 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Species differences in physiology and unique active human metabolites contribute to the limited predictive value of preclinical rodent models for many central nervous system (CNS) drugs. In order to explore possible drivers for this translational disconnect, we developed a computer model of a dopaminergic synapse that simulates the competition among three agents and their binding to pre- and postsynaptic receptors, based on the affinities for their targets and their actual concentrations. The model includes presynaptic autoreceptor effects on neurotransmitter release and modulation by presynaptic firing frequency and is calibrated with actual experimental data on free dopamine levels in the striatum of the rodent and the primate. Using this model, we simulated the postsynaptic dopamine D2 receptor activation levels of bifeprunox and aripiprazole, two relatively similar dopamine D2 receptor agonists. The results indicate a substantial difference in dose–response for the two compounds when applying primate calibration parameters as opposed to rodent calibration parameters. In addition, when introducing the major human and rodent metabolites of aripiprazole with their specific pharmacological activities, the model predicts that while bifeprunox would result in a higher postsynaptic D2 receptor antagonism in the rodent, aripiprazole would result in a higher D2 receptor antagonism in the primate model. Furthermore, only the highest dose of aripiprazole, but not bifeprunox, reaches postsynaptic functional D2 receptor antagonism similar to 4 mg haloperidol in the primate model. The model further identifies a limited optimal window of functionality for dopamine D2 receptor partial agonists. These results suggest that computer modeling of key CNS processes, using well-validated calibration paradigms, can increase the predictive value in the clinical setting of preclinical animal model outcomes.
Introduction
All currently marketed drugs in schizophrenia reduce the effects of the dopaminergic striatal pathological hyperactivity by either directly or indirectly reducing the activity of the postsynaptic D2 receptor.Citation1 Recently, partial D2 receptor agonists have been proposed as a therapeutic approach. Their clinical efficacy is hypothesized to be dependent both on the stimulation of presynaptic D2 autoreceptors that reduce synaptic dopamine (DA) release and on the substitution of a full agonist (dopamine) with a partial agonist, with both effects being functionally equivalent to a postsynaptic D2 receptor block. This is an interesting approach as recent imaging studies in an at-risk for mental state cohorts suggest a pre-synaptic pathology in schizophrenia patients.Citation2 Therefore, drugs acting on presynaptic D2 autoreceptors are supposed to act more closely to the actual pathology. Despite a substantial amount of research, only one partial agonist, aripiprazole, has shown successful clinical efficacy,Citation3 while the clinical effect of bifeprunox,Citation4 a partial agonist very similar to aripiprazole, is much more limited, leading to its premature clinical development halt. This suggests that it is difficult to identify the correct range of pharmacology parameters for partial agonism.
Because partial agonists at the postsynaptic receptor tend to counterbalance somewhat the effect at the presynaptic receptor, it is crucial to obtain an optimal balance between binding affinity and potency. In addition, increasing evidence suggests a substantial difference in quantitative biological parameters between rodents and primates in key brain areas important for psychiatric diseases. For instance, the coupling of presynaptic dopamine D2 autoreceptor to DA release in striatal synapses is lower in primatesCitation5 than that in rodents.Citation6 This difference can have important consequences for the effect of antipsychotics in humans in the treatment of schizophrenia.
Detailed in vitro studiesCitation7 show a small difference between aripiprazole and bifeprunox with regard to binding affinity and maximal partial agonist effect. The question arises whether this small difference could actually lead to substantially different functional antagonism at the postsynaptic D2 receptor, which drives a large part of the clinical response.Citation8 Because bifeprunox has been shown to be equivalent to, if not better than, aripiprazole in preclinical animal models,Citation9 the ability to estimate more quantitatively the global effect in a humanized situation becomes mandatory.
In order to explore these important questions, we developed a computer model of the striatal dopaminergic synapse, which includes the physiology of time-dependent presynaptic autoreceptor activation and its subsequent modulation of DA release. The model allows for different presynaptic firing regimens, the effect of facilitation and depression on DA release, and simulates the competition between DA and up to three different agents for the same binding site, dependent on their affinity and functionality. Our intention was to develop a model that focuses on the competition between endogenous DA and other agents and takes into account the modulation of free DA by firing frequencies and presynaptic mechanisms that is based on a large population of molecules. This article illustrates how calibration of this model with experimental data from rodents can lead to different conclusions, as compared to the calibration with experimental primate data, and the possible consequences with regard to clinical predictions.
Methods
Receptor competition model
The receptor competition model (see ) consists of a set of ordinary differential equations, describing the time-dependent competition of neurotransmitter and up to three different agents for a presynaptic and postsynaptic receptor, based on the relative affinities and concentrations of each of the different agents. The model runs for a maximum run time of 10,000 msec (10 sec). A detailed mathematical description of the model is given in the Appendix.
Figure 1 General description of the striatal dopaminergic synapse and receptor competition model. The model allows user-defined presynaptic firing patterns for neurotransmitter release and simulates the effect of presynaptic D2 autoreceptor negative feedback on presynaptic neurotransmitter release, facilitation and depression of synaptic release, the decay of DA in the cleft due to diffusion, transporters and enzymes, the competition between four agents (the neurotransmitter, up to two drugs, and a tracer), and the dynamics of kon/koff binding of each of these agents to their respective receptors using ordinary differential equations (see Appendix) at millisecond time resolution. The output is the time-dependent activation level of pre- and postsynaptic dopamine D2 receptors, the fraction of each agent bound to these receptors in the low- and high-affinity state, as well as the concentration of free DA in the cleft.
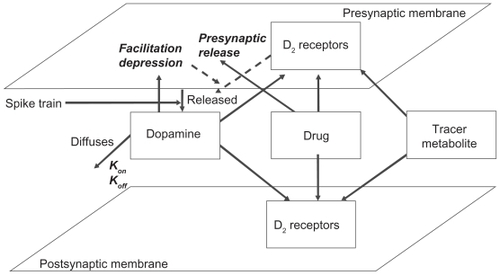
The activation of postsynaptic dopamine D2 receptor can be calculated based on the competition between DA and the different agents for both pre- and postsynaptic receptors over time. DA levels are determined by firing-related presynaptic DA release, its half-life in the synaptic cleft, and the activation of presynaptic D2 receptor. In addition, the model considers both high and low DA affinity receptor populations. All outcomes are averages over a 10-sec timescale in which realistic firing patterns of burst and tonic firing are presented to the system (see Appendix).
