 Moreover, the risks associated with powered exoskeletons and the possible approaches to mitigate those risks are neither reported adequately nor systematically. Although reviews of clinical trials deem exoskeletons safe for gait assistance or gait rehabilitation, these studies 1) often differ on their inclusion and exclusion criteria, 2) lack details on reported adverse events, or 3) do not explicitly state that adverse events did not occur.Citation17,Citation18,Citation21–Citation23 Without a clear understanding and comprehensive knowledge of adverse events and the risks users face, it is difficult to evaluate the risk-to-benefit ratio of powered exoskeletons. Some researchers admit this limitation. For example, one study noted that safety is usually overlooked in the current literature as the authors “did not find any specific mention of falls or the prevention of falls” in some studies.Citation12 Another study noted that “little [information] concerning safety and satisfaction was documented in the selected studies”.Citation17 The authors believe user safety is the top concern for exoskeleton developers, researchers, end users, and regulators. Therefore, in this manuscript, we systematically reviewed risk management in current lower limb exoskeletons and how existing regulations address risk assessment and mitigation.
Lower limb powered exoskeletons
This review focuses on powered, over-ground lower limb exoskeletons for medical use. That is to say, exoskeletons used in military and industry applications are outside the scope of this review. Upper limb robotic arms/exoskeletons have been widely studied as a medical device in rehabilitation.Citation24 They are generally safer compared to their lower limb counterparts due to a stationary setup which is free from the dangers associated with falling; they are thus excluded from this review. Throughout this article, we used the word “exoskeleton” interchangeably with the phrases “lower limb/extremity/body exoskeleton” for brevity, unless explicitly specified otherwise. The US Food and Drug Administration (FDA) identifies a powered lower extremity exoskeleton as “a prescription device that is composed of an external, powered, motorized orthosis that is placed over a person’s paralyzed or weakened limbs for medical purposes” (Regulation 21 CFR 890.3480).Citation25 On the other hand, all current FDA-cleared exoskeletons are recognized under product code PHL, which describes a powered lower extremity exoskeleton as “a prescription device that is composed of an external, powered, motorized orthosis used for medical purposes that is placed over a person’s paralyzed or weakened limbs for the purpose of providing ambulation”. The ability to provide over-ground ambulation is a distinct feature compared to stationary robotic devices that are generally attached to treadmills. Ambulation also imposes significant challenges and risks in terms of maintaining balance and preventing falls. In treadmill-based systems, a body weight support tether is often used as an easy and robust countermeasure to prevent falls.
In this review, we focus on exoskeletons that enable users to ambulate over-ground. As we followed the definition of product code PHL, treadmill-based robotic devices such as the Lokomat™ (Hocoma, Zurich, Switzerland) were excluded. In fact, Lokomat is categorized by the FDA as an isokinetic evaluation and testing system under Regulation 21 CFR 890.1925.Citation26 Ankle-foot orthoses and reciprocating gait orthoses were also excluded.
While there are many research laboratories and private companies with exciting prototypes in development, the developers usually release only limited specifications until they file for marketing clearance. Moreover, their designs are also more likely to be subject to change. Therefore, we discuss exoskeletons that have been cleared for marketing by the FDA, devices that have been indexed in the FDA’s database and have related peer-reviewed publications, and those registered in clinical trials. Currently, there are three devices in the USA that have been cleared by the FDA since this device category was established in 2014: ReWalk™ Personal (Argo Medical Technologies Ltd, Yokneam Ilit, Israel), Indego™ (Parker Hannifin Corp., Cleveland, OH, USA), and Ekso GT (Ekso™ Bionics Ltd., Richmond, VA, USA). ReWalk was cleared for marketing in the USA as a de novo Class II medical device by the FDA in 2014Citation27 and became the first of its kind. Indego was confirmed to be substantially equivalent (SE) to ReWalk in March 2016 by the FDA, and therefore also cleared for marketing in the USA.Citation28 Ekso was approved by the FDA in the same manner shortly after.Citation29
Risk management for powered exoskeletons
Risk management is defined as the “systematic application of management policies, procedures, and practices to the tasks of analyzing, evaluating, and controlling risk”.Citation30 In the context of designing medical exoskeletons, the focus is on identifying and mitigating physical risks introduced to device users (including assistants, who are required to be near most systems) and possible risk mitigation strategies. These risks typically include, but are not limited to, falls, joint misalignment, skin damage, software malfunction leading to uncontrolled behaviors, electrical and fire hazard, and user error. We examined risk management methods in the medical exoskeleton industry by first reviewing current industrial standards from the International Organization for Standardization (ISO).
ISO is the world’s largest developer of voluntary international standards, and many ISO standards are recognized by the FDA. The FDA recognizes the standard for risk management in medical devices (ISO 14971), which was last revised in 2007. In 2012, the European Committee for Standardization (CEN) adopted a harmonized version of the ISO standard as EN ISO 14971:2012. Compliance with ISO 14971 and EN ISO 14971:2012 is an integral step in the process of bringing a medical device to the USA and the European consumer markets. ISO 14971 outlines the process of risk management and provides guidelines for evaluating, reducing, and documenting risks. The standard lists required qualifications for involved personnel, defines management responsibilities, and outlines the contents of a risk management file in which all documentation is collected and stored.Citation31 The FDA has formally recognized powered lower extremity exoskeletons as a Class II medical device with special controls. It requires 510(k) submission from manufacturers who seek marketing approval in the USA.Citation32
Gap between the industry and regulations
With three devices already approved by the FDA and more reportedly in the process, it is plausible that this new field will soon become crowded with competitors from both large companies and lab spin-off startups around the world. However, it appears that the risk management and regulations of exoskeletons have not been addressed adequately. Fall is a major risk of using exoskeletons, yet there is not enough information on this issue in existing FDA-cleared devices. Each device has a different strategy for dealing with potential falls, but their effectiveness is still unclear (). In addition, the definition of exoskeleton is somewhat confusing. ReWalk, Indego, and Ekso are all regulated primarily under Product Code PHL according to their de novo/510(k) submission.Citation27,Citation28,Citation33 Product Code PHL states that “the control of the device (exoskeleton) is achieved through a wrist-worn, user-operated wireless communicator…”,Citation32 but Indego and Ekso do not have any wrist-worn accessories. This definition is only applicable to ReWalk, the first system approved by the FDA in 2014. FDA’s different definition of “exoskeleton” in Product Code PHL and CFR 890.3480 also suggests the expansiveness of the term. To address the gap between the fast developing robotic technology and its regulations, in this manuscript the authors review the risk management strategies in current devices as well as related standards and regulations.
Table 1 Safety features related to falling in ReWalk™, Indego™, and Ekso™ exoskeletons
Risks of powered exoskeletons
Devices
Having a clear understanding of the risks users face while operating these devices is critical. There is evidence suggesting benefits with the use of exoskeletons,Citation17,Citation20,Citation34 but without adequate information on potential risks, it remains difficult to properly assess the risk–benefit ratio. We reviewed all devices that have been cleared for marketing as a medical device in the USA, arguably one of the most stringent markets in the world. This list includes ReWalk, Indego, and Ekso. Additionally, we included Rex™ (Rex Bionics, Auckland, New Zealand) and hybrid assistive limb (HAL™; Cyberdyne, Tsukuba, Japan) systems. Rex has been cleared for marketing in the European Union (EU). HAL has been cleared in both Japan and the EU. They are both listed in multiple FDA and National Institutes of Health (NIH) databases and have related clinical studies. The current marketing status of these five devices is summarized in . For a technical review of current lower limb medical exoskeletons, readers are referred to recent studies.Citation16–Citation18,Citation20,Citation34,Citation35 In the following sections, we review related studies on the risk and its mitigations for these powered exoskeletons.
