Abstract
Background and methods
A double-blind, parallel-group, controlled study was performed to investigate if milnacipran was noninferior to paroxetine in terms of improvement in symptoms of depression in Japanese patients with major depressive disorders in a fixed-dose design. The efficacy and safety of milnacipran 200 mg/day were also assessed in comparison with those at the standard dose of 100 mg/day.
Results
Changes in 17-item Hamilton depression rating scale (HAM-D) total score (mean ± standard deviation) for group M1 (milnacipran 100 mg/day), group M2 (milnacipran 200 mg/day), and group PX (paroxetine 30 or 40 mg/day) were −12.9 ± 5.8, −12.8 ± 6.1, and −13.1 ± 6.2, respectively, and the estimated differences in total score for group PX (Dunnett’s 95% simultaneous confidence interval) were 0.1 (−1.1 to 1.3) for group M1 and 0.3 (−0.9 to 1.5) for group M2. The noninferiority of groups M1 and M2 to group PX was thus confirmed, because the upper confidence limit of differences between groups M1 and PX and between groups M2 and PX was less than 2.0. The estimated mean difference of change in HAM-D total score (95% confidence interval) between groups M2 and M1 was 0.2 (−0.9 to 1.2), indicating a comparable change in total score for both groups. The incidence of treatment-related adverse events was 71.7% for group M1, 68.8% for group M2, and 69.3% for group PX, indicating no significant difference between the three groups.
Conclusion
These results demonstrate that milnacipran 100 mg/day and 200 mg/day is not inferior to paroxetine in terms of efficacy and safety.
Introduction
Milnacipran is an antidepressant that inhibits the reuptake of serotonin and norepinephrine, and is thus classified as a serotonin-norepinephrine reuptake inhibitor. Unlike tricyclic and tetracyclic antidepressants, studies have shown that milnacipran has no affinity for neurotransmitter receptors.Citation1 Unlike selective serotonin reuptake inhibitors, milnacipran has a potent inhibitory action on the reuptake of norepinephrine in addition to that of serotonin,Citation1 as shown by the ratio of the serotonin reuptake inhibitory Ki value to norepinephrine reuptake inhibitory Ki value, which is close to unity.Citation2
In Japan, the usefulness of milnacipran as an antidepressant has been demonstrated in clinical studies with a fixed-flexible dose design, and the drug was launched in 2000 as the first serotonin-norepinephrine reuptake inhibitor. In studies carried out in Europe and the US, the antidepressant effect of milnacipran has been shown to be superior to placebo.Citation3 Further, the antidepressant effect and tolerability have been shown to be comparable or superior to comparators, such as selective serotonin reuptake inhibitors (fluoxetine, fluvoxamine, paroxetine),Citation4–Citation8 a serotonin-norepinephrine reuptake inhibitor (venlafaxine),Citation9 and tricyclic antidepressants (amitriptyline, imipramine).Citation10–Citation13 However, no study has been conducted in Japan with the aim of confirming the noninferiority of milnacipran to previously introduced drugs using a fixed-dose scheme.
We considered that it is important to verify the antidepressant effect of milnacipran using a fixed-dose design, and conducted a double-blind, controlled study to determine the noninferiority of milnacipran in patients with major depressive disorders against the selective serotonin reuptake inhibitor, paroxetine, which is the most extensively used antidepressant in Japan.
Use of antidepressants at adequate doses is required to achieve the goal of treatment for depression, ie, remission. In recent years, some studies have demonstrated the usefulness of milnacipran at doses higher (150–200 mg/day) than that used routinely (100 mg/day).Citation14–Citation16 Therefore, we set the dose levels of milnacipran at the standard dose of 100 mg/day and a higher dose of 200 mg/day, in order to assess the significance of high-dose milnacipran therapy.
Materials and methods
Subjects
The target disease for this study was major depressive disorder (single episode or recurrent) under the classification specified in the DSM-IV-TR (Diagnostic and Statistical Manual of Mental Disorders Fourth Edition, Text Revision). Patients who met the following conditions at the time of giving their informed consent were included in the study: age 20–64 years, a 17-item Hamilton depression rating scale (HAM-D) total score ≥ 18 with a HAM-D “depressed mood” score ≥ 2, and duration of illness of 1–12 months. Outpatients and inpatients of either gender were eligible for the study.