The neurotransmitter is released following a user-defined set of firing patterns. Dopaminergic neurons tend to switch between low-frequency tonic firing frequencies and high-frequency burst firing patterns.Citation10,Citation11
The presynaptic autoreceptor modulates the release of the neurotransmitter based on actual physiological processes and can be calibrated using experimental data on free DA. Because the D2 receptor uses a G-coupled protein pathway, a short time delay is introduced by basing the effect on how many receptors were bound 150 msec before release (see Appendix, Eq. 6). Fast cyclic voltammetry experiments in rats indeed suggest that the effect of autoreceptor activity on DA release is complete within a few hundred milliseconds.Citation6
The release dynamics can be described by a depression or facilitation mechanismCitation12 (see Appendix, Eq. 7). Instead of using a detailed model of internal Ca++ levels to determine DA release, we consider the facilitation and depression of DA release based on the amount of time elapsed since the previous firing using a phenomenological equation.
The parameters that govern presynaptic release in the rodent are then calibrated by correlating the effects of high doses of haloperidol with real-time striatal DA voltammetry data obtained in vivoCitation6 and by microdialysis using D2 agonistsCitation13 in the rat. We use previously published data on sulpiride in the marmoset to calibrate a more primatized dopaminergic synapseCitation5 (see Results).
Free DA removal from the cleft is modeled by an exponential decay. The decay rate is adjusted so as to correspond to the specific DA kinetics in various brain regions and to take into account the removal of DA from the cleft, not only via diffusion but also via transporter and/or enzyme mechanisms. In the striatum, DA is mostly taken up by the dopamine transporter (DAT) and is much less degraded by Catechol-O-methyl Transferase (COMT). The half-life of free DA from rodent striatal areas such as n. accumbensCitation14,Citation15 is in the range of 30–50 msec. We further assume that the binding on rate for DA and the compounds is diffusion limited and that this depends on the size and molecular weight of the molecules using the Stokes–Einstein equation. The fraction of receptors with bound DA, tracer, drug, or metabolite can be calculated by solving ordinary differential equations that describe the binding and unbinding processes (see Appendix, Eqs. 1–4). We use a value of 10 nM for the affinity of DA for the high-affinity signal-transducing D2 receptor.Citation16
The simulation is initiated by running for a period of 5 sec at the tonic firing rate of 4 Hz. The simulation then runs with a firing regime of 4 Hz for 2 sec, 40 Hz for 0.5 sec, 1 Hz for 5 sec, 4 Hz for 1 sec, and 80 Hz for 0.125 sec, all together for 8.625 sec, based on the fact that the subcortical areas are usually silent and only fire in bursts when salient stimuli are presented.Citation10 For calibration purposes, we apply the specific (artificial) firing frequencies, as outlined in the experimental papers.
Calibration of the striatal DA receptor in rodents and marmosets
We calibrated the presynaptic effect on striatal dopaminergic release using previously published, experimental, fast-cyclic voltametry in vivo rodent data with high doses of the D2 receptor antagonist, haloperidol.Citation6 These parameter settings were further verified with experimental data in wild-type and D3 KO mice from PD128907, a D2/D3 agonist.Citation13 For a calibration on the primate striatal dopaminergic synapse, we used in vitro fast-cyclic voltametry data from experiments on marmosets.Citation5
Rapid cyclic voltammetry was used in ratsCitation6 that were pretreated with RTI-76, an irreversible DAT inhibitor, to monitor the free caudate putamen and n. accumbens DA after a high dose of haloperidol (0.5 mg/kg, i.p.) and at different stimulation frequencies in the ventral tegmentum area (10–50 Hz for 2 sec). This haloperidol concentration corresponds to an extremely high D2 receptor occupancy well above 80%; for instance, tenfold lower concentrations (0.04–0.08 mg/kg) correspond to clinically relevant D2 receptor occupancies of 70%–80% in rodents.Citation17 Haloperidol increased free DA with a maximum of ninefold increase over the no-haloperidol condition at 30-Hz frequency. Blocking DA uptake with RTI76 ensured that only the effect on presynaptic D2 autoreceptors was measured. The forced VTA firing paradigm ensured that any feedback effect of D2 receptor modulation at the midbrain dopaminergic neurons was overruled and that the resulting change in free DA in the striatum was almost exclusively driven by the effect of haloperidol on presynaptic autoreceptors.
We reproduced the outcomes of this experiment by using 50 nM of haloperidol in the computer model (leading to over 90% block at the D2 receptor) at different firing frequencies and increasing the half-life of free DA threefold to mimic the block of DAT. This allowed us to identify the best settings for the presynaptic autoreceptor-related parameters. shows the outcome of the model and the experiment as a function of the firing frequency for the best parameters ().
Figure 2 A) Calibration of striatal dopaminergic synapse model in rodents (open shapes) using the experimental data (closed shapes) on the ratio of free DA levels with fast cyclic voltametry in vivo in the presence and the absence of high haloperidol treatment and forced dopaminergic firing and in the absence or presence of RTI-76 a potent DAT inhibitor.Citation6 Using these experimental data, we calibrated the appropriate values in Appendix Eq. 6 that led to the observed ratios of free DA in haloperidol treated versus nontreated situations in all four conditions. B) Calibration of striatal dopaminergic synapse model using the experimental data in marmosets on free DA levels with fast cyclic voltametry in vitro and forced dopaminergic firing.Citation5 There is an additional data point with 10-Hz firing with 1 μM of sulpiride, a specific D2 receptor antagonist. Using these experimental data, we calibrated the appropriate values in Appendix Eq. 6 that led to the model outcomes similar to experimentally observed free DA levels.
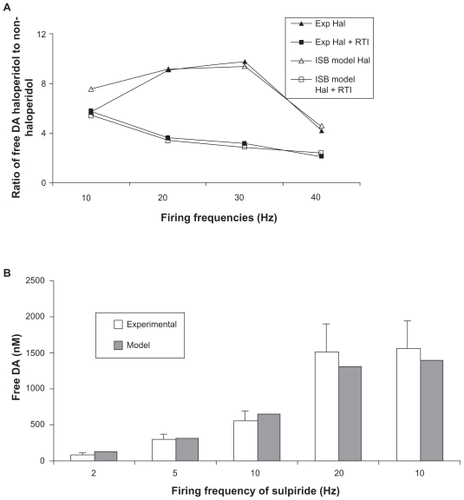
Table 1 Different parameters for fitting the striatal dopaminergic synapse to experimental rodent and primate data suggest that presynaptic autoreceptor coupling to dopaminergic release is weaker in the primate case, as well in absolute size as in the extent of temporal relationship
A different experiment in rodents documented the decrease of free DA in n. accumbens, measured by microdialysis, after application of a D2/D3 receptor agonist, PD128907.Citation13 These authors reported a maximal effect of 65% decrease in free DA, and this effect was similar in D3 KO mice, suggesting a predominant D2 receptor mechanism. Because free DA was quantified in this study using the slow detection process of microdialysis, we used a more normal in vivo firing pattern of tonic low-frequency firing (1–4 Hz) interspersed with rare high-frequency bursts of 40–80 Hz (see Methods). The reported affinity of PD128907 for the human D2 receptor is 340 nM,Citation18 but for the rodent D2 receptor it is 18 nM,Citation19 and the compound has a partial agonist effect of 25% relative to DA. Using the parameters derived from the haloperidol experiment, for a partial agonist effect of 0.25, the model results in a 70% decrease of free DA, comparable to the observed decrease of 65%.