Table 2 Marketing status of some commercially available devices
ReWalk
ReWalk is the first exoskeleton cleared by the FDA for both personal use as well as use in a rehabilitation setting in the USA. The FDA approved ReWalk as a de novo Class II device with special controls. Along with the de novo approval, the FDA issued Order PS140001,Citation36 requiring postmarket surveillance because the device’s failure to prevent a fall would be reasonably likely to cause serious injury or death to the users and assisting individuals.Citation27 Argo Medical Technologies Ltd received a warning letter from the FDA after failing to conduct such a postmarket surveillance program regarding the risk of fall.Citation37 ReWalk announced the launch of the required postmarket study in August 2016.Citation38
In addition to commonly accepted ISO standards and FDA guidelines to mitigate risks, ReWalk features the following safety measures concerning software and hardware.Citation27
“Graceful collapse/sitting”: In the event of a major system failure, such as complete loss of power, the weight of the patient causes the ReWalk unit to enter into a graceful collapse, ie, the body’s weight rotates the inner rotor of the motor and moves the exoskeleton joints into a slowly achieved collapsed sitting position.
Battery: The main battery is a Lithium ion battery that allows the user to walk continuously for >2 hours on a charge. The secondary is a Lithium polymer battery that allows at least an additional 15 minutes of continuous walking. The user is alerted when the charge of the battery is low by a short vibration (buzz) repeated every 10 seconds. Warnings are provided to the user not to use the device while charging.
Excessive joint angles: There is a threshold in the software that limits the movement through a safe range of motion and a fixed mechanical stop, which also prevents movement beyond a safe joint angle trajectory.
Software: The system does a self-check at start up and disables the system until the problem is corrected. The system will default over to manual control when a main computer failure occurs.
Misstep or obstacle: If the user contacts an obstacle with one of the limbs, the movement restriction generates excess torque. A torque threshold limit and alert is issued by the buzzer and vibrator, and the leg then moves back to a standing position.
Loss of communication between remote and main computer: The system can enter a bypass mode where the device can be controlled with buttons on the hip actuation unit.
ReWalk has been tested in several clinical studies to examine its efficacy and safety.Citation12,Citation21,Citation23,Citation39–Citation41 It has received mixed reviews. Positive opinions include that ReWalk is a safe device for in-hospital ambulationCitation23 and that it provides potential for functional gainCitation40 and improved fitness because of higher heart rate and oxygen demand than standing or sitting.Citation39 There are no reports of serious adverse events,Citation18 and participants had generally positive opinions regarding the use of the system.Citation12 ReWalk has been the most studied powered exoskeleton for the SCI population, as evidenced by the number of peer-reviewed publications compared to that of its competitors.Citation20 However, one studyCitation21 reported a high incidence of skin aberrations when using the ReWalk exoskeleton: 5 out of 10 enrolled subjects experienced at least one mild skin aberration, and two of them were withdrawn from the study owing to recurring skin breakdown problems. Also, one subject had a hairline fracture of the talus after using the device, possibly because of inaccurate joint alignment.Citation42 No further treatment was needed, but the subject was excluded for the rest of the study because of this near-serious adverse event. The administrators of that study recommended prescreening subjects with osteopenia or osteoporosis in future studies.Citation21
Indego
The Indego exoskeleton is deemed SE to ReWalk, and thus, most of its identified risks and associated risk mitigation approaches are similar.Citation28 It has passed various tests such as durability testing and software verification. There are some differences when compared to ReWalk. Small variations exist in the size of components, weight, allowable height and weight of users, control method, and battery, all of which are deemed similar without additional safety or efficacy concerns. Of interest is Indego’s fall detection feature: it detects forward, backward, and sideways falling as it is happening, with the device making adjustments during the course of the fall to position the user for minimal risk of injury, although the details of this maneuver are not publicly available. In the event of power failure, the knees become locked and hips free. This mechanism allows the user to remain standing in the event of malfunction. There are a few clinical studies to date that utilize Indego. It is reported that Indego outperforms knee–ankle-foot orthosis,Citation43 enables acute cardiorespiratory and metabolic responses,Citation44 and enabled persons with tetraplegia and paraplegia learn to use it quickly.Citation45 No adverse events have been reported.
Ekso
Similar to ReWalk and Indego, Ekso also has bilateral powered hip and knee joints in the sagittal plane. However, it is the first and, currently, the only exoskeleton cleared by the FDA for use by stroke patients.
After the announcement that the FDA would regulate exoskeletons as Class II medical devices with special controls, Ekso Bionic filed a 510(k) notification to the FDA in December 2014. In the meantime, the company was allowed to continue marketing under Class I registration while the application was under review. The company received clearance for marketing by the FDA in April 2016, on the basis that it is SE to ReWalk.Citation29
Ekso has been evaluated in several clinical studiesCitation22,Citation46–Citation48 and has been shown to improve gait speed and step length of SCI subjects after 20 sessions of training.Citation47 A pilot study explored incorporating transcutaneous spinal cord stimulation in daily training with Ekso.Citation49 In a study using a prototype version of Ekso, multiple falls were recorded without any physical adverse conditions.Citation22 Note that an overhead tether was used in this study to prevent actual falls. A “fall” was therefore defined as an event when the tether was triggered to function, not when the user actually fell down. It is unclear whether these “falls” would have been prevented if the tether had not been used, and instead would have required assistants to provide more support. Except from several falls due to the subjects losing balance, mechanical and software errors also contributed to multiple falls. In particular, a faulty featureCitation50 that was initially designed to help trigger steps via contact sensors on the crutch was removed in later versions of the device because of frequent malfunctioning. There are six adverse events registered in the Manufacturer and User Facility Device Experience (MAUDE, an FDA’s database of device-related adverse events; https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/search.cfm) database. None of them resulted in the injury of users or anyone else. All are mechanical issues, such as a footplate separating from the device and broken epoxy bond allowing the ankle joint to rotate. It is not publicly available what improvements have been made to the system to address these issues, but no accidents have been reported in later studies.
