Abstract
This review summarizes Phase III clinical trial data available for fingolimod. The main purpose is to evaluate the benefit-risk profile of fingolimod, the first oral compound available for treatment of multiple sclerosis (MS) and just recently approved by the European authorities. The authors place this evaluation in the context of the known safety and efficacy profile of established compounds for therapy of MS to outline the current and future potential of fingolimod. The authors conclude that only long-term safety data from post-marketing surveillance plans, together with additional head-to-head studies, would allow evidence-based treatment decisions. Furthermore, risk-profile analyses including patient history, exposure data to certain pathogens, and genetic analyses may potentially help to choose the right drug for individual patients in the future. Until these approaches toward an individualized medicine have been validated, treatment decisions for one or the other compound will have to be based partly on class IV evidence. Therefore, a close dialog with the well-informed patient, secured by effective risk mitigation plans, is required to choose the compound.
Introduction
Multiple sclerosis (MS) is a chronic and debilitating immune-mediated disease of the central nervous system (CNS). Recent epidemiological data supports the established view that the incidence of MS peaks at about 30 years of age and that it is a disease with a positive female-to-male ratio.Citation1 Widespread axonal pathology has already been reported in early stages of MS, including clinically isolated syndrome (CIS).Citation2,Citation3 Thus, treatment initiation at an early stage in the disease seems crucial; this is supported by positive clinical trial data for first-line disease-modifying drugs (DMDs) in delaying conversion of CIS to MS in patients.Citation4 Paraclinical surrogate parameters are currently under investigation to predict disease progression and conversion from CIS to MS, including magnetic resonance imaging (MRI) criteria at initial presentation and the presence of oligoclonal bands or levels of CXCL-13 in cerebrospinal fluid (CSF).Citation3–Citation5 However, these surrogates are not yet able to predict the future grade of disability on an individual basis with certainty.Citation6 In the European Union (EU), the three different interferon-beta (IFNβ) formulations available, as well as glatiramer acetate (GA), are considered as first-line treatments based on class I evidence for similar efficacy and a positive safety profile.Citation7 Recent head-to-head studies did not detect differences in the primary endpoints between IFNβ and GA. When selecting a treatment from among these injectable drugs, individual decisions will be based mainly on the preferred route of application (subcutaneous or intramuscular [IM]) and the individual tolerability of the compound used.
Until recently, natalizumab was the established second choice for patients failing first-line DMDs. In addition, it has been approved as a primary treatment for patients with highly active relapsing-remitting MS (RRMS). Natalizumab is a humanized monoclonal IgG4 antibody, designed to target the alpha-4 integrin, and is most relevant for leukocyte migration across endothelial barriers.Citation8,Citation9 Class I evidence is available for superiority regarding clinical outcome measures of natalizumab compared with placebo and for the combination of natalizumab and IM IFNβ-1a compared with IM IFNβ-1a alone.Citation10,Citation11 However, no class I evidence is available directly comparing efficacy of natalizumab with first-line DMDs. When looking at the efficacy of natalizumab and first-line DMDs across different clinical trials on clinical outcome measures such as the annualized relapse rate (ARR), the data available suggest superiority of natalizumab; this is supported by clinical experience (class IV evidence).Citation4 Therefore, restricted approval as a second-line treatment is not explained by inferiority compared with first-line DMDs or by study design of trials relevant for approval; it is explained mainly by the occurrence of cases of progressive multifocal leukoencephalopathy (PML) in around 1/1000 patients treated with natalizumab.Citation12–Citation15 The risk of this potentially lethal or highly disabling adverse event (AE) increases to up to 8.1/1000 patients (95% confidence interval: 5.4–11.6/1000 patients) among a subset of patients with prior immunosuppressant treatment who have been treated with natalizumab for more than 2 years, and who show evidence of JC virus exposure as assessed by JC virus serology.Citation16
Furthermore, mitoxantrone was licensed in 2002 for treatment of patients with secondary progressive MS or where progressive relapsing MS is failing or not tolerating previous immunomodulatory therapy.Citation7 However, the risk of cardiomyopathy and secondary leukemia, overall observed in 1/250 to 1/800 patients treated, limits the use of this drug and makes it a third-line treatment for patients with relapsing forms of MS.Citation7