In contrast, in marmosets, in vitro preparations of caudate putamenCitation5 and application of high concentrations of sulpiride, a specific presynaptic D2 autoreceptor antagonist, lead to only a 285% increase in free DA for ventromedial striatum when stimulated at 10 Hz, rather than the sixfold increase seen in rodents at the same firing frequencies. Although not directly comparable, these data already suggest that the coupling between presynaptic D2 receptor activation and DA release in nonhuman primates might be lower than that in the rodent case. The available data on free DA after different firing frequencies were used to determine the best-fit parameters for the primate striatal dopaminergic synapse. This leads to a different set of calibration parameters (see and ).
For a realistic set of burst and tonic firing frequencies, the average postsynaptic D2 receptor occupancy by DA is in the range of 30%–45% for the rodent calibration set and 35%–50% for the primate calibration set. The results of the simulations are therefore mechanistically determined.
In the following sections, we will illustrate the consequences of these differences.
Interpreting radiotracer experiments
The effect of different compounds in vivo depends on the dose, brain penetration, and target engagement of the compound. Therefore, the best way to compare the effects of compounds is to normalize the concentration of the compounds against the apparent D2 receptor occupancy displacement of specific radiotracers such as 11C-raclopride and 125I-IBZM. Ideally, one would like to quantify the binding of a specific radiotracer before and after neuroleptic treatment to correct for any individual baseline variability of the D2 receptor. Although this is possible with our model, it is usually difficult in the clinical setting; hence, many studies define a binding index (Eq. 1) compared to a normal control population.
where Am and Cerm are the specific radioactive signals in the region of interest, ie, in the striatum and cerebellum, respectively.
We used a radiotracer at a concentration of 1 pM. The Kd of raclopride, IBZM, and FLB457 for the D2 receptor are 1.3 nM, 0.6 nM, and 0.018 nM, respectively.Citation20,Citation21 We defined the apparent D2 receptor occupancy as
where Rdrug and Rcontrol are the receptor tracer occupancies, respectively, in the presence and the absence of the D2 receptor modulator. Because the density of radioactive-sensitive D2 receptor binding sites is about 100-fold lower in the cerebellum than that in the striatum,Citation22 our simulations indicate that applying Eq. 2 results in between 1% and 1.5% error, compared to the correct use of Eq. 1 (data not shown).
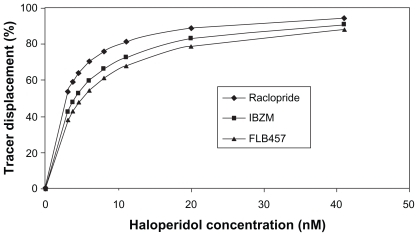
shows the effect of different radiotracers on the calculated receptor occupancy for a range of haloperidol doses. The observation that the calculated D2 receptor occupancy level decreases when higher-affinity tracers are used is in line with reported differences between apparent receptor occupancy measured with raclopride and IBZM in the same subjects.Citation23 This can be partially explained by the fact that the greater the affinity a tracer has for a receptor, the more difficult it is for the drug to compete with it and dislodge it.
Figure 3 Effect of tracer molecules with different affinities on the apparent D2 receptor occupancy at fixed haloperidol concentrations (nM). All tracers are applied at 1 pM concentration, and the appropriate affinity constants were used (the Kd of raclopride, IBZM, and FLB457 for the D2 receptor are 1.3 nM, 0.6 nM, and 0.018 nM, respectively). For each haloperidol concentration, the apparent D2 receptor occupancy level, measured by the radioactive tracers, decreases in the order raclopride > IBZM > FLB457, with the same order of their Kd binding affinity constants against D2 receptor.
Results
Effect of partial agonists and full antagonists on functional postsynaptic D2 receptor activation
In order to compare the effect of partial agonists with full antagonists on postsynaptic receptor physiology, we introduce the concept of functional D2 receptor antagonism, FA. This is calculated as the reduced postsynaptic D2 receptor activation, normalized to the no-drug situation:
where Actdrug and Actcontrol are actual postsynaptic D2 receptor activation levels in the presence and absence of drug, respectively.
These functional antagonist levels reflect the complex consequences of both an indirect effect of the partial agonist on presynaptic D2 receptor-mediated DA release and its direct effect on inhibiting the postsynaptic receptor. The rationale for using partial agonists in schizophrenia is partly based on the experimental observation that stimulating presynaptic D2 autoreceptors reduces free DA release, which is functionally equivalent to a postsynaptic D2 receptor block. In addition, a partial agonist at the postsynaptic receptor can compete with endogenous DA (a full agonist) to reduce the activation level. As these two processes require vastly different degrees of partial agonism, the ideal profile is a balance of affinity for the pre- and postsynaptic receptor and the degree of partial activation. We will now compare the effect of aripiprazole and bifeprunox on functional postsynaptic D2 receptor antagonism in function of the tracer-reported D2 receptor occupancy.
A recent studyCitation7 reports a low partial agonism for aripiprazole (25%), whereas the affinity of aripiprazole for both pre- and postsynaptic receptors is identical (Kd of 7.1 nM). To further complicate matters, in humans, the major metabolite dehydro-aripiprazole OPC14857 has a lower partial agonist effect (18% maximal effect) and a higher affinity (Kd = 3.5 nM); whereas, the major rat metabolite 4-hydrox-phenylpiperazine DM1451 is a full antagonist at the rodent receptor with a Kd of 1.5 nM.Citation24 In humans, this metabolite contributes to between 25% and 40% of the active moiety.Citation25 We assume an identical level of the metabolite in rodents.