HAL
HAL for Medical Use (Lower Limb Type) is a bilateral lower limb exoskeleton with two active degrees of freedom at the hip and knee, and a passive degree of freedom at the ankle joint of each leg. Its control system processes data from surface electromyography (EMG) sensors, angle/acceleration sensors, and force sensors to estimate the necessary forces to assist-as-needed the user’s intended actions. The use of EMG signals in HAL’s shared control system to help detect the user’s intent represents a type of hybrid peripheral neural interface.Citation51
Its European model has been certified under the European Medical Device Directive (CE 0197). HAL was granted approval to manufacture and sell Japan’s first robot therapeutic device HAL for Medical Use (Lower Limb Type) by the Japanese Ministry of Health, Labour, and Welfare on November 25, 2015. In December, the company applied for national health insurance coverage for HAL for medical use. The company has reportedly applied for the FDA approval in the USA, but its status in the regulatory path is unclear.Citation52
HAL has been widely tested in clinical trials. A systematical review of clinical applications of HAL for gait training for 140 subjects with stroke or SCI suggested that minor and transient side effects occurred, but no serious adverse events were reported.Citation53 HAL is thus considered safe and feasible for stroke patients to use. One study examined the feasibility and safety issues of using HAL in acute-phase rehabilitation after stroke.Citation54 Though no serious incident happened, 4 out of the 22 subjects experienced orthostatic hypotension (Orthostatic hypotension was defined as a decrease in systolic blood pressure of >20 mmHg immediately after sitting or standing.), resulting in one subject withdrawing from the experiment. This is noteworthy because blood pressure responses after using exoskeletons are usually not measuredCitation43 or reported as within normal limitsCitation23 in other studies. A similar acute phase rehabilitation feasibility and safety study recruited eight patients and thoroughly reported all adverse reports from the participants.Citation55 Moderate discomfort from tight straps and heavy weight of the device were reported by several subjects, although these did not last after the training. Moderate pain due to pressure was reported at the cuff over the knee and over the malleolus. This was solved by readjusting the device. Although minor and solvable, this adverse effect highlighted the importance of proper alignment of the device. Chafed feet were reported in one subject due to wrong shoe size. This was not a technical error, but better labeling and rigorous staff training will minimize the chance of user errors. Stumbling due to impaired weight shifting occurred from time to time. Subjects were secured by an overhead tether and also supported by two therapists when needed. However, such protective procedures are usually not available in normal usage with HAL (and with other similar devices).
Rex
The Robotic Exoskeleton (Rex) carries the CE mark and is available in the EU market. It distinguishes itself from other exoskeletons because it has the ability to self-balance, albeit under restricted conditions, without the need for any extra balancing instruments (cane, walker, etc.). Rex thus offers a “hands-free” experience to users.
To date, there are no published clinical trials to demonstrate the clinical effectiveness and risks of Rex. However, a serious adverse event was reported to the MAUDE database.Citation56 One user with SCI suffered bilateral symmetrical fractures after finishing a supervised session. The patient noticed swelling in the ankles and knees in the evening. An appointment was organized for the following day, and bone fractures were identified by X-ray. According to the manufacturer, there are three likely contributing factors: 1) the patient was found to have osteoporosis; 2) the patient had a spasm, and so inadvertently kicked the heel stops back, resulting in misaligned ankles and knees; or 3) two ankle braces were used to support the user’s ankles. They restricted the range of motion such that the user no longer met the required range of motion specified for the device. This adverse event highlights the possibility of bone fracture due to misalignment of joints. The event was probably the result of a series of unfortunate mistakes: poor, or lack of, training of physiotherapists; lack of clarity in the exclusion criteria; and lack of countermeasures for unsecured joints.
Inclusion and exclusion criteria in clinical trials
Medical exoskeletons have the potential to help people with a wide spectrum of lower limb motor impairments, including weakened legs due to advanced aging. Initial deployment of the exoskeleton technology – by manufacturers like ReWalk and Indego – has focused on individuals with paraplegia due to SCI, and clinical trials reflect this trend.Citation20 Other exoskeleton systems, such as Ekso, HAL, and Rex, target a broader population. A systematic review of 27 studies of powered lower limb exoskeletons that included 144 participants reported that 35% of the participants were stroke patients, 58% were SCI patients, 3% were subjects with gait disorders of unspecified etiology, and the rest, 4%, were healthy controls.Citation34 To the best of our best knowledge, no study has compared the risks and effectiveness of powered exoskeletons across SCI and stroke user groups.
One of the challenges in prescribing powered exoskeletons is how best to match the technology to the user. This challenge is reflected in the wide-ranging inclusion and exclusion criteria found in clinical trials of powered exoskeletons (). The data in the table were aggregated from ClinicalTrials.gov (https://clinicaltrials.gov/), an online NIH database that compiles information of clinical studies involving human participants.
Table 3 Inclusion and exclusion criteria in clinical trials
The popularity of all criteria categories was compared based on the percentage of clinical studies (in the cohort of 28 identified studies) that explicitly used each type of criteria. Range of motion (ROM)-related deficits such as contractures are most commonly used as an exclusion criterion, with a leading rate of 79% (22/28). This is followed by criteria related to skin issues such as pressure sores or ulcers in the areas that come into contact with the device (75%, 21/28). Other categories include muscle and spasticity issues (71%, 20/28), bone health (71%, 20/28), hypertension (71%, 20/28), mentality (64%, 18/28), cardiology (64%, 18/28), and pregnancy (61%, 17/28). These percentages are similar in quantity, but the studies they each include differ. Moreover, different studies often evaluate the same category differently.
The Ashworth Scale was used in most studies to measure the spasticity of muscles. However, some chose to use a modified version (denoted as M.Ashworth in ). And the exclusion bar differs across studies, for example, Study #8 included patients with Ashworth score of 4 while most others did not. ROM was usually quantitatively measured as the contracture limitations, although Study #7 measured the full range of motion of joints. As shown in , the values of these contracture limitations also differ across studies, and even across studies that used the same device. The difference therefore cannot be explained by hardware differences.
Severe osteoporosis is a risk factor for bone fractures in individuals using powered exoskeletons due to the potentially high external forces applied to the user’s limbs. However, the threshold varied between studies. Bone mineral density in different bones were used for examination and different t-scores were required. Study #1 only required “healthy bone density” without further details. Study #19 excluded subjects if severe osteoporosis affecting the hip and spine was documented. Future studies should investigate the effects of exoskeleton use on bone health and use a standard metric for defining osteoporosis.
Subjects should also be mentally able to understand the risk, communicate with the experimenter, and correctly follow instructions. Some of the studies that included mentality check used the Mini-Mental State Examination (MMSE) scores, for example, study #23 using Ekso required MMSE scores >17, whereas study #4 using ReWalk required MMSE scores > 26. Some other studies that mentioned the mentality requirement left the determination to on-site physicians, for example, study #12 excluded subjects with “inability to follow instructions”.
There were no studies that used a quantitative scale or metric to measure the condition of the skin. Those studies that did explicitly check skin condition usually required the area of skin in contact with the device to be intact and free from pressure sore or ulcers. The inadequate attention and knowledge about skin conditions may lead to frequent skin and soft tissue damage in seemingly normal usage. Remarkably, the exclusion criteria did not include metrics to assess the risk of falls. These topics will be discussed in later sections.
Reported adverse events
An adverse event is defined by ISO standard 62366-2 as an event associated with a medical device that led to death or serious injuries of a patient, or may lead to such if event recurs. In this review, we extended this definition to include minor injuries as well. Adverse events that have been reported in the peer-reviewed literature or public databases were reviewed. Our sources include all publications related to lower limb exoskeleton from PubMed and the MAUDE, up to November 17, 2016. The FDA requires a manufacturer to report to this database within 30 days after being aware of the adverse events. We used the name of the previously discussed devices as keywords in search. The events have been compiled into , sorted by the type of accident.
Table 4 Published adverse events involving exoskeletons
Identified risks
The FDA identified nine risks when it reviewed ReWalk’s de novo application.Citation27 It is likely that similar risks exist in other devices as well since they usually claim to be SE to ReWalk. Based on previous information of device features, studies, and reported adverse events, we have identified additional risks that are not explicitly mentioned by the FDA. These risks are compiled in .