On September 22, 2010, the US Food and Drug Administration approved fingolimod 0.5 mg, a sphingosine 1-phosphate (S1P) receptor modulator, as the first oral medication for treatment of RRMS.Citation17 In March 2011, the European Medicines Agency (EMA) approved fingolimod in Europe. However, the European authorities restricted the approval to patients with high disease activity despite treatment with IFNβ (nonresponder) and to patients with rapidly evolving RRMS.Citation18 The EMA defined nonresponders to IFNβ as those having failed to respond to “normally at least one year of treatment of IFNβ.”Citation18 According to EMA recommendations, these patients should have experienced a minimum of one relapse in the previous year while on IFNβ, and at least nine T2-hyperintense lesions in cranial MRI or at least one gadolinium-enhancing lesion, or should have had an “unchanged or increased relapse rate or ongoing severe relapses, as compared to the previous year.”Citation18 The second group of patients with rapidly evolving severe RRMS was defined “by 2 or more disabling relapses in 1 year, and with one or more Gadolinium enhancing lesions on brain MRI or a significant increase in T2 lesion load as compared with a previous recent MRI.”Citation18
Thus, a restricted approval in the EU is given for two compounds, natalizumab and fingolimod, available for the same indication: first-line therapy for highly active MS or second-line therapy in patients not tolerating or not responding to first-line DMDs. In this review, the authors give an overview of clinical trial efficacy and safety data available that is relevant for the approval of fingolimod by the US and the European authorities; the authors then evaluate the current and future potential of fingolimod within the treatment algorithms for MS.
Fingolimod and its mode of action
The molecular structure of fingolimod is shown in . Fingolimod is derived from myriocin, which has been primarily isolated from Isaria sinclairii, a fungus used in traditional herbal medicine.Citation19 Fingolimod is predominately metabolized in the liver by sphingosine kinase to the active metabolite fingolimod-phosphate.Citation20 Knock-out studies in S1P receptor-deficient mice suggest S1P receptors as key receptors relevant to the therapeutic effects in experimental autoimmune encephalitis, an animal model of MS. Accordingly, fingolimod-phosphate binds to four of the five known S1P receptors: S1P1, S1P3, S1P4, and S1P5. Expression of these receptors varies throughout tissues, being differently expressed on lymphocytes, in various different peripheral organs, and cells of the CNS or the peripheral nervous system.Citation21 Coupling to G proteins, S1P receptors regulate complex processes such as growth and survival, cell motility, cell invasion, angiogenesis, and trafficking of immune cells. Upon binding (eg, to the membrane-bound cell surface S1P1 receptor) fingolimod-phosphate induces internalization of the receptor, which then leads to a sustained downregulation of the receptor on the gene expression level.Citation22,Citation23 In lymphatic tissue, fingolimod-phosphate thereby blocks the capacity of certain subpopulations of lymphocytes to egress lymph nodes, causing redistribution, rather than depletion, of lymphocytes. The redistribution of certain lymphocyte subpopulations is considered to reduce the infiltration of pathogenic cells into the CNS and to mediate the main therapeutic effect on the clinical course of MS. Naïve and memory T-cells, expressing the chemokine receptor CCR7 on their surface, are preferentially retained. In contrast, effector memory T-cells, which are capable of downregulating their surface CCR7, are less dependent on S1P signaling and thus are considered to preserve important immunological functions for antiviral or antineoplastic defense.Citation21 In addition to reducing infiltration of pathogenic cells into the CNS, a direct neuroprotective and regenerative potential of fingolimod has been extensively discussed in recent literature. Fingolimod-phosphate is highly protein bound and is able to cross the blood–brain barrier. Thus, it could potentially directly interact with receptors on neurons and glia cells, and mediate the neuroregenerative effects proposed to be associated with fingolimod treatment.Citation22 However, many of these immunological data derive from rodent models,Citation24,Citation25 and expression of S1P receptors in a variety of different tissues may mediate not only its therapeutic but also its adverse effects so far observed in clinical trials. Overall, the lymphocyte count decreases to approximately 60% of baseline within 4–6 hours after the first dose, with normal counts usually reached within 1–2 months after treatment discontinuation.Citation20
Figure 1 The chemical structure of fingolimod (2-amino-2-[2-(4-octylphenyl)ethyl] propan-1,3-diol hydrochloride).