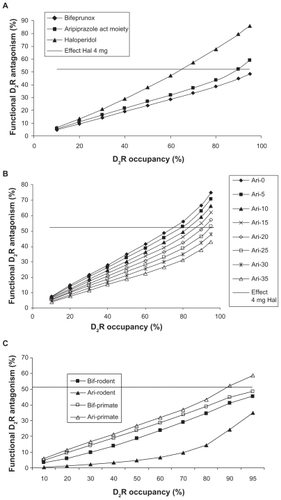
We simulated the effect of bifeprunox versus the active moiety of aripiprazole in comparison to haloperidol on the functional antagonism dose–responses under primate calibration parameters (). Bifeprunox has a slightly higher partial agonist effect (range 27%–35%, average 31%) than aripiprazole (average 25%).Citation7,Citation26–Citation28 We also indicated the postsynaptic D2 receptor antagonism of 4 mg haloperidol (corresponding to 65% D2 receptor occupancy), a dose which is known to have a minimal antipsychotic effect. The modeling results suggest that the dose–response of haloperidol is steeper than that of both partial agonists. In addition, only the active human moiety of aripiprazole at the highest doses achieves a postsynaptic D2 receptor antagonism comparable to haloperidol at 70% D2 receptor occupancy. Bifeprunox fails to reach the minimum level of functional postsynaptic D2 receptor antagonism, corresponding to 4 mg haloperidol, at any dose likely because in the primate dopaminergic synapse there is a smaller effect on presynaptic DA release so that the increased partial agonism of bifeprunox doesn’t pay off presynaptically and ends up hurting more postsynaptically.
Figure 4 A) Functional antagonism (ie, the normalized decrease in actual D2 receptor activation) for different doses of haloperidol, bifeprunox, and the active moiety of aripiprazole and OPC-14857 using primate-derived calibration parameters. The x-axis (dose) is expressed as measured D2 receptor occupancy in a positron emission tomography (PET) tracer imaging experiment with raclopride for each drug. The clinically relevant functional antagonism corresponding to a 4-mg haloperidol dose of about 52% is indicated by the horizontal line. The data suggest that the dose–response of the partial agonists is not as pronounced as haloperidol’s dose response and that aripiprazole at the highest dose, but not bifeprunox, can achieve D2 receptor functional antagonist levels comparable to clinically active levels of haloperidol. B) Functional antagonism at the postsynaptic D2 receptor of hypothetical partial agonists with the same 7.1 nM Kd for the D2 receptor as aripiprazole but with different maximal agonists effects (0%–35%) in a firing frequency paradigm similar to an in vivo situation. The x-axis is expressed as the apparent D2 receptor occupancy measured with raclopride for each hypothetical molecule. A 4-mg haloperidol dose corresponds to a functional D2 receptor antagonism of about 52% (the horizontal line). The figure suggests that there is only a limited degree of partial agonism (0%–25%) that results in sufficient functional D2 receptor antagonism at the highest achievable dose. C) Comparison of functional postsynaptic D2 receptor antagonism between the relevant active moiety of aripiprazole and bifeprunox using both rodent and primate calibration parameters. The major human metabolite of aripiprazole is a partial agonist with a Kd of 3.5 nM and a maximal effect of 18%, while the rodent metabolite is a full antagonist with a Kd of 1.9 nM; both of them account for 25% of the active moiety. The functional postsynaptic D2 receptor antagonism is plotted against the apparent D2 receptor occupancy measured with raclopride. The data suggest that for the rodent case, bifeprunox is superior to the active moiety of aripiprazole, while the opposite is true for the primate situation, where bifeprunox does not achieve the same functional D2 receptor antagonism with 4 mg haloperidol.
Balance of pre- and postsynaptic effect of partial agonists
The previous data suggest that the balance of pre- and postsynaptic effects is critical to achieve sufficient postsynaptic functional D2 receptor antagonism. We simulated the effects of hypothetical analogs of a partial agonist with the same high affinity as that of aripiprazole (7.1 nM) for the D2 receptor but with different degrees of partial agonism (equal at both the pre- and postsynaptic receptors) in a dopaminergic synapse calibrated with the primate parameter set. suggests that there is an optimal window of partial agonism (<25%) that can result in sufficient functional D2 receptor antagonism similar to clinically relevant doses of haloperidol. This can account for the balance between presynaptic autoreceptor activation and postsynaptic competition with endogenous DA. Note that for higher levels of partial agonism, an increasingly higher dose is needed. However, for a partial agonist with a maximal effect >30%, the functional antagonism at the postsynaptic D2 receptor never gets in the range of 4-mg-haloperidol-induced functional postsynaptic D2 receptor antagonism.
Consequence of rodent and primate settings for partial agonists
We further illustrate the difference between rodent and primate parameter settings by comparing bifeprunox and the active moiety of aripiprazole in dopaminergic synapses in rodent synapses. As mentioned above, they do have a similar affinityCitation26–Citation28 for the D2 receptor (a Kd of 1.9 nM for bifeprunox and 7.1 nM for aripiprazole), but the functional activity of bifeprunox is slightly higher than partial agonism (36% versus 25%).Citation7 In addition, the major rat metabolite of aripiprazole, DM-1457, is a potent full D2 receptor antagonist with a fivefold higher affinity and accounts for about 25% of the active moiety.
In vivo preclinical animal studies suggest that bifeprunox is at least as active as, if not more active than, aripiprazole,Citation9 whereas the clinical efficacy of bifeprunox in schizophrenia is substantially lower.Citation6 We used our receptor competition model to address the difference of these compounds in the rodent animal models and the primate setting.
shows the functional antagonism for rodent versus primate calibration parameters. The model outcome suggests that in terms of functional postsynaptic D2 receptor antagonism, bifeprunox outperforms the active moiety of aripiprazole under rodent calibration settings, but that the opposite is true for the primate settings. The major human metabolite of aripiprazole has a somewhat lower maximal partial receptor effect than the parent molecule, ensuring that the active moiety is well within the range of sufficient postsynaptic D2 receptor antagonism for the primate calibration setting (see ), whereas the full antagonism of the rodent metabolite tends to raise inadvertently presynaptic DA levels in the rodent calibration setting.
Recently new data suggest that aripiprazole is a full antagonist in cell systems expressing the long isoform, D2L.Citation29 As D2L is likely the postsynaptic D2 receptor,Citation30 this would even increase the level of functional antagonism at the postsynaptic D2 receptor (data not shown). However, in the absence of similar data for bifeprunox, we wanted to limit our analysis to two very similar agents with similar pharmacology and explore the parameter space of a partial agonist that would lead to a robust postsynaptic functional D2 receptor antagonism.