Table 5 Identified risks to health and mitigation measures
Falls
Injuries resulting from falls are a major public health concern for the elderly, representing one of the main causes of long-standing pain, functional impairment, disability, and death in this population.Citation57 Using exoskeletons impose an additional risk of falls to users who already suffer from motor deficiency. When a person walks freely, the body interacts with the environment (ground), which is predictable unless obstacles catch the person off guard. When the person uses an exoskeleton, however, this direct interaction is reduced or distorted. Feedback is inevitably distorted because of the extra layer of media. Sometimes, there are additional sources of feedback such as vibrations and sound. Users may need some time to adjust to these extrinsic stimulations. Additionally, users may attempt to execute certain movements, yet cannot correctly achieve it because of the physical constraint imposed by the exoskeleton. Last but not least, depending on the control scheme, the exoskeleton may incorrectly react to body movement, resulting in the triggering of an unexpected command that may contribute to the occurrence of a fall.
ReWalk, Indego, and Ekso all have their own strategies to mitigate the risk of falls.Citation27,Citation28,Citation33 summarizes them in three categories: 1) to detect and actively mitigate falling, 2) to seek minimal damage should power failure and/or a fall become inevitable, and 3) to deploy assistants who stand beside the user according to indications for use. Indego is the only device that has an active fall detection and mitigation feature: it detects falls and adjusts itself during the course of a fall to minimize the risk of injury. The method by which Indego detects falls, its responsiveness, and what kind of adjustments are to be made during the course of the fall are unclear. Nonetheless, such a feature is important in controlling an exoskeleton. In the unlikely event of a power failure when falling is inevitable, ReWalk will collapse to a sitting position slowly, which is called “graceful collapse”.Citation27 Although this is a more controlled fall, it is not free from hazards, because most likely there will not be a chair right behind the user to sit on when the accident occurs. Indego and Ekso lock the knee joint and allow free movement of hip when power failure occurs. Although it provides a better solution if the user is in a standing posture when power fails, it is still a risk to lock the knee should power failure occur during the swing phase. Another difference among the three devices is their indications for use. All require a trained person to stand beside and supervise the user at all times. While ReWalk and Indego offer training programs and certificates to allow trained companions to give supervision, Ekso only allows users to be supervised by trained physical therapists in rehabilitation institutions.
Although many exoskeleton-related studies mention the risk of falls, most of them arrive at the conclusion that there is no risk of falls just because no falls were observed in data collection.Citation12 It is likely that the safeguards and task conditions followed in those clinical trials will be distinct from those imposed by settings outside the clinic. Past studies were usually conducted in hospitals or rehabilitation centers where professionally trained personnel guided the protocols. However, ReWalk and Indego are not required to be operated in professional facilities. Therefore, the low number of falls and fractures in current clinical studies, where users have continuous expert supervision, may underestimate the real risk of adverse events among exoskeleton users in the community.Citation21 As previously discussed, multiple “falls” (tether was triggered) were registered in a study using a prototype of Ekso.Citation22 Changes have been made since this prototype to address the design flaws leading to falls. In a feasibility study of HAL, stroke patients occasionally stumbled during walking. The risk of actual falls in these studies was completely mitigated because of the overhead harness. The FDA has requested ReWalk to conduct a postmarket surveillance program to further monitor and study the risks of falls.Citation37
Skin and soft tissue injury
As shown in , skin and soft tissue injury is the most frequent type of injury. It is universal as it happened to all listed devices and is repetitive, happening to several subjects in the same study. In some cases, skin damage happened repeatedly to the same subject,Citation21 suggesting that the researchers could not avoid this problem even after noticing its occurrence.
Despite the prevalence of this type of injury, it has attracted little attention. shows that only half of the clinical trials considered skin condition as an exclusion criterion. Skin and tissue injuries were usually dismissed as minor issues and did not affect the safety evaluation of exoskeletons.Citation18 Many studies were able to prevent further damage by adding additional padded support to the area where the damage occurred.Citation23,Citation45 These easy but postinjury mitigations suggest that it is sometimes possible to avoid such injuries given prior knowledge of which part of the body is likely to get hurt. It is important that manufacturers systematically examine the cause and location of skin and soft tissue damages. Currently, it is difficult to summarize any pattern of skin and tissue damages given the information available and the lack of sensors in padded braces that could monitor forces at the physical interface. Even in the studies that do report them, they are usually briefly mentioned without specifying the frequency, cause, location, and seriousness of the damage. Of interest, exoskeletons with integrated pressure sensors have been proposed as a novel method to prevent skin injuries related to excessive pressure in mobility-impaired exoskeleton users.Citation58 Force sensing arrays, integrated with wireless transmitters, were placed at exoskeleton joints to give alarm when excessive pressure occurs.
User error
User error is defined in ISO Standard 62366-1 as user’s unexpected action or lack of action that could lead to a different result than intended by the manufacturer or user. It can occur during normal use. For instance, a user may accidentally rotate their pelvis and shift their body weight. This may trigger false alarm movement in exoskeletons because they use such movements as a trigger to initiate a new gait cycle. Other possible errors include tripping over obstacles, wrong menu selection, and operating with low battery life. Thus, it is important to design a human–machine interface that is friendly and robust, independent of the technological fluency or attentional state of the user.
Caregivers and physical therapists accompanying the user during training may also make errors. They may choose wrong commands in settings, incorrectly fasten straps, or fail to support the user when he or she loses balance. In a bone fracture accident, the manufacturer suspected that one of the causes was that the therapist did not correctly secure the user’s ankles.Citation56 Human factor validation testing is an important step in risk management according to FDA’s guideline for medical devices.Citation59 While there are many wearable exoskeletons in development, there is little published information on this topic. Training sessions and labeling seem to be the only countermeasure from ReWalk, Indego, and Ekso. Concrete consideration and plans over this issue are still absent.
Bone fracture
The exoskeleton must be precisely aligned with the user’s joint so that there is minimal incorrect torque forced onto the joints. Misalignment can result in skin abrasion, sores,Citation55 hairline fractures,Citation21 or bone fractures.Citation56 Currently, there are two reported bone fracture accidents. Both injured users had SCIs, and therefore did not notice the fracture immediately and continued to finish their scheduled training sessions. They both reported abnormal appearance of their legs later in the day, and discovered bone fracture through X-ray exams on the following day.
Medical exoskeleton users are particularly vulnerable to bone damage because it is common for SCI patients to experience osteoporosis, thus resulting in higher likelihood of bone fracture.Citation60 More importantly, SCI patients lack the feedback of pain from lower limbs when excessive torque is applied. Contrary to the conclusion from a previous review study,Citation18 we consider bone fracture to be a serious possible risk of using exoskeletons. Possible mitigation strategies include better strap design to minimize the chance of any misalignment and improving participant screening by excluding patients with severe osteoporosis by requiring DXA scans and X-rays to evaluate this risk.
Osteoporosis screening is only implemented in some studies. Unfortunately, there is no consensus in the guidelines with regard to what level of osteoporosis shall be considered an indicator to exclude subjects. More research and regulatory science are needed to find the optimal exclusion criteria.