![Figure 1 The chemical structure of fingolimod (2-amino-2-[2-(4-octylphenyl)ethyl] propan-1,3-diol hydrochloride).](/cms/asset/a38ff5ef-133e-4b3d-bdd7-b1a696b4a67a/dndt_a_10481_f0001_b.jpg)
Pharmacokinetics of fingolimod
The oral bioavailability of fingolimod is 93% and the average terminal half-life is 6–9 days.Citation20 As fingolimod is primarily metabolized by hepatic enzymes of the CYP family (mainly CYP4F2), inhibitors or inducers of these isozymes alter the exposure of fingolimod or fingolimod-phosphate. Patients with severe hepatic dysfunction should be closely monitored when receiving fingolimod as the risk of adverse reactions is potentially higher.Citation20
Efficacy of fingolimod in Phase III clinical trials
Fingolimod vs placebo
Study design
Patients 18–55 years of age with RRMSCitation26 and a score from 0 to 5.5 on the Expanded Disability Status Scale (EDSS)Citation27 were eligible for the FTY720 Research Evaluating Effects of Daily Oral therapy in Multiple Sclerosis (FREEDOMS) double-blind clinical trial, which aimed to assess the efficacy of two different doses of oral fingolimod compared with placebo for 24 months.Citation28 Prior to randomization, patients were required to have an active course of disease (one or more relapses in the previous year or two or more in the previous 2 years) and IFNβ or GA was to be stopped at least 3 months before the trial. Randomization was conducted in a 1:1:1 ratio to the high-dose (fingolimod 1.25 mg) or the low-dose (fingolimod 0.5 mg) treatment group or to placebo, each administered once daily. The primary clinical outcome measure was the ARR, defined as the number of confirmed relapses per year. A confirmed relapse needed to be associated with an increase of at least 0.5 points in the EDSS score, 1 point in each of two EDSS functional system (FS) scores, or 2 points in one EDSS FS score. The key secondary clinical outcome measure was time to confirmed disability progression after 3 months, as measured in an increase of 1 point in the EDSS score (or 0.5 points if the baseline EDSS score was 5.5) confirmed after 3 months.Citation28 In addition, clinical endpoints (time to first relapse, time to disability progression after 6 months, changes in the EDSS score and the Multiple Sclerosis Functional Composite z-score between baseline and 24 months) and paraclinical endpoints (number of gadolinium- enhancing lesions, proportion of patients free from gadolinium-enhancing lesions, number of new or enlarged lesions on T2-weighted MRI scans, proportion of patients free from new or enlarged lesions on T2-weighted scans, volumes of hyperintense lesions on T2-weighted scans and hypointense lesions on T1-weighted scans, change in brain volume between baseline and 24 months) were assessed.Citation28
Study results
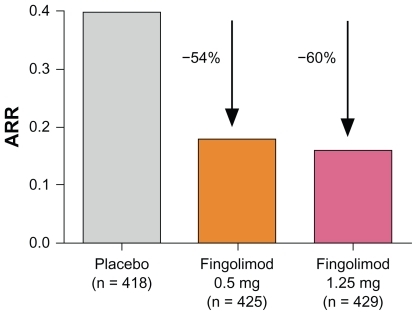
In a total of 1033 patients completing the 24-month study, all clinical and paraclinical endpoints demonstrated superiority for both the low and the high dose of fingolimod over placebo. No significant differences in efficacy were observed when comparing the two different doses of fingolimod. Interestingly, the overall ARR observed in the fingolimod Phase III clinical trials was low compared with studies relevant for approval of interferons, for example. Thus, although the absolute reduction of the ARR appeared to be low, the relative reduction of the ARR (primary endpoint: 0.18 for fingolimod 0.5 mg; 0.16 for fingolimod 1.25 mg; 0.40 for placebo) compared with placebo was 54% and 60%, respectively (), independent of previous treatment with other DMDs.Citation28 In addition, the percentage of patients without relapse was around 70% in the fingolimod 0.5 mg group and 75% in the fingolimod 1.25 mg group, compared with 46% in the placebo group. The key secondary outcome measure, the time to disability progression, confirmed after 3 months, demonstrated a hazard ratio for the risk of disability progression of 0.70 for the 0.5 mg dose and 0.68 for the 1.25 mg dose compared with placebo, showing that fingolimod may be able to delay disability progression. All paraclinical outcome measures were in favor of both of the treatment groups compared with placebo, including surrogates for inflammatory activity or scar formation on MRI (eg, number of contrast-enhancing lesions or number of new or enlarged lesions on T2-weighted scans), as well as measures for tissue loss (eg, change in volume of hypointense lesions on T1-weighted images or change in brain volume).Citation28