Major exclusion criteria included the following: a depressive episode refractory to treatment with two or more antidepressants pronounced suicidality with a HAM-D “suicide” score of 4, a history of or concurrent schizophrenia or other psychotic disorder, a history of or concurrent alcohol dependence or drug dependence, concurrent personality disorder, treatment with any antipsychotic drug at the time of giving informed consent, use of a monoamine oxidase inhibitor within 2 weeks before the day of giving informed consent, being pregnant or possibly pregnant, and nursing mothers.
Each subject, after receiving a full explanation of the nature and details of the clinical study as well as the rights of study subjects, gave written consent of his/her own free will to participate in the study.
Study design
The study was a multicenter, double-blind (double-dummy design), parallel-group, controlled trial conducted at 75 medical institutions in Japan with the approval of the institutional review board of each participating institution. The design was accepted by the Japanese regulatory authorities. The study treatment scheme is shown in . Patients were randomized to receive milnacipran 100 mg/day (group M1), milnacipran 200 mg/day (group M2), or paroxetine at 30 or 40 mg/day (group PX). Patients in groups M1 and M2 were administered the test drug in two divided doses given orally after breakfast and after supper, while those in group PX took a placebo orally after breakfast and the comparator orally after supper.
Figure 1 Study treatment scheme.
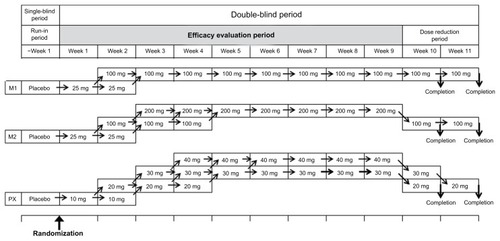
Before starting the study treatment, all subjects received placebo orally twice daily during a run-in period of one week. Only subjects whose rate of decrease in HAM-D total score at the end of the run-in period was less than 25% of the total score at the start of the run-in period, whose total HAM-D score was ≥18, and whose HAM-D “depressed mood” score was not <2 were allowed to continue in the study. Randomization to treatment groups used a dynamic randomization scheme with gender, severity of depressive symptoms (HAM-D total score), frequency of depressive episodes (initial or plural), and any previous use of antidepressant(s) for the current episode as stratification variables. Dynamic randomization methods allocate patients to a treatment group by checking the allocation of similar patients already randomized, and allocating to a treatment group so as to best balance the treatment groups across all stratification variables.Citation17
The test drug or the comparator was administered daily during the efficacy evaluation period of 9 weeks under the double-blind scheme. Patients in group M1 received milnacipran 25 mg/day for one week, and the dose was then increased to 100 mg/day unless there was a tolerability concern. In group M2, milnacipran was administered at 25 mg/day for one week, and the dose was then increased to 100 mg/day and to 200 mg/day unless there was a tolerability concern. Patients in group PX received paroxetine 10 mg/day for one week, and the dose was then increased to 20 mg/day and to 30 mg/day unless there was a tolerability concern. The dose of paroxetine was further increased to 40 mg/day if the patient failed to respond (with a reduction of less than 50% of the HAM-D total score at the end of the run-in period). In all cases, the final dose level was maintained for at least the final 4 weeks of the study.
The dose reduction before stopping the drug at the end of the study was conducted as shown in . In group M1, patients continued at 100 mg for 1–2 weeks after the end of the evaluation phase. In group M2, the dose of milnacipran was reduced to 100 mg for 1–2 weeks after the end of the evaluation phase. For patients in group PX, the paroxetine dose level was reduced stepwise to 30 mg/day and 20 mg/day at weekly intervals after the end of the efficacy evaluation period, to complete the study.