Long-term secondary effects
With some conflicting opinions, it is hypothesized that exoskeletons have the potential to bring clinical change to users with disability. As shown in systematic reviews,Citation20,Citation34 recovery is observed after weeks of training in many studies. However, it is not clear how this change is compared to other traditional rehabilitation procedures.Citation34 Meanwhile, other physical and mental conditions of the users may also change after using exoskeletons. Some subjects reported improvements in pain and bowel and bladder function after using ReWalk.Citation40 One of the advantages of using exoskeletons for mobility rehabilitation is that they provide repetitive practice session after session. However, very few clinical studies have protocols longer than 6 months. So it remains unknown what the long term effect is of using an exoskeleton. A review of protocols of body weight-support treadmill training interventions with robotic orthosis found that, in general, longer treatments provide better outcomes.Citation61 Yet, the opposite conclusion was made in a review of exoskeletons used in stroke rehabilitation.Citation16 A recent study found that there is neurological recovery after chronic SCI patients train with an exoskeleton for 12 months.Citation62
Besides, osteoporosis/osteopenia is viewed as a relative contraindication for SCI patients being considered for exoskeleton ambulatory training because of a risk in bone fracture.Citation18 Thus far, it is unclear what would happen when a person with low bone density repeatedly uses an exoskeleton. Unfortunately, there are only few long-term studies on the risks and benefits of powered exoskeletons. More studies are needed to understand how exoskeletons may change patients’ neurological status and biomechanical condition over time.
Regulations and standards
Manufacturers, users, and regulators
Exoskeleton manufacturers, users, and regulators are the three major players in the exoskeleton industry (). “Manufacturers” are companies (ReWalk, Indego, Ekso, etc.), institutions (NASA X1,Citation14 Kinesis,Citation63 etc.), and many other labs and startups whose prototypes are still in early development. Currently, there are few evaluations across devices to compare their advantages and disadvantages.
Figure 1 Entities involved in the risk management of exoskeleton.
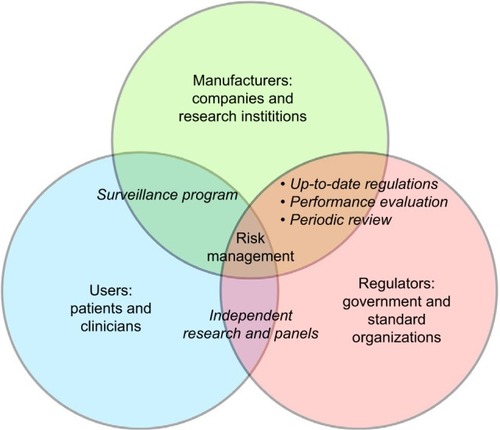
“Regulators” consist of government agencies and third-party organizations that create and update industry standards. In the USA, this occurs mostly between the FDA and the ISO. The FDA has directly recommended many ISO standards. For example, FDA’s guideline to the biological valuation of medical devices suggests following standard ISO 10993-1.Citation64 Aside from interacting with manufactures, it is important for the FDA to include end users in its regulatory path. The FDA recognized the importance of patient-centric assessment and patient-reported outcomes in its recently issued guidance.Citation65 Systematic studies in a top-down approach to collect users’ feedback across devices will help regulators and the industry to better understand the need and problems end users face.
The “user” group consists of individuals who use and potentially benefit from the device directly. There are also clinicians, who indirectly interact with the device and oversee its performance. Clinicians, especially physical therapists, benefit from the device as it decreases the amount of the physical labor required by them, making their work less physically intense and decreasing their risk of injury. However, the benefit compared to the imposed risk for the exoskeleton user remains to be seen. There is no strong evidence suggesting the usefulness of exoskeletons yet.Citation17 There are cases where significant clinical improvement is found, but they are not consistent across studies.Citation20 In ReWalk’s de novo application to the FDA, it is stated that “the probable benefits outweigh the probable risks” when the target population uses the device.Citation27
As ordered by the FDA, a postmarket surveillance program for ReWalk is already underway.Citation38 We encourage this type of communication between manufacturers and regulators to be carried out to a greater extent, because ultimately only data from significantly large user groups can reveal drawbacks and show the industry the right direction to proceed. We also expect clinical studies funded by the government to evaluate and compare commercially available exoskeletons. Independent studies from third-party research groups and discussions from independent panels can provide reliable information to the regulators. They can then make more informed regulations to provide safer devices. Currently, there is no consensus for evaluating the medical potential of exoskeletons as different metrics are typically used to assess benefits.Citation20 Systematic evaluation of different devices is needed for a better and more transparent market.
Global overview of medical device regulations
While this review focuses on the USA market and regulations, the regulations in FDA’s counterparts in Europe and Japan are also summarized. Research labs in Europe have been developing several exoskeleton prototypes for years, such as H2Citation15 and Mindwalker.Citation66 It is also common for exoskeletons to obtain CE clearance in the EU before obtaining FDA’s clearance in the USA, as has been the case for ReWalk, Ekso, HAL, and Rex. Japan is already famous for their innovation in various humanoid robots. HAL is arguably its most prestigious lower limb exoskeleton. Its mass production began before the founding of many other companies: more than 20 sets of HAL exoskeletons were in use at hospitals and rehabilitation centers in 2009.Citation67 As opposed to the USA and Europe where exoskeletons are usually developed by labs and startups, there is a trend for industry giants in Asia to directly jump into this market. Honda’s R&D team is developing their stride management assistive device that helps the rotation of the hip joint. It has gone through a clinical trial.Citation68 Samsung has applied for a USA patent for their wearable robot that features EMG control.Citation69 Panasonic also announced development of several assistive robots, with plans to use them in elder care.Citation70
In the USA, powered exoskeletons have been officially classified by the FDA as a Class II device with special controls.Citation71 The special controls include 1) biocompatibility, 2) electromagnetic compatibility, 3) software validation, 4) geometry and material composition, 5) various nonclinical performance testing, 6) clinical testing, 7) training program, and 8) labeling.Citation71 When a product is to be marketed in the USA, the company should first classify the device (Class II for exoskeletons) and prepare a premarket submission (de novo if it is a new category or 510(k) to claim SE to a currently approved medical device) for the FDA to review. The product has to wait for FDA clearance before marketing.
In the EU, there is no central government organization to issue certificates. Instead, medical devices are required to obtain the CE mark. CE marking on a product is a manufacturer’s declaration that the product complies with the essential requirements of the relevant European regulations. Products with CE can be legally placed on the EU market. The CE certificate of an exoskeleton can be obtained from a notified body, which is a third-party, independent group that specializes at the conformity assessment of a product. A comparison between the procedures in the USA and the EU is shown in the top panel in . Except for the fact that certificates are issued by different bodies, their procedures are generally similar.
Figure 2 Procedures for obtaining medical device approval in the USA, EU, and Japan. The USA requires applications to be approved by a federal agency, namely the FDA, whereas the EU distributes the responsibility to many independent notified bodies. Japan’s government reviews reliability of the manufacturers both on site and via documents, while the USA and the EU leave that responsibility to manufacturers themselves. The bottom panel about Japan was adapted from the diagram on Page 11 in the materials of the 2011 AHC Workshop on Medical Devices. Tamura A. Understanding Japanese medical device requirements. 2011. Available from: https://www.pmda.go.jp/files/000164006.pdf.Citation73
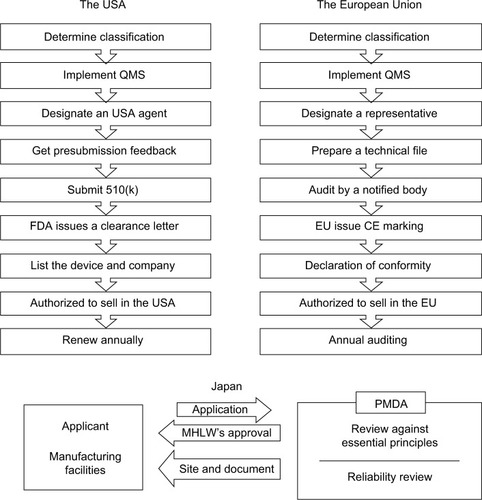
The Japanese Ministry of Health, Labour, and Welfare (MHLW) is responsible for the device classification and issuing marketing approval for medical devices in Japan. Pharmaceuticals and Medical Devices Agency (PMDA) is the technical branch that performs the actual review, examination, data analysis, and so on to help MHLW’s measure. While third-party certification is allowed for low-risk medical devices, MHLW’s approval on the basis of PMDA review is required for high-risk medical devices. There is no direct information on how MHLW classifies powered exoskeletons. Yet according to its database, HAL was approved by MHLW in 2015.Citation72 Therefore, MHLW’s review is the likely path for future exoskeletons seeking marketing approval in Japan.