Figure 2 Annualized relapse rate (ARR) from baseline to month 24 in the FTY720 Research Evaluating Effects of Daily Oral therapy in Multiple Sclerosis (FREEDOMS) study.Citation28
Fingolimod vs IFNβ
Study design
A 12-month, multicenter, randomized, double-blind, head-to- head core study (Trial Assessing Injectable Interferon vs FTY720 Oral in Relapsing-Remitting Multiple Sclerosis [TRANSFORMS]) assessed the efficacy and safety of fingolimod compared with IM IFNβ-1a.Citation29 Patients were randomized to receive either the high or the low dose of oral fingolimod once daily or IM IFNβ-1a once a week as active comparator. The study was performed in a double-dummy fashion, with all patients receiving matching placebo in addition to the active treatment to ensure blinding. In a 12-month extension of the TRANSFORMS study, patients originally assigned to receive the high or the low dose of fingolimod continued with the same treatment, whereas patients originally receiving IFNβ-1a were re-randomized to either the high or the low dose of fingolimod.Citation30 Inclusion criteria and definitions of outcome measures were similar to those described for the FREEDOMS study. The primary endpoint was the ARR. Time to confirmed disability progression and the number of new or enlarged hyperintense lesions on T2-weighted MRI scans were key secondary outcome measures.Citation29
Study results
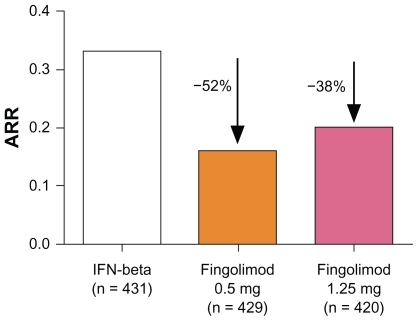
A total number of 1153 patients completed the core study, whereas 882 patients completed the total of 24 months on treatment including the extension phase. The primary outcome measure after 12 months showed superiority of both doses of fingolimod compared with IM IFNβ-1a: there was a significantly greater reduction in the ARR (primary endpoint: 0.16 for fingolimod 0.5 mg; 0.20 for fingolimod 1.25 mg; 0.33 for IM IFNβ-1a), with a relative reduction compared with IFNβ-1a of 52% and 38%, respectively ().Citation29 Furthermore, patients initially randomized to receive IM IFNβ-1a in the core study demonstrated a lower ARR after switching to fingolimod (IFNβ-1a to fingolimod 0.5 mg: ARR 0.31 [first year] vs 0.22 [second year]; IFNβ-1a to fingolimod 1.25 mg: ARR 0.29 [first year] vs 0.18 [second year]). The percentage of patients without relapse was 83% after 12 months in patients treated with fingolimod 0.5 mg, 80% for the fingolimod 1.25 mg group, and 69% in the placebo group. In contrast to the FREEDOMS study, the TRANSFORMS study failed to show beneficial effects on disability progression. As TRANSFORMS was not designed to primarily demonstrate the beneficial effect of fingolimod on disability progression, this discrepancy could potentially be explained by the TRANSFORMS study being under-powered with an active comparator and a very low proportion of patients with disability progression overall in the study cohort. Regarding MRI outcome measures, patients in the two fingolimod groups demonstrated fewer new or enlarged hyperintense lesions on T2-weighted images at month 12 (mean: 1.7 for fingolimod 0.5 mg; 1.5 for fingolimod 1.25 mg; 2.6 for placebo). Whereas the mean percent reduction in brain volume from baseline to 12 months was significantly reduced for the fingolimod groups, changes in the volume of lesions on unenhanced T2- or T1-weighted images at 12 months were similar among the different study arms.Citation29 In this respect the results again differ from the FREEDOMS study data, most likely attributed to a lack of power as discussed for the disability progression.