Correlation with the clinical situation
Because the calibration conditions for rodent and primate were determined in different conditions, we next addressed the question “How will the two calibration sets compare to the clinical situation?” We therefore simulated the four clinical doses of aripipirazole and the three clinical doses of bifeprunox together with the reference drug haloperidol at 4 mg in the computer model with both calibration sets and compared the outcome to the reported clinical effects on the Positive and Negative Syndrome Scale (PANSS) total scale. The actual compound concentrations for all the conditions were determined by simulating the reported positron emission tomography (PET) imaging displacement studies with raclopride.
In stable treatment-responsive patients who were switched from other antipsychotics, it was reported that a dose of 5 mg aripiprazole resulted in a PANSS total improvement of 9.4 points,Citation31 whereas a dose of 15 mg,Citation32,Citation33 20 mg,Citation34,Citation35 and 30 mgCitation33,Citation35,Citation36 improved the PANSS total by 15.5 points, 14.5 points, and 12.6 points, respectively. These doses corresponded to receptor occupancies of 83%, 91%, 95%, and 97%, respectively.Citation37,Citation38 Similarly, the improvements for bifeprunox on the PANSS total in a 6-week study were reported to be 9.7 points, 5 points, and 11.3 points with 5 mg, 10 mg, and 20 mg of bifeprunox, respectively.Citation6 Unfortunately, no PET imaging data are publicly available for bifeprunox; however, from the reported clinical data and side effects,Citation6 we inferred that bifeprunox readily crossed the blood–brain barrier and results in receptor occupancies of 80% and above.
The clinical data for 4 mg haloperidol were derived as weighted averages from 4 studies covering 623 patients.Citation39–Citation42 Actual haloperidol concentration for this dose was simulated to be 70% using the reported PET displacement studies with raclopride.Citation43–Citation45
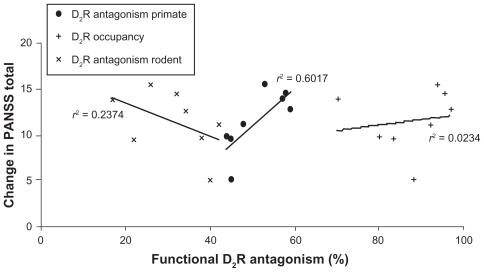
shows the correlation between the clinical outcomes (improvement in PANSS total), the postsynaptic functional D2 receptor antagonism, and the primate calibration sets. The primate calibration sets result in a correlation of r2 = 0.601 or r = 0.77 (P = 0.016), while a correlation coefficient of r2 = 0.023 or r = 0.15 (P = 0.70) is obtained using the D2 receptor occupancy as independent variable and a correlation of r2 = 0.237 or r = −0.45 (P = 0.20) when using the functional postsynaptic D2 receptor antagonism calibrated with rodent data, suggesting that the computer model with the primate calibration set is able to explain much more of the variance. Moreover, the correlation with the rodent calibration set would lead to a negative slope, ie, more clinical efficacy with lower postsynaptic D2 receptor antagonism. This is clearly not in line with the clinical observations.
Figure 5 Correlation between reported clinical efficacy of 4 mg haloperidol, four doses of aripiprazole (5 mg, 10 mg, 20 mg, and 30 mg), and three doses of bifeprunox (5 mg, 10 mg, and 20 mg) and the functional postsynaptic D2 receptor antagonism in the dopaminergic synapse computer model with primate calibration. The y-axis value for each point is the weighted average of PANSS total for a specific dose of the drug; the x-axis value is the functional postsynaptic D2 receptor antagonism as simulated in the DA synapse model using the primate calibration set (•), the rodent calibration set (×), and the postsynaptic D2 receptor occupancy (+). The observed correlation coefficient for the primate calibration set (•) of r2 = 0.602 or r = 0.77 (P = 0.015) contrasts with a correlation coefficient of r2 = 0.023 or r = 0.15 (P = 0.70) when using the D2 receptor occupancy as independent variable (+) and a correlation of r2 = 0.234 or r = −0.45 (P = 0.20) when using the functional postsynaptic D2 receptor antagonism calibrated with rodent data (×), suggesting that the computer model with the primate calibration set is able to explain much more of the variance. In addition, the computer model with the rodent calibration set would suggest a negative correlation between postsynaptic D2 receptor antagonism and clinical efficacy.
Discussion
The actual competition among the neurotransmitter, the tracer, the drug, and the active drug metabolite in a typical CNS synapse for a receptor binding site is complex and hard to understand quantitatively without systematic computer modeling. Neurotransmitters are released in well-defined firing patterns, and their release is modulated by presynaptic autoreceptor activation. Time-dependent facilitation and depression can further alter the quantity of neurotransmitter released, while the dynamics of free neurotransmitter in the synapse are determined by transporters, diffusion, and catabolic enzymes. The ability to quantitatively simulate the interactions between different physicochemical processes is one of the main arguments for this approach of computer modeling.
This report describes a computer model and its calibration and application to the striatal dopaminergic synapse. As our objective was to compare partial agonists with full antagonists, we introduced the concept of functional D2 receptor antagonism at the postsynaptic D2 receptor. This is calculated as the reduction in receptor activation, normalized to the no-drug situation. Interestingly, for a full antagonist, this functional D2 receptor antagonism, although proportional to the D2 receptor occupancy level calculated from the imaging experiments, is somewhat smaller and reflects the complex interaction between the drug and the endogenous neurotransmitter in such a synapse. For instance, inhibition of the presynaptic receptor by a full antagonist leads to an increase in the release of free DA, thereby countering its effect at the postsynaptic receptor.
The model has been calibrated using published experimental data that quantified the strength of presynaptic DA receptor feedback on DA release with high concentrations of dopamine D2 receptor antagonists and forced firing frequencies of dopaminergic neurons in rodentsCitation6 or in primates.Citation5 Both calibration sets lead to an average D2 receptor occupancy by endogenous DA in drug-free conditions between 30% and 50%, with the primate data being slightly higher. This is in the experimental range observed in humans,Citation46 baboons,Citation22 and rodents.Citation47 In primates at least, the experimental data suggest that the effect of presynaptic receptor coupling to DA release is substantially lesser, leading to a different dose dependency of a partial agonist on the functional postsynaptic D2 receptor antagonism.
Marmosets are increasingly being used for biomedical research; their usefulness is likely to be limited as they can differ quite substantially from old-world primates.Citation48 However, they have characteristic anthropoid primate traits. It is of interest to note that similar measurements in striatal slices of squirrel monkeys result in low DA levels that are at least in qualitative agreement with data from the marmoset.Citation49
Although we don’t have similar data for the human situation, such disconnect between rodent and primate physiology suggests that extrapolations from preclinical rodent models to the human clinical situation need to proceed cautiously.