The bottom panel in is an overview of the medical device approval process in Japan, adapted from materials of a PMDA workshop.Citation73 A distinctive difference between MHLW’s and FDA’s approval processes is that PMDA performs on-site and document reviews of various good practice evaluations. These evaluations include Good Laboratory Practice/Good Clinical Practice/Good Post-Marketing Surveillance Practice/Good Postmarketing Study Practice compliance assessments and Good Manufacturing Practice/Quality Management System/Good Gene, Cellular, and Tissue-based Products Manufacturing Practice inspections, which are required by the FDA to be established and guaranteed solely by manufacturers themselves.Citation74 The Japanese agency continues to monitor the compliance of these regulations after the market approval.
Standards recognized by the FDA
Although there are three devices cleared by the FDA for marketing in the USA, there are currently no standards that apply specifically to powered lower limb exoskeletons. contains a list of existing standards recognized by the FDA for either Class II medical devices or lower limb pros-theses. The standards applicable to prostheses are included as regulations for durability testing and cyclic loading testing.
Table 6 Standards recommended by the FDA
There are also Joint Working Groups that are currently working toward developing standards for rehabilitation robots. For example, Joint Working Group 36’s mission is to develop particular requirements for the basic safety and essential performance of medical robots.Citation75
Special topic: pediatric exoskeletons
SCI and gait deficits are not restricted to the adult population. Pediatric SCI affects ~500 children aged <15 years every year in the USA.Citation76 Cerebral palsy (CP) is a neurodevelopmental disease related to nonprogressive cerebral abnormalities that occur before birth or early in life, and it affects 2–3 children out of every 1,000.Citation77 Effective interventions that can preserve or augment strength on a continuous basis for those with crouch gait from CP are needed.Citation78 Passive orthoses have been studied and showed no significant benefits.Citation79 It is hypothesized that the benefits obtained from a system like the Lokomat, in which the child is constrained between parallel bars and suspended above a treadmill by an upper body harness, will translate to a lower limb exoskeleton in which the child is granted more freedom of mobility. A single-blind randomized controlled trial provides the first evidence that upper limb robot-assisted therapy is effective in children with CP.Citation80
Some exoskeleton developers have started to design special devices for the pediatric population. These designs are either explicitly for children or incorporate flexible aspects (eg, detachable segments) into the adult-scale designs, allowing children to wear the devices, for example, Lokom-atPro (https://www.hocoma.com/usa/us/products/lokomat/lokomatpro/pediatric-orthoses/): Atlas 2030 (http://www.marsibionics.com/products/?lang=en#/atlas-2030). a device developed by the Spanish National Research Council currently in preclinical phase (http://www.roboticstrends.com/article/worlds_first_kid_sized_exoskeletonhelps_chil_dren_walk): and a customized exoskeleton built by the NIH.Citation78 As devices are developed and enter clinical trials, data will begin to emerge from these trials so that the risks and the benefits associated with pediatric exoskeletons can be better understood.
Pediatric exoskeletons, like their adult counterparts, inherently pose risks to users, such as falls and skin-related damage. Additionally, designing pediatric exoskeletons can often be more complex as additional challenges exist. Adult users of the normal exoskeletons are often individuals who possessed the ability to walk at one point in their lives. In the case of many children with CP, however, medical practitioners would be tasked with teaching the child to walk. CarefUl consideration is required about whether user interfaces and labeling of devices used for adults are applicable to children. Lastly, there are technical difficulties when it comes to adapting the structure and control approach for exoskeletons to match the cognitive–motor development of the child. The size, weight, and power of the motors used in current exoskeletons are inadequate for the pediatric population, especially small children.Citation81
Conclusion
The industry of powered lower limb exoskeletons is emerging. Manufactures, users, and regulators need to adapt quickly. Since ReWalk was cleared by the FDA as a de novo device in the USA, Indego and Ekso have proved SE to ReWalk and have successfully obtained clearance for marketing from the FDA. It is likely that new exoskeleton systems will join the mobile powered exoskeleton industry in the near future. However, currently there is no clear definition of the term powered exoskeleton and no standards that directly apply to this type of device. We compiled related standards that the FDA recommends and encourage regulators and the industry to work together to release practical guidelines and standards for this field.
Meanwhile, in order for the FDA to make informed decisions, regulatory science of powered exoskeletons must advance. In this regard, more computational modeling, bench testing, preclinical and clinical studies, and postsurveillance reports from both manufacturers and independent groups are encouraged. Besides the need for more data, it is important that data are collected and reported in more systematical manner. Presently, there are vastly different inclusion and exclusion criteria and outcome measures used among the published clinical studies, even with the same brand of device. Adverse events are also reported with different levels of detail and methods.
Safety is the top concern in medical device development and regulation. More work is needed to address risk concerns such as falls, fractures, and long-term adverse effects of using exoskeletons. It is alarming that multiple accidents have caused bone fracture of the users. Risks such as user error and skin/tissue damage, although identified, have been often overlooked in past studies. Moreover, in order for powered exoskeletons to be deployed safely outside the clinic, it is critical that potential risks of deploying exoskeletons in rich, highly dynamic environments cohabited by humans and machines be identified and such risks mitigated. We hope this review will help spark constructive discussions leading to better exoskeleton designs and regulations and greater opportunities for this industry, and ultimately significant improvements in the quality of life of the users it serves.