Figure 3 Annualized relapse rate (ARR) from baseline to month 12 in the Trial Assessing Injectable Interferon vs FTY720 Oral in Relapsing-Remitting Multiple Sclerosis (TRANSFORMS) study.Citation29
Ongoing phase III and IV clinical trials
Currently, the search on ClinicalTrials.gov for the terms “fingolimod AND multiple sclerosis” lists 14 ongoing or planned clinical trials ().Citation31 Among these, the extension trials, as well as post-marketing surveillance studies including a pregnancy registry, focus on long-term efficacy and safety of the approved low dose of fingolimod. The FTY720 in Patients With Primary Progressive Multiple Sclerosis study evaluates the efficacy of fingolimod in patients with primary progressive MS, and additional studies focus on biomarker changes during fingolimod treatment, the immune response during vaccination, or the beneficial effect of fingolimod on cognitive function compared with IFNß-1a ().
Table 1 Ongoing Phase II–IV clinical trials: summary of ClinicalTrials.gov search results for the terms “fingolimod AND multiple sclerosis,” selecting only ongoing or planned studiesCitation31
Safety and tolerability of fingolimod
Most of the safety data available derive from the two completed Phase III clinical trials, FREEDOMS and TRANSFORMS. Upper respiratory tract infections, headache, fatigue, nausea, and gastrointestinal dysfunction were among the most frequently observed AEs.Citation28,Citation29 As in previous clinical trial experience, dose-dependent decreases in the heart rate commencing 1 hour after the first dose of fingolimod were observed; thus, bradycardia and atrioventricular block were more frequently reported in patients receiving fingolimod than in controls.Citation28,Citation29 Therefore, according to the manufacturer’s recommendations, patients are required to be observed for signs and symptoms of bradycardia for at least 6 hours after administration of the first dose. However, a recent report of a patient with MS who developed a delayed 7.5-second asystole and sustained bradycardia 21 hours after the first dose of fingolimod stresses the importance of cardiac monitoring during treatment initiation.Citation32 This is of particular importance if additional risks for cardiac conduction abnormalities exist, such as risperidone comedication in the latter case mentioned.Citation32 Within the clinical trials, the therapeutic low dose of fingolimod caused an increase of approximately 2 mmHg systolic and 1 mmHg diastolic blood pressure. Thus, blood pressure should be monitored throughout treatment with fingolimod.
Three deaths occurred in the FREEDOMS study: two in the placebo group (pulmonary embolism, traffic accident) and one in the high-dose fingolimod group (suicide). However, in the TRANSFORMS study, the two deaths observed both occurred in the high-dose fingolimod group: one due to disseminated primary varicella zoster infection (with corticosteroids as concomitant medication) and the other due to herpes simplex encephalitis. Two additional patients of this study arm died after the study (aspiration pneumonia, metastatic breast cancer). Infections were reported equally throughout the groups (69%–72% for FREEDOMS, 51%–53% for TRANSFORMS), with serious infections in 1.6%–2.6% and 0.2%–1.7% of patients, respectively. Bronchitis and pneumonia were more common in patients treated with fingolimod.Citation28,Citation29
Macular edema in patients treated with low-dose fingolimod occurred in 0.5% of patients in the TRANSFORMS study but did not occur for this dose in the FREEDOMS study. However, macular edema was more frequently observed in the high-dose fingolimod group (1% and 0.7%, respectively). Patients with diabetes mellitus or history of uveitis seem to be at increased risk. Macular edema improved or resolved with or without treatment in most of the patients when the drug was discontinued. Nevertheless, some patients had residual visual acuity loss even after resolution.Citation28,Citation29
Modest dose-dependent reductions in forced expiratory volume over 1 second (FEV1) and diffusing capacity of the lung for carbon monoxide were observed in patients treated with fingolimod, requiring spirometric evaluation if clinically indicated.
Increases in liver enzymes such as alanine aminotransferase were more frequently observed in the treatment groups of the FREEDOMS and TRANSFORMS studies (8.5% and 8%, respectively, for fingolimod 0.5 mg; 12.5% and 7%, respectively, for fingolimod 1.25 mg; 1.7% for placebo; 2% for IFNβ).Citation28,Citation29 In general, serum transaminase levels returned to normal within 2 months after treatment discontinuation.