Concomitant use of the following drugs was not allowed during the study period: other antidepressants, antianxiety agents, antipsychotic drugs, antiparkinsonian agents, central stimulants, psychoanaleptics, hypnotics, mood stabilizers, monoamine oxidase inhibitors, antihypertensive drugs, serotonin 1B/1D receptor agonists, digoxin, epinephrine, norepinephrine, metoprolol tartrate, timolol maleate, propafenone hydrochloride, flecainide acetate, linezolid, investigational drugs, and any drugs not approved in Japan. Concomitant use of a single ultrashort-acting or short-acting hypnotic was allowed, where necessary, during the study period. Electroconvulsive therapy, sleep deprivation therapy, bright light phototherapy, magnetic stimulation therapy, and psychotherapies other than those commonly prescribed as routine medical practice were also prohibited.
Efficacy and safety evaluation
The severity of depressive symptoms was evaluated in each patient using the HAM-D and Montgomery–Åsberg Depression Rating Scale (MADRS) at the start and end of the run-in period and weekly at weeks 1–9 of the efficacy evaluation period. The primary efficacy endpoint consisted of changes in HAM-D total score, ie, the score at completion of the efficacy evaluation period or at discontinuation (endpoint) minus the score at the end of the run-in period (baseline). The secondary efficacy endpoints included HAM-D response rate (percentage of patients achieving a decrease in total baseline HAM-D score ≥ 50%), HAM-D remission rate (percentage of patients achieving a decrease in HAM-D17 total score to ≤7), and changes in MADRS total score. The changes in MADRS total score were handled in the same manner as were those on HAM-D.
All adverse events that occurred from the start of the efficacy evaluation period until the end of the study treatment period were reported. Laboratory investigations were performed, along with measurement of blood pressure and heart rate, at the start of the run-in period and at weeks 4 and 9 of the efficacy evaluation period.
Statistical analysis
Efficacy
A full analysis set and a per protocol set were defined for evaluation of efficacy. Because the primary aim of the present study was to confirm (or not) drug noninferiority, the per protocol set was taken as the primary analysis set. Patients for whom efficacy data were not available after treatment with the study drug at the final dose for at least 4 weeks were not included in the per protocol set, because this study was conducted as a fixed-dose design.
The primary efficacy objective of this study was to confirm (or not) drug noninferiority in groups M1 and M2 to group PX. The main analysis consisted of calculation of the 95% simultaneous confidence interval based on Dunnett’s multiple comparison as to differences between groups M1 and PX and between groups M2 and PX in respect of changes in HAM-D total score. The test drug was considered to be noninferior to the comparator if the upper confidence limit was less than 2.0, using the noninferiority margin set at 2.0 based on two placebo-controlled paroxetine trials.Citation18,Citation19
The sample size necessary to demonstrate noninferiority of milnacipran 100 mg/day or 200 mg/day versus paroxetine was estimated from the change in HAM-D total score. The clinical equivalence margin of 2.0 was based on the results of the two placebo-controlled clinical trials of paroxetine.Citation18,Citation19 The calculated sample size, based on a power of 80%, was 705 subjects (n = 235 in each group), assuming the difference between groups of zero with 7.0 SD. The type I error was fixed at one-sided 1.25% considering multiplicity.
The secondary objective of this study was to assess the efficacy of milnacipran in group M2 in comparison with that in group M1. In the main analysis, differences in changes in HAM-D total score between groups M1 and M2 and the 95% confidence intervals thereof were calculated.
The secondary analyses were performed to calculate differences in HAM-D response rate, HAM-D remission rate, and changes in MADRS total score between groups M1 and PX and between groups M2 and PX and the 95% confidence interval thereof (primary objective), as well as differences in the above items between groups M1 and M2 and the 95% confidence interval thereof (secondary objective).
Safety
The safety analysis set comprised all patients randomized to the treatment groups, except those who failed to take any of the test drug or comparator and those for whom no safety data were obtained. The incidence of adverse events was compared between groups M1 and M2, between groups M1 and PX, and between groups M2 and PX using Fisher’s Exact test.