Acknowledgments
This work was supported in part by Mission Connect – a TIRR Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
- Centers for Disease Control and PreventionTable A-10a. Difficulties in physical functioning among adults aged 18 and over, by selected characteristics: United States, 2014CDC2014 Available from: http://www.cdc.gov/nchs/fastats/disability.htm
- Belda-LoisJMMena-del HornoSBermejo-BoschIRehabilitation of gait after stroke: a review towards a top-down approachJNeuroeng Rehabil2011816622165907
- National Institute of Neurological Disorders and StrokeWhat you need to know about stroke Available from: https://stroke.nih.gov/materials/needtoknow.htmAccessed December 6, 2016
- National Institute of Neurological Disorders and StrokeSpinal cord injury: hope through research Available from: http://www.ninds.nih.gov/disorders/sci/detail_sci.htmAccessed December 6, 2016
- GodwinKMWassermanJOstwaldSKCost associated with stroke: outpatient rehabilitative services and medicationTop Stroke Rehabil201118Suppl 167668422120036
- MehrholzJPohlMElsnerBTreadmill training and body weight support for walking after stroke (Review)Cochrane Database Syst Rev20141CD00284024458944
- MehrholzJPohlMElectromechanical-assisted gait training after stroke: a systematic review comparing end-effector and exoskeleton devicesJ Rehabil Med20124419319922378603
- HubliMDietzVThe physiological basis of neurorehabilitation – locomotor training after spinal cord injuryJ Neuroeng Rehabil2013101523336934
- TsukaharaAHasegawaYSankaiYGait support for complete spinal cord injury patient by synchronized leg-swing with HALIEEE Int Conf Intell Robot Syst201117371742
- Kolakowsky-HaynerSASafety and feasibility of using the Ekso Bionic exoskeleton to aid ambulation after spinal cord injuryJ Spine2013S43
- FarrisRJQuinteroHAGoldfarbMPerformance evaluation of a lower limb exoskeleton for stair ascent and descent with ParaplegiaConf Proc IEEE Eng Med Biol Soc20121908191123366287
- ZeiligGWeingardenHZweckerMDudkiewiczIBlochAEsquenaziASafety and tolerance of the ReWalk™ exoskeleton suit for ambulation by people with complete spinal cord injury: a pilot studyJ Spinal Cord Med20123529610122333043
- KilicarslanAPrasadSGrossmanRGContreras-VidalJLHigh accuracy decoding of user intentions using EEG to control a lower-body exoskeletonConf Proc IEEE Eng Med Biol Soc201320135606560924111008
- HeYNathanKVenkatakrishnanAAn integrated neuro-robotic interface for stroke rehabilitation using the NASA X1 powered lower limb exoskeletonConf Proc IEEE Eng Med Biol Soc20143985398825570865
- BortoleMVenkatakrishnanAZhuFThe H2 robotic exoskeleton for gait rehabilitation after stroke: early findings from a clinical studyJ Neuroeng Rehabil20151215426076696
- LouieDREngJJPowered robotic exoskeletons in post-stroke rehabilitation of gait: a scoping reviewJ Neuroeng Rehabil20161315327278136
- LajeunesseVVincentCRouthierFCareauEMichaudFExoskeletons’ design and usefulness evidence according to a systematic review of lower limb exoskeletons used for functional mobility by people with spinal cord injuryDisabil Rehabil Assist Technol201611753554726340538
- MillerLZimmermannAHerbertWClinical effectiveness and safety of powered exoskeleton-assisted walking in patients with spinal cord injury: systematic review with meta-analysisMed Devices20169455466
- DijkersMPAkersKGalenSSPatzerDEVuPTLetter to the editor regarding “Clinical effectiveness and safety of powered exoskeleton-assisted walking in patients with spinal cord injury: systematic review with meta-analysis”Med Devices20169419421
- Contreras-VidalJLA BhagatNBrantleyJPowered exoskeletons for bipedal locomotion after spinal cord injuryJ Neural Eng201613331001
- BensonIHartKTusslerDvan MiddendorpJJLower-limb exoskeletons for individuals with chronic spinal cord injury: findings from a feasibility studyClin Rehabil2016301738425761635
- Kolakowsky-HaynerSASafety and Feasibility of using the Ekso™ bionic exoskeleton to aid ambulation after spinal cord injuryJ Spine2013S43
- YangAAsselinPKnezevicSKornfeldSSpungenAAssessment of in-hospital walking velocity and level of assistance in a powered exoskeleton in persons with spinal cord injuryTop Spinal Cord Inj Rehabil201521210010926364279
- Norouzi-GheidariNArchambaultPSFungJEffects of robot-assisted therapy on stroke rehabilitation in upper limbs: Systematic review and meta-analysis of the literatureJ Rehabil Res Dev201249447922773253
- Food and Drug Administration21 CFR 890.3480 Available from: http://www.ecfr.gov/cgi-bin/retrieveECFR?gp=1&SID=b37c513bf62af085f32e31c0a1310078&ty=HTML&h=L&mc=true&r=SECTION&n=se21.8.890_13480Accessed November 8, 2016
- Food and Drug Administration§890. 1925: Isokinetic testing and evaluation system Available from: http://www.ecfr.gov/cgi-bin/text-idx?SID=a01aed88a2ea548118ec143023ae264d&mc=true&node=se21.8.890_11925&rgn=div8Accessed November 22, 2016
- Food and Drug AdministrationEvaluation of automatic class III designation (De Novo) for Argo Rewalk™2014 Available from: https://www.accessdata.fda.gov/cdrh_docs/pdf13/DEN130034.pdfAccessed September 1, 2016
- Food and Drug AdministrationIndego 510(k) summary2016 Available from: http://www.accessdata.fda.gov/cdrh_docs/pdf15/K152416.pdfAccessed September 1, 2016
- Food and Drug AdministrationEkso classification Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn_template.cfm?id=k143690Accessed August 30, 2016
- SchmulandCValue-added medical-device risk managementIEEE Trans Device Mater Reliab200553488493
- ISO 14971:2007International Organization for Standardization2007
- Food and Drug AdministrationProduct classification: powered exoskeleton Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?ID=PHLAccessed August 28, 2016
- Food and Drug AdministrationEkso 510(k)2016 Available from: http://www.accessdata.fda.gov/cdrh_docs/pdf14/K143690.pdfAccessed September 1, 2016
- FedericiSMeloniFBracalentiMDe FilippisMLThe effectiveness of powered, active lower limb exoskeletons in neurorehabilitation: a systematic reviewNeuroRehabilitation201537332134026529583
- TuckerMROlivierJPagelAControl strategies for active lower extremity prosthetics and orthotics: a reviewJ Neuroeng Rehabil2015121125557982
- Food and Drug Administration522 Postmarket Surveillance Studies Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMA/pss.cfm?t_id=347&c_id=2675Accessed December 7, 2016
- Food and Drug AdministrationWarning Letter to ARGO Medical Technologies2015 Available from: http://www.fda.gov/ICECI/EnforcementActions/WamingLetters/2015/default.htmAccessed September 1, 2016
- ReWalk Announces Launch of 522 Post-Market Study with Stanford University School of Medicine as Lead Investigator Site http://rewalk.com/rewalk-announces-launch-of-522-post-market-study-with-stan-ford-university-school-of-medicine-as-lead-investigator-site/Accessed August 26, 2016
- AsselinPKnezevicSKornfeldSHeart rate and oxygen demand of powered exoskeleton-assisted walking in persons with paraplegiaJ Rehabil Res Dev201552214715826230182
- EsquenaziATalatyMPackelASaulinoMThe ReWalk powered exoskeleton to restore ambulatory function to individuals with thoracic-level motor-complete spinal cord injuryAm J Phys Med Rehabil2012911191192123085703
- FinebergDBAsselinPHarelNYVertical ground reaction force-based analysis of powered exoskeleton-assisted walking in persons with motor-complete paraplegiaJ Spinal Cord Med20133631332123820147