Interestingly, the model outcome suggests that the calculated D2 receptor occupancy from a PET imaging study can be different when different radiotracers are used in the same patient treated with antipsychotic medication. This is in line with reported clinical data that systematic lower occupancy readout was found with IBZM when compared to raclopride for all subjects and brain regions in the same schizophrenia patients.Citation23 The authors concluded that the reasons for the radiotracer differences were due to the degradation by high-energy photons in the IBZM images, but that they were not due to the choice of reference region or the assumption of pseudo-equilibrium. Our results suggest that the differential competition among the tracer, the neurotransmitter, and the drug at the level of the binding site can contribute additionally to this difference. Failure to appreciate this effect can lead to misinterpretation with regard to receptor occupancies when using different radiotracers. Fortunately, the large majority of clinical imaging studies with antipsychotics have been performed with the same radiotracer, raclopride, making comparisons between drugs easier.
Partial D2 receptor agonists are an interesting option to achieve functional D2 receptor inhibition, because they tend to reduce the amount of presynaptic DA released by stimulating the negative-feedback D2 autoreceptor. These drugs are supposed to act more closely to the actual pathology as recent imaging studies in an at-risk for mental state cohorts suggests a presynaptic pathology in schizophrenia patients.Citation4 The differences in calibration of this presynaptic effect also suggest that partial agonists in the rodent calibration will lead to greater functional postsynaptic antagonism, as compared to the primate calibration setting, because they reduce the levels of released DA more strongly. This might lead to an overestimation of the effect of partial agonists in rodent models.
It is important to note that the model outcome is presented as the level of postsynaptic D2 receptor activation and does not take into account any further physiological intracellular effect or pathway activation. Recently, the concept of functional selectivity has been proposed to account for some of the in vivo effects of aripiprazole.Citation29 The aripiprazole effect can range from partial agonism to full antagonism, depending on the cell system, the latter mostly in cell systems overexpressing human D2L receptors. These D2L receptors are most likely located postsynaptically,Citation30 and an antagonistic effect of aripiprazole can therefore explain the clinical observations of worsening motor symptoms in the treatment of patients with Parkinson’s.Citation50 Substituting the partial agonist effect of aripiprazole at the postsynaptic D2 receptor by a full antagonist effect will increase the functional postsynaptic D2 receptor antagonism, as free DA is now replaced by a compound that, rather than partially activating, completely blocks the receptor, whereas the partial agonist effect at the presynaptic receptor continues to reduce levels of released DA.
The simulation further suggests that D2 receptor functional antagonism of the active moiety of aripiprazole at high D2 receptor occupancy levels (>90%) can achieve similar levels as clinically relevant doses of haloperidol. We compared the effects of the partial agonists on postsynaptic D2 receptor antagonism in the presence of 4 mg haloperidol. Although often higher doses are used in clinical practice, a 4-mg dose gives the minimal clinical benefitCitation43 and is therefore a good benchmark. Although the pharmacology of both aripiprazole and bifeprunox is quite complex, including effects on other dopaminergic, serotonergic, and noradrenergic receptors,Citation29 functional postsynaptic D2 receptor antagonism drives a substantial part of the clinical response.Citation8 The fact that high doses are needed is in line with the reported proportional dose–response for aripiprazole in clinical trials,Citation35,Citation36 with clinically efficacious doses that correspond to >95% D2 receptor occupancy.Citation37
The model simulations further suggest that there is a limited therapeutic window for partial agonists, because of the delicate balance between pre- and postsynaptic effects. Having too strong an affinity and maximal agonist effect might indeed lead to a lower postsynaptic inhibition that outweighs the benefits of presynaptic D2 receptor stimulation.
Such computer models can help to better extrapolate findings from preclinical rodent models to the clinical situation. Because of the stronger coupling of presynaptic D2 autoreceptor to DA release in the rodent case, bifeprunox outperforms the active moiety of aripiprazole, a result observed experimentally in preclinical rodent models.Citation9 In contrast, the model predicts a superiority of the active moiety of aripiprazole in primate conditions, which is partially due to the different pharmacological activities of its major metaboliteCitation24 and partially due to the lower coupling of presynaptic D2 autoreceptor activation to subsequent DA release.Citation5 The results of the model suggest that aripiprazole has a substantial clinical advantage over bifeprunox at least in functional D2 receptor antagonism. Obviously, it is possible that clinical efficacy is also driven by other processes as discussed above, but it is of interest to note that the bifeprunox clinical effect on PANSS total is limitedCitation6 to the point that the clinical development of the compound has been halted.
In this study, we assumed that, unlike aripiprazole, bifeprunox has no major active metabolite in the human/primate or the rodent situation, as there are no published data on such a metabolite. It could still be the case that human metabolites of bifeprunox, if present, could have a pharmacological activity that is sufficiently different to change dramatically the functional D2 receptor antagonism. Notwithstanding this, the data illustrate the power of the model to assess the effect of unique human metabolites.
As with any model, there are limitations on this specific modeling approach. Because we don’t take into account spatial dimensions, we assume that all of the agents have perfect access to each other. This could lead to an overestimation of the amount of bound receptors. However, in terms of understanding the relative effects, such as inhibition fraction, we expect the model to behave reasonably well. Because we are dealing with synaptic effects where the distance between the membranes is very small, there is also a greater probability that the different agents will come into contact with a receptor, making it more similar to the well-mixed solution that the equations describe.
Although the model is based on biological properties of the dopaminergic synapse (using realistic values for DA dynamics), some of the relations, for instance those between presynaptic dopamine D2 autoreceptor and DA release after stimulation, are introduced in a phenomenogical, rather than an exact biological, way. The biology of presynaptic DA release is quite complex, and there are still many unresolved issues (for a complex computational model see Qi et alCitation51). This particular computational model has focused on the biochemistry of processes we have modeled in a phenomenological way but calibrated using actual experimental data with a limited set of parameters. In contrast to rodent data, the primate values are measured from ex vivo preparationsCitation5 that obviously might lack a number of physiological processes that are active in vivo. However, the relative effect and the dose–response for the active moiety of aripiprazole with primate parameters corresponds to observed clinical effects, again assuming that the D2 receptor block drives most of the clinical outcome.Citation8,Citation52
Unlike other models dealing with the interpretation of PET imagingCitation22 that simulate the binding kinetics of tracers over many minutes, our model takes into account detailed physiological processes on a millisecond timescale.