Results
Analysis sets
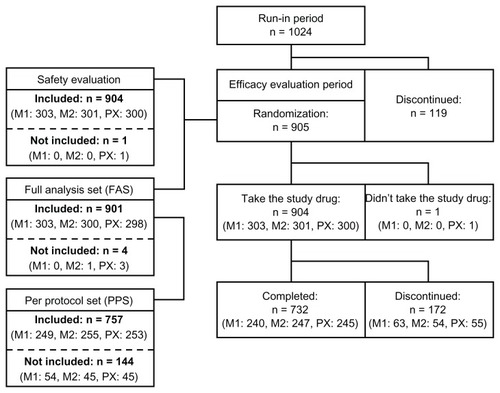
A total of 1024 patients entered the run-in period (), of whom 905 patients started the study treatment in the efficacy evaluation period. With the exception of one patient in group PX who did not take the study drug, 904 patients (303 in group M1, 301 in group M2, and 300 in group PX) were included in the safety evaluation. A total of 901 patients were included in the full analysis set (303 in group M1, 300 in group M2, and 298 in group PX), and of these patients, 757 were included in the per protocol set (249 in group M1, 255 in group M2, and 253 in group PX). For the majority of the 144 patients not included in the per protocol set, the reason for their exclusion was “lack of evaluable efficacy data” due to discontinuation of study treatment during the efficacy evaluation period (125 patients, 51 in M1, 33 in M2, and 41 in PX). A total of 732 patients (240 in M1, 247 in M2, 245 in PX) completed the study, and a total of 172 patients (63 in M1, 54 in M2, and 55 in PX) withdrew during the efficacy evaluation period. Major reasons for withdrawal included adverse events in 67 patients (26 in M1, 18 in M2, and 23 in PX), missed treatment for 3 days or more in 55 patients (15 in M1, 24 in M2, and 16 in PX) and patient’s own request for withdrawal in 39 patients (16 in M1, 10 in M2, and 13 in PX). Some of the patients in the per protocol set withdrew from the study after treatment with the study drug at final dose for at least 4 weeks.
Figure 2 Flow chart of recruited patients.
Patient demographic and clinical characteristics
Demographic and other baseline clinical characteristics of patients included in the per protocol set, ie, the main analysis set for efficacy evaluation, are presented in . There were slightly more female patients than male patients, and almost all were outpatients and had no concomitant psychiatric disorder. Approximately 75% of patients had not previously used antidepressants for the current depressive episode, and more than 60% of patients had no previous depressive episodes. Baseline HAM-D and MADRS total scores were approximately 22 and 29, respectively. There was a uniform distribution of patients among the treatment groups.
Table 1 Baseline patient demographic and clinical characteristics of the per protocol set
Efficacy
HAM-D total scores throughout study
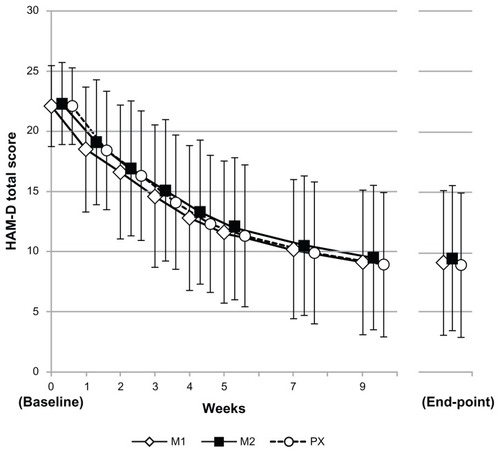
Changes in HAM-D total scores and MADRS total scores over time from baseline to the end of the efficacy evaluation period are shown in . In all three treatment groups, the HAM-D total score decreased progressively, with an essentially comparable trend throughout the efficacy evaluation period.
Figure 3 HAM-D17 total scores throughout the study.
Abbreviations: HAM-D, 17-item Hamilton Rating Scale for Depression; M1, milnacipran 100 mg/day group; M2, milnacipran 200 mg/day group; PX, paroxetine group.