Oral carcinogenicity studies in mice and rats with doses higher than the equivalent recommended human dose resulted in an increased incidence of malignant lymphomas.Citation20 Malignancies occurring in patients treated with fingolimod within the FREEDOMS and TRANSFORMS core studies were basal cell carcinoma (ten cases in total), breast cancer (five cases), malignant melanoma (two cases), and Bowen’s disease (one case); however, overall occurrence of malignancies did not differ between the fingolimod-treated patients and the control groups.
Data on teratogenicity and embryolethality in humans is limited, but fetal malformations including persistent truncus arteriosus and ventricular septal defect were observed in rats. Within the Phase II and III clinical trials, 47 pregnancies occurred and 15 healthy children were born.Citation33 However, one case of tibia malformation was reported, five spontaneous abortions occurred, and 14 patients decided to undergo elective abortion (among these, one case of tetralogy of Fallot was seen). Acrania was detected by ultrasonography in one of the twelve pregnancies reported as still ongoing.Citation33 Thus, due to the calculated elimination time, women of childbearing potential have to use effective contraception during and for 2 months after treatment discontinuation.Citation20 In addition, a pregnancy registry has been initiated to monitor outcomes of pregnancies in patients treated with fingolimod.
Conclusion and perspective: potential of oral fingolimod in treatment of MS
There are obvious reasons why the approval of fingolimod should be highly appreciated by MS patients and their physicians: First, in one head-to-head clinical trial,Citation29 fingolimod has been proven to be superior to approved first-line DMDs as regards efficacy. This seems to hold true also for patients initially treated with IM IFNβ-1a and then switched to fingolimod.Citation30 This is of particular relevance, as this will mimic clinical practice when fingolimod is given as a second-line treatment.Citation34 Second, oral administration could potentially further improve treatment acceptance in patients (no evident lifestyle restrictions, no further self-injections with local adverse effects or flu-like symptoms, no risk of developing antidrug antibodies). This could increase the compliance rate with an additional positive effect on long-term outcome in the individual patient.Citation35
However, as a new compound without any post-marketing experience from other fields or conditions, only long-term surveillance data after approval will allow us to judge the future position of fingolimod within the current treatment regimens for MS.
The following two key questions are still to be answered before the treating neurologist is able to make evidence- based decisions: (1) Has fingolimod a positive benefit-risk ratio compared with the established first-line DMDs such as IFNβ or GA? or, in other words, considering the situation in the US: Can fingolimod equally be offered to the young and otherwise healthy patients of childbearing potential? (2) Considering the approval in the EU: which compound to choose for patients with highly active MS or patients not tolerating or not responding to first-line DMDs, natalizumab, or fingolimod?
With regard to the first question, we learned from natalizumab that phase III clinical trials are unable to predict the risk of rare complications such as PML. Targeting lymphatic S1P1 receptor is considered to preferentially target naïve and central memory T-cells, sparing the effector memory T-cell population.Citation36 This could potentially result in a selective mode of action targeting autoimmunity and preserving key responses of the adaptive immunity relevant for viral defence.Citation37 However, these concepts and treatment rationales still have to be confirmed by data from the post-marketing period. The first data available on changes to the immune response and cellular composition of peripheral blood and CSF from patients in clinical trials indicate not only significant peripheral cell depletion in patients treated with fingolimod but also potential changes to the immune surveillance in the CNS; fingolimod seems to decrease the absolute cell counts in CSF compared with pretreatment values, but it also leads to a reversion of the CD4:CD8 ratio in the CSF,Citation38 a finding discussed previously as having potential relevance to the PML pathogenesis in MS patients treated with natalizumab.Citation39 Vaccination studies imply that these changes to the adaptive immunity may be of clinical relevance; although a recent study was not suggestive for relevant differences of the humoral immune response to influenza vaccines in patients treated with fingolimod compared with placebo,Citation40 data from studies involving healthy volunteers treated with fingolimod 0.5 mg assessing immunogenicity of keyhole limpet hemocyanin and the 23-valent pneumococcal polysaccharide vaccine indicate a decrease of antigen-specific IgM titers by 91% and 25%, respectively, and of IgG titers by 45% and 50%, respectively.Citation20 Therefore, the observation of serious herpes virus infectious complications in the TRANSFORMS study leading to death in two cases may not be by chance,Citation29 although causality to the treatment with fingolimod still has to be established. Furthermore, data on teratogenicity is limited and although effective contraceptive measures are mandatory during and for 2 months after treatment discontinuation, cases of pregnancy in this cohort of patients will most likely occur, with uncertain consequences for the unborn child. So, will the benefits outweigh the risks in a long-term perspective of several decades of treatment in the individual patient? The honest answer is this is not known yet. However, the established safety profile of long-term IFNß or GA treatment is known. Therefore, in the authors’ opinion, these issues will need to be discussed with the well-informed patient before considering fingolimod as a first-line choice in treatment of MS.