- Food and Drug AdministrationMAUDE Adverse Event Report: ReWalk Powered Exoskeleton2013 Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/detail.cfm?mdrfoi_id=4645875Accessed April 1, 2016
- FarrisRJQuinteroHAMurraySAHaKHHartiganCGoldfarbMA preliminary assessment of legged mobility provided by a lower limb exoskeleton for persons with paraplegiaIEEE Trans Neural Syst Rehabil Eng201422348249023797285
- EvansNHartiganCKandilakisCPharoEClessonIAcute cardiorespiratory and metabolic responses during exoskeleton-assisted walking overground among persons with chronic spinal cord injuryTop Spinal Cord Inj Rehabil201521212213226364281
- HartiganCKandilakisCDalleySMobility outcomes following five training sessions with a powered exoskeletonTop Spinal Cord Inj Rehabil2015212939926364278
- KozlowskiAJBryceTNDijkersMPTime and effort required by persons with spinal cord injury to learn to use a powered exoskeleton for assisted walkingTop Spinal Cord Inj Rehabil201521211012126364280
- SalePRussoEFRussoMEffects on mobility training and de-adaptations in subjects with spinal cord injury due to a wearable robot: a preliminary reportBMC Neurol20161611226818847
- KresslerJThomasCKField-FoteECUnderstanding therapeutic benefits of overground bionic ambulation: exploratory case series in persons with chronic, complete spinal cord injuryArch Phys Med Rehabil201495101878188724845221
- GadPNGerasimenkoYPZdunowskiSIron “ElectriRx” man: overground stepping in an exoskeleton combined with noninvasive spinal cord stimulation after paralysisConf Proc IEEE Eng Med Biol Soc201520151124112726736463
- StrausserKAKazerooniHThe development and testing of a human machine interface for a mobile medical exoskeleton. Presented at: Intelligent Robots and Systems (IROS)IEEE/RSJ International ConferenceSeptember 25–30, 2011San Francisco, CA, USA
- Contreras-VidalJLKilicarslanAHuangHHuman-centered design of wearable neuroprostheses and exoskeletonsAI Magazine20153641222
- The Daily BeastThe mechanical exoskeleton shaping the future of health care Available from: http://www.thedailybeast.com/articles/2015/08/03/the-mechanical-exoskeleton-shaping-the-future-of-health-care.htmlAccessed August 29, 2016
- WallABorgJPalmcrantzSClinical application of the Hybrid Assistive Limb (HAL) for gait training – a systematic reviewFront Syst Neurosci201594825859191
- UebaTHamadaOOgataTInoueTShiotaESankaiYFeasibility and safety of acute phase rehabilitation after stroke using the hybrid assistive limb robot suitNeurol Med Chir (Tokyo)201353528729023708218
- NilssonAVreedeKSHäglundVKawamotoHSankaiYBorgJGait training early after stroke with a new exoskeleton – the hybrid assistive limb: a study of safety and feasibilityJ Neuroeng Rehabil2014119224890413
- Food and Drug AdministrationMAUDE adverse event report: REX BIONICS LTD REX REHAB. MAUDE Database2016 Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/detail.cfm?mdrfoiid=5793129Accessed October 10, 2016
- KannusPSievanenHPalvanenMJärvinenTParkkariJPrevention of falls and consequent injuries in elderly peopleLancet200536695001885189316310556
- Tamez-duqueJCobian-ugaldeRKilicarslanAVenkatakrishnanASotoRContreras-vidalJLReal-time strap pressure sensor system for powered exoskeletonsSensors (Basel)20151524550456325690551
- Food and Drug AdministrationApplying human factors and usability engineering to medical devices2016 Available from: http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm259760.pdf
- DolbowDRGorgeyASDanielsJAAdlerRAMooreJRGaterDRJrThe effects of spinal cord injury and exercise on bone mass: a literature reviewNeuroRehabilitation201129326126922142760
- SchwartzIMeinerZRobotic-assisted gait training in neurological patients: who may benefit?Ann Biomed Eng20154351260126925724733
- DonatiARCShokurSMoryaELong-term training with a brain-machine interface-based gait protocol induces partial neurological recovery in paraplegic patientsSci Rep201663038327513629
- del-AmaAJGil-AgudoAPonsJLMorenoJCHybrid FES-robot cooperative control of ambulatory gait rehabilitation exoskeletonJ Neuroeng Rehabil2014112724594302
- Food and Drug AdministrationUse of International Standard ISO 10993-1, “Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process”2016 Available from: http://www.fda.gov/downloads/medicaldevices/deviceregulationand-guidance/guidancedocuments/ucm348890.pdf
- Food and Drug AdministrationFactors to consider when making benefit-risk determinations in medical device premarket approval and de novo classificationsFed Regist2016 Available from: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM517504.pdf
- Estec ESAMINDWALKER : a brain controlled lower limbs exoskeleton for rehabilitation. Potential applications to spacePresented at: ASTRA 2011April 12–14, 2011Noordwick, The Netherlands
- IEEE SpectrumExoskeletons are on the March Available from: http://spectrum.ieee.org/robotics/medical-robots/exoskeletons-are-on-the-marchAccessed September 1, 2016
- BuesingCFischGO’DonnellMEffects of a wearable exoskeleton stride management assist system (SMA®) on spatiotemporal gait characteristics in individuals after stroke: a randomized controlled trialJ Neuroeng Rehabil2015126926289955
- Chang HyunROHWearable robot and method for controlling the same2015 Available from: https://patents.google.com/patent/US20150134080A1/en
- Panasonic Newsroom globalNo more power barriers with Panasonic assist robots Available from: http://news.panasonic.com/global/stories/2016/44969.htmlAccessed September 1, 2016
- Food and Drug AdministrationMedical devices; physical medicine devices; classification of the powered lower extremity exoskeleton; republication. Final order; republicationFed Regist201580852522625230 Available from: http://www.ncbi.nlm.nih.gov/pubmed/2598547825985478
- Pharmaceuticals and Medical Devices AgencyPharmaceuticals and medical devices agency2015 http://www.pmda.go.jp/review-services/drug-reviews/review-information/devices/0025.htmlAccessed September 22, 2016
- TamuraAUnderstanding Japanese medical device requirements2011 Available from: https://www.pmda.go.jp/files/000164006.pdfAccessed September 22, 2016
- Food and Drug AdministrationQuality System (QS) Regulation/Medical Device Good Manufacturing Practices Available from: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/PostmarketRequirements/QualitySystemsRegulations/Accessed September 22, 2016
- International Electrotechnical CommissionJWG 36: Medical robots for rehabilitation Available from: http://www.iec.ch/dyn/www/f?p=103:14:0::::FSP_ORG_ID,FSP_LANG_ID:12533,25Accessed November 11, 2016
- ParentSMac-ThiongJ-MRoy-BeaudryMSosaJFLabelleHSpinal cord injury in the pediatric population: a systematic review of the literatureJ Neurotrauma20112881515152421501096
- BlairEWatsonLEpidemiology of cerebral palsySemin Fetal Neonatal Med200611211712516338186
- LernerZDamianoDParkH-SGravunderABuleaTA robotic exoskeleton for treatment of crouch gait in children with cerebral palsy: design and initial applicationIEEE Trans Neural Syst Rehabil Eng Epub2016727
- RossiSColazzaAPetrarcaMCastelliECappaPKrebsHIFeasibility study of a wearable exoskeleton for children: is the gait altered by adding masses on lower limbs?PLoS One20138919
- GilliauxMRendersADispaDUpper limb robot-assisted therapy in cerebral palsy: a single-blind randomized controlled trialNeurorehabil Neural Repair201529218319225015650
- SamadiBAchicheSParentABallazLChouinardURaisonMCustom sizing of lower limb exoskeleton actuators using gait dynamic modelling of children with cerebral palsyComput Methods Biomech Biomed Engin201619141519152426980164