We anticipate that this method becomes even more valuable in the cases where simple occupancy rules/formulae do not apply in conditions such as (1) in the absence of tracers, (2) with multiple drugs (and metabolites) acting on the same receptor, (3) when dealing with partial agonists, and (4) when the rodent physiology is different from the human physiology. In principle, such a generic receptor competition model could also be applied to different CNS synapses, such as cortical DA synapses or serotonergic synapses, using appropriate affinity and physiology parameters.
Acknowledgments
We thank In Silico Biosciences, Inc. for the financial support. We thank Patrick Roberts and John Dani for valuable discussions and comments on the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- HowesODKapurSThe dopamine hypothesis of schizophrenia: version III – the final common pathwaySchizophr Bull200935354956219325164
- HowesODMontgomeryAJAsselinMCElevated striatal dopamine function linked to prodromal signs of schizophreniaArch Gen Psychiatry2009661132019124684
- KomossaKRummel-KlugeCSchmidFAripiprazole versus other atypical antipsychotics for schizophreniaCochrane Database Syst Rev200974CD00656919821375
- CaseyDESandsEEHeisterbergJYangHMEfficacy and safety of bifeprunox in patients with an acute exacerbation of schizophrenia, results from a randomized, double-blind, placebo-controlled, multicenter, dose-finding studyPsychopharmacology (Berl)2008200331733118597078
- CraggSJHilleCJGreenfieldSADopamine release and uptake dynamics within nonhuman primate striatum in vitroJ Neurosci200020218209821711050144
- WuQReithMEWalkerQDKuhnCMCarrollFIGarrisPAConcurrent autoreceptor-mediated control of dopamine release and uptake during neurotransmission, an in vivo voltammetric studyJ Neurosci200222146272628112122086
- TadoriYForbesRAMcQuadeRDKikuchiTReceptor reserve-dependent properties of antipsychotics at human dopamine D2 receptorsEur J Pharmacol200960713354019326565
- PaniLPiraLMarcheseGAntipsychotic efficacy, relationship to optimal D2-receptor occupancyEur Psychiatry200722526727517419008
- DahanLHusumHMnie-FilaliOArntJHertelPHaddjeriNEffects of bifeprunox and aripiprazole on rat serotonin and dopamine neuronal activity and anxiolytic behaviourJ Psychopharmacol200923217718918515444
- BrownPOlivieroAMazzonePInsolaATonaliPDi LazzaroVDopamine dependency of oscillations between subthalamic nucleus and pallidum in Parkinson’s diseaseJ Neurosci20012131033103811157088
- ZhangLDoyonWMClarkJJPhillipsPEDaniJAControls of tonic and phasic dopamine transmission in the dorsal and ventral striatumMol Pharmacol200976239640419460877
- MontaguePMcClureSBaldwinPDynamic gain control of dopamine delivery in freely moving animalsJ Neurosci20042471754175914973252
- KoeltzowTEXuMCooperDCAlterations in dopamine release but not dopamine autoreceptor function in dopamine D3 receptor mutant miceJ Neurosci1998186223122389482807
- PovlockSLSchenkJOA multisubstrate kinetic mechanism of dopamine transport in the nucleus accumbens and its inhibition by cocaineJ Neurochem1997693109311059282932
- WaymentHSchenkJSorgBCharacterization of extracellular dopamine clearance in the medial prefrontal cortex, role of monoamine uptake and monoamine oxidase inhibitionJ Neurosci2001211354211150317
- SeemanPTargeting the dopamine D2 receptor in schizophreniaExpert Opin Ther Targets200610451553116848689
- KapurSVanderSpekSCBrownleeBANobregaJNAntipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancyJ Pharmacol Exp Ther2003305262563112606608
- SautelFGriffonNLévesqueDPilonCSchwartzJCSokoloffPA functional test identifies dopamine agonists selective for D3 versus D2 receptorsNeuroreport1995623293327756621
- PugsleyTADavisMDAkunneHCNeurochemical and functional characterization of the preferentially selective dopamine D3 agonist PD 128907J Pharmacol Exp Ther19952753135513668531103
- HalldinCFardeLHögbergTCarbon-11-FLB 457, a radioligand for extrastriatal D2 dopamine receptorsJ Nucl Med1995367127512817790956
- BrückeTTsaiYFMcLellanCIn vitro binding properties and autoradiographic imaging of 3-iodobenzamide ([125I]-IBZM), a potential imaging ligand for D-2 dopamine receptors in SPECTLife Sci19884221209721043260318
- DelforgeJBottlaenderMPappataSLoc’hCSyrotaAAbsolute quantification by positron emission tomography of the endogenous ligandJ Cereb Blood Flow Metab200121561363011333372
- CatafauAMSuarezMBullichSWithin-subject comparison of striatal D2 receptor occupancy measurements using [123I] IBZM SPECT and [11C]Raclopride PETNeuroimage200946244745819233294
- WoodMDScottCClarkeKAripiprazole and its human metabolite are partial agonists at the human dopamine D2 receptor, but the rodent metabolite displays antagonist propertiesEur J Pharmacol200654613889416914136
- KimJRSeoHBChoJYPopulation pharmacokinetic modeling of aripiprazole and its active metabolite, dehydroaripiprazole, in psychiatric patientsBr J Clin Pharmacol200866680281019032724
- CosiCCarilla-DurandEAssiéMBPartial agonist properties of the antipsychotics SSR181507, aripiprazole and bifeprunox at dopamine D2 receptors, G protein activation and prolactin releaseEur J Pharmacol20065351313514416527269
- BurrisKDMolskiTFXuCAripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptorsJ Pharmacol Exp Ther2002302138138912065741
- EtievantABétryCArntJHaddjeriNBifeprunox and aripiprazole suppress in vivo VTA dopaminergic neuronal activity via D2 and not D3 dopamine autoreceptor activationNeurosci Lett20094601828619450663
- ShapiroDARenockSArringtonEAripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacologyNeuropsychopharmacology20032881400141112784105
- UsielloABaikJHRougé-PontFDistinct functions of the two isoforms of dopamine D2 receptorsNature2000408680919920311089973
- SalzmanCRosenbergJFeldmanJJSchizophrenia symptoms remain stable during decreases from 2 antipsychotics to aripiprazoleJ Clin Psychiatry200768697017592928
- KaneJMCarsonWHSahaAREfficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorderJ Clin Psychiatry200263976377112363115
- ChristensenAFPoulsenJNielsenCTBorkBChristensenAChristensenMPatients with schizophrenia treated with aripiprazole, a multicentre naturalistic studyActa Psychiatr Scand2006113214815316423167
- TandonRMarcusRNStockEGA prospective, multicenter, randomized, parallel-group, open-label study of aripiprazole in the management of patients with schizophrenia or schizoaffective disorder in general psychiatric practice: Broad Effectiveness Trial with Aripiprazole (BETA)Schizophr Res2006841778916483745
- PotkinSGSahaARKujawaMJAripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorderArch Gen Psychiatry200360768169012860772
- CaseyDECarsonWHSahaARSwitching patients to aripiprazole from other antipsychotic agents, a multicenter randomized studyPsychopharmacology (Berl)2003166439139912610718
- YokoiFGründerGBiziereKDopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): a study using positron emission tomography and [11C]racloprideNeuropsychopharmacology200227224825912093598
- KegelesLSSlifsteinMFrankleWGDose-occupancy study of striatal and extrastriatal dopamine D2 receptors by aripiprazole in schizophrenia with PET and [18F]fallyprideNeuropsychopharmacology2008333111312518418366
- ZimbroffDLKaneJMTammingaCAControlled, dose-response study of sertindole and haloperidol in the treatment of schizophrenia. Sertindole Study GroupAm J Psychiatry199715467827919167505
- GomezJCCrawfordAMSuperior efficacy of olanzapine over haloperidol: analysis of patients with schizophrenia from a multicenter international trialJ Clin Psychiatry200162Suppl 261111232753
- SikichLHamerRMBashfordRASheitmanBBLiebermanJAA pilot study of risperidone, olanzapine, and haloperidol in psychotic youth: a double-blind, randomized, 8-week trialNeuropsychopharmacology200429113314514583740
- ZipurskyRBChristensenBKDaskalakisZTreatment response to olanzapine and haloperidol and its association with dopamine D receptor occupancy in first-episode psychosisCan J Psychiatry200550846246916127964
- KapurSRemingtonGJonesCHigh levels of dopamine D2 receptor occupancy with low-dose haloperidol treatment: a PET studyAm J Psychiatry199615379489508659621
- KapurSZipurskyRBRemingtonGClinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophreniaAm J Psychiatry199915622862939989565
- BernardoMParelladaELomenaFDouble-blind olanzapine vs haloperidol D2 dopamine receptor blockade in schizophrenic patients: a baseline-endpointPsychiatry Res20011072879711530275
- LaruelleMD’SouzaCDBaldwinRMImaging D2 receptor occupancy by endogenous dopamine in humansNeuropsychopharmacology19971731621749272483
- FisherREMorrisEDAlpertNFischmanAJIn vivo imaging of neuromodulatory synaptic transmission using PET, a review of relevant neurophysiologyHum Brain Mapp199532434
- AbbottDHBarnettDKColmanRJYamamotoMESchultz-DarkenNJAspects of common marmoset basic biology and life history important for biomedical researchComp Med200353433935014524409
- PerezXAParameswaranNHuangLZO’LearyKTQuikMPresynaptic dopaminergic compensation after moderate nigrostriatal damage in non-human primatesJ Neurochem200810551861187218248617
- FriedmanJHBermanRMGoetzCGOpen-label flexible- dose pilot study to evaluate the safety and tolerability of aripiprazole in patients with psychosis associated with Parkinson’s diseaseMov Disord200621122078208117013906
- QiZMillerGWVoitEOComputational systems analysis of dopamine metabolismPLoS One200836e244418568086
- TauscherJKüfferleBAsenbaumSTauscher-WisniewskiSKasperSStriatal dopamine-2 receptor occupancy as measured with [123I] iodobenzamide and SPECT predicted the occurrence of EPS in patients treated with atypical antipsychotics and haloperidolPsychopharmacology (Berl)20021621424912107616
- LandBRSalpeterEESalpeterMMKinetic parameters for acetylcholine interaction in intact neuromuscular junctionProc Natl Acad Sci U S A19817811720072046947281
Appendix
Here, we present a more detailed description of the receptor competition model.
Dynamical binding and unbinding of the different agents to the receptor sites are calculated as follows. If [dop] is the free DA concentration and [Df] is the concentration of free receptors, then the change in receptors bound by DA, [Dn], is governed by the following ordinary differential equation (ODE):
with the initial condition that all receptors begin in the free state. With sub- and superscripts n, d, m, and t referring to neurotransmitter DA, drug, metabolite, and tracer, respectively, the change in receptors for drug, metabolite, and tracer binding is governed by the following coupled ODEs:
Alternatively, the metabolite can be substituted by a second drug in simulation of polypharmacy. We assume that the association to the receptor is diffusion limited, giving us a value for the forward binding rate constant kon of 140–150 μM−1 sec−1 for a molecule with MW in the range of 200–400 (as an example for the case of AchCitation53). In general, Kd = koff/kon so that Koff = Kd ×Kon. Furthermore,
where Do is the concentration of receptors. All differential equations are solved with a fourth-order Runge–Kutta method with a time step of 0.01 msec.
The amount of free DA depends on two processes, exponential decay and quantal release. Exponential decay is classically defined as [dop](t)=exp(−t ln (2)/half-life,) where half-life is the half-life of the decay process. At times of release, [dop] is immediately updated by adding the release amount.
The amount of presynaptic receptor activation which occurred 150 msec before the current release event then determines the amount of new release as follows
where release0 is the base release amount, relScale is the maximum relative change for release, recAct is the actual presynaptic receptor activation 150 msec earlier, relSens is the sensitivity to the presynaptic receptor (lower values create a shallow response and higher values create a sharp difference between activation levels), and normBound is the amount of normal presynaptic binding that one would expect in the tonic case (ie, when recAct equals normBound, the new release equals the baseline release amount). The dynamical independent variable recAct is calculated from earlier time points and is initiated using a run-in time period of 4 sec. We calibrate the parameters so that the coupling of presynaptic D2 receptor activation to DA release reflects actual experimental data (as seen in Results).
In addition, the release can be modulated by a depression or facilitation mechanism.Citation12 Instead of using internal Ca++ levels to determine DA release, we consider the facilitation and depression of DA release based solely on the amount of time elapsed since the previous firing using a simple transfer equation. Thus, the amount of DA released is based both on the history of firing and the activation level of the presynaptic D2 autoreceptors. If we denote the time of the nth firing by tn, then the release amount is modified based on all previous firings as follows
where wf is the facilitation weight, wd is the depression weight, kf is the decay rate of facilitation, and kd is the decay rate of depression. These four parameters will be calibrated to experimental data.
The simulation is initiated by first finding the equilibrium, given a constant amount of free DA at 500 nM. The simulation is then run for a transitory time of 5 sec at the tonic firing rate of 4 Hz. Finally, the simulation runs for an additional 2.5 sec during which time average binding levels are determined and then the simulation runs with the predefined firing pattern.