On the grounds of these considerations, the authorities in the EU restricted the approval of fingolimod to cases of highly active MS or patients not tolerating or not responding to first-line DMDs. However, this leads to a situation of two compounds approved in the EU for the same indication: natalizumab and fingolimod. Both have been proven to be highly effective, although direct comparative head-to-head clinical trials have not yet been undertaken. So, in regard to the second key question – which compound to choose for this indication, natalizumab or fingolimod? – again, the final decision will be based on benefit-risk considerations in the individual patient. The first prerequisite for an evidence-based decision for the individual is head-to-head clinical trials comparing the two compounds in efficacy. While these should be performed, they most likely will never be conducted. While only post-marketing experience will be able to demonstrate the safety profile of fingolimod, first attempts to stratify patients at risk of PML treated with natalizumab are on the way. These approaches may in the future lead toward an individualized medicine, where data from a patient’s history, genetic factors such as HLA status, functional measures for the innate and adaptive immunity, and exposure to specific infectious agents (such as JC virus serology) could be taken into account to find the best treatment for the individual patient. Until these promising future tools are validated and have been proven to be applicable in daily clinical practice, the correct individual treatment decisions can only be found in dialog with the well-informed patient, supported by high standards of post-marketing safety surveillance programs.
Acknowledgment
A European Committee for Treatment and Research in Multiple Sclerosis fellowship stipend to CW supported this work.
Disclosures
BCK and H-PH have received honoraria for lecturing, travel expenses for attending meetings, and financial support for research from Bayer HealthCare, Biogen Idec, Merck Serono, Novartis, sanofi-aventis, and Teva Neuroscience. AF-H has received unrestricted research support from Biogen Idec, Merck Serono, and Bayer Schering. OS and CW report no conflicts of interest in this work.
References
- Koch-HenriksenNSorensenPSThe changing demographic pattern of multiple sclerosis epidemiologyLancet Neurol20109552053220398859
- FilippiMBozzaliMRovarisMEvidence for widespread axonal damage at the earliest clinical stage of multiple sclerosisBrain2003126Pt 243343712538409
- CalabreseMRinaldiFMattisiIThe predictive value of gray matter atrophy in clinically isolated syndromesNeurology201177325726321613600
- HartungHPMontalbanXSorensenPSVermerschPOlssonTPrinciples of a new treatment algorithm in multiple sclerosisExpert Rev Neurother201111335136221375441
- KhademiMKockumIAnderssonMLCerebrospinal fluid CXCL13 in multiple sclerosis: a suggestive prognostic marker for the disease courseMult Scler201117333534321135023
- ChardDTGeurtsJJPredicting the development of multiple sclerosis: is gray matter a missing piece of the puzzle?Neurology201177321021121613598
- WiendlHToykaKVRieckmannPGoldRHartungHPHohlfeldRBasic and escalating immunomodulatory treatments in multiple sclerosis: current therapeutic recommendationsJ Neurol2008255101449146319005625
- RansohoffRMNatalizumab for multiple sclerosisN Engl J Med2007356252622262917582072
- RiceGPHartungHPCalabresiPAAnti-alpha4 integrin therapy for multiple sclerosis: mechanisms and rationaleNeurology20056481336134215851719
- RudickRAStuartWHCalabresiPANatalizumab plus interferon beta-1a for relapsing multiple sclerosisN Engl J Med2006354991192316510745
- PolmanCHO’ConnorPWHavrdovaEA randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosisN Engl J Med2006354989991016510744
- WarnkeCAdamsOGoldRProgressive multifocal leukoencephalopathy under natalizumab. Initial possibilities for risk stratification?Nervenarzt2011824475480 [German]21240604
- WarnkeCMengeTHartungHPNatalizumab and progressive multifocal leukoencephalopathy: what are the causal factors and can it be avoided?Arch Neurol201067892393020697042