Primary endpoint analysis
HAM-D total scores at baseline and at endpoint, as well as changes in HAM-D total scores with differences between the three treatment groups, are presented in . Changes in mean HAM-D total scores in groups M1, M2, and PX were −12.9 ± 5.8, −12.8 ± 6.1, and −13.1 ± 6.2, respectively. The estimated differences between groups M1 and PX and between groups M2 and PX (with Dunnett’s 95% simultaneous confidence interval) were 0.1 (−1.1 to 1.3) and 0.3 (−0.9 to 1.5), respectively. The upper limits of the 95% simultaneous confidence interval of the differences between groups M1 and PX and between groups M2 and PX were thus less than 2.0, confirming the noninferiority of group M1 and group M2 to group PX. Furthermore, the estimated difference between groups M1 and M2 (with the 95% confidence interval) was 0.2 (0.9 to 1.2), so there was no significant difference in changes in HAM-D total score between groups M1 and M2 either.
Table 2 Results of the primary endpoint analysis
Changes in mean HAM-D total scores in the full analysis set were −11.6 ± 6.7 for group M1, −12.0 ± 6.6 for group M2, and −11.8 ± 6.9 for group PX. The estimated differences between groups M1 and PX and between groups M2 and PX (with Dunnett’s 95% simultaneous confidence interval) were 0.2 (−1.0 to 1.5) and −0.1 (−1.4 to 1.1), respectively. The upper limit of the confidence interval of the differences between groups M1 and PX and that between groups M2 and PX were less than 2.0; thus, the noninferiority of group M1 and group M2 to group PX was confirmed.
Secondary endpoint analysis
HAM-D response rates, HAM-D remission rates, and changes in MADRS total scores for the treatment groups, as well as differences in these respective parameters among the groups are presented in . The response rates were 67.1%, 67.1%, and 67.6% and the remission rates were 46.2%, 41.6%, and 43.9% for groups M1, M2, and PX, respectively. Mean changes in MADRS total scores were −16.7 ± 8.6, −16.6 ± 8.4, and −17.1 ± 8.7 for groups M1, M2, and PX, respectively. There was no significant difference in any of these parameters between groups M1 and PX or between groups M2 and PX, or between groups M1 and M2. Similarly, there were no significant differences in scores on individual items of the respective scales between groups at endpoint. However, the study was not powered for such an analysis.
Table 3 Results of the secondary endpoint analysis
Safety
No deaths were reported during the study. A serious treatment-related adverse event (headache) was noted in one patient in group M1. The patient was hospitalized due to this adverse event, subsequently recovered, and was discharged on the second day of hospitalization. Treatment-related adverse events were reported in 71.6% (217/303) of patients in group M1, 68.8% (207/301) in group M2, and 69.3% (208/300) in group PX. The incidence of adverse events in group M2 did not significantly differ from that in group M1 (P = 0.477, Fisher’s Exact test). The incidence of adverse events in group M1 and in group M2 were not significantly different from that in group PX (P = 0.592 and P = 0.930, Fisher’s Exact test).
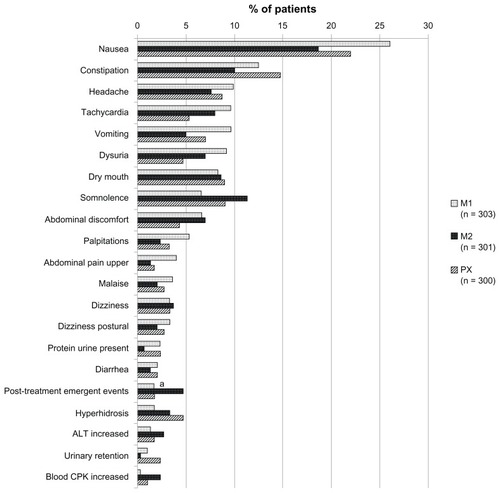
Adverse events that occurred with an incidence of 2% or more in any treatment group are shown in . Nausea, constipation, headache, and tachycardia were the most frequent adverse events reported in groups M1 and M2, whereas nausea, constipation, dry mouth, and somnolence were the most frequent in group PX.
Figure 4 Common treatment-related adverse events (with an incidence of ≥2%).
Abbreviations: M1, milnacipran 100 mg/day group; M2, milnacipran 200 mg/day group; PX, paroxetine group; ALT, alanine aminotransferase; CPK, creatine phosphokinase.