- CliffordDBDe LucaASimpsonDMArendtGGiovannoniGNathANatalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 casesLancet Neurol20109443844620298967
- YousryTAMajorEORyschkewitschCEvaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathyN Engl J Med2006354992493316510746
- SandrockAHCRichmanSNatarajanARisk stratification for progressive multifocal leukoencephalopathy (PML) in MS patients: role of prior immunosuppressant use, natalizumab-treatment duration, and anti-JCV antibody statusPaper presented at: The 63rd Annual Meeting of the American Academy of Neurology2011 Apr 9–16Honolulu, Hawaii
- US Food and Drug Administration (FDA)FDA approves first oral drug to reduce MS relapses [press release]Sep 222010 Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm226755.htmAccessed June 1, 2011
- European Medicines Agency (EMA)Assessment report: Gilenya. Doc Ref: EMA/108602/2011February 172011 Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002202/WC500104529.pdfAccessed June 1, 2011
- FujitaTInoueKYamamotoSFungal metabolites: Part 11, a potent immunosuppressive activity found in Isaria sinclairii metaboliteJ Antibiot (Tokyo)19944722082158150717
- Novartis Pharmaceutical CorporationPrescribing information Available at: http://www.pharma.us.novartis.com/product/pi/pdf/gilenya.pdfAccessed June 1, 2011
- HlaTBrinkmannVSphingosine 1-phosphate (S1P): physiology and the effects of S1P receptor modulationNeurology2011768 Suppl 3S3821339489
- AktasOKuryPKieseierBHartungHPFingolimod is a potential novel therapy for multiple sclerosisNat Rev Neurol20106737338220551946
- MatloubianMLoCGCinamonGLymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1Nature2004427697235536014737169
- BrinkmannVDavisMDHeiseCEThe immune modulator FTY720 targets sphingosine 1-phosphate receptorsJ Biol Chem200227724214532145711967257
- MandalaSHajduRBergstromJAlteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonistsScience2002296556634634911923495
- PolmanCHReingoldSCEdanGDiagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”Ann Neurol200558684084616283615
- KurtzkeJFRating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS)Neurology19833311144414526685237
- KapposLRadueEWO’ConnorPA placebo-controlled trial of oral fingolimod in relapsing multiple sclerosisN Engl J Med2010362538740120089952
- CohenJABarkhofFComiGOral fingolimod or intramuscular interferon for relapsing multiple sclerosisN Engl J Med2010362540241520089954
- KhatriBBarkhofFComiGComparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: a randomised extension of the TRANSFORMS studyLancet Neurol201110652052921571593
- ClinicalTrials.gov [database online]Bethesda, MDUS National Institutes of Health Available at: http://clinicaltrials.gov/ct2/results?term=fingolimod+AND+multiple+sclerosisAccessed June 1, 2011
- EspinosaPSBergerJRDelayed fingolimod-associated asystoleMult Scler2011In press
- CollinsWFrancisGKorenLack of interaction between fingolimod (FTY720) and oral contraceptives, and pregnancy experience in the clinical program of fingolimod in multiple sclerosisPaper presented at: The 63rd Annual Meeting of the American Academy of Neurology2011 Apr 9–16Honolulu, Hawaii
- KieseierBCWiendlHTransforming multiple sclerosis trials into practical realityLancet Neurol201110649349421571592
- TurnerAPWilliamsRMSloanAPHaselkornJKInjection anxiety remains a long-term barrier to medication adherence in multiple sclerosisRehabil Psychol200954111612119618711
- PinschewerDDBrinkmannVMerklerDImpact of sphingosine 1-phosphate modulation on immune outcomesNeurology2011768 Suppl 3S151921339486
- MehlingMJohnsonTAAntelJKapposLBar-OrAClinical immunology of the sphingosine 1-phosphate receptor modulator fingolimod (FTY720) in multiple sclerosisNeurology2011768 Suppl 3S202721339487
- KowarikMCPellkoferHLCepokSDifferential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MSNeurology201176141214122121464424
- StüveOMarraCMBar-OrAAltered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosisArch Neurol200663101383138717030653
- MehlingMHilbertPFritzSAntigen-specific adaptive immune responses in fingolimod-treated multiple sclerosis patientsAnn Neurol201169240841321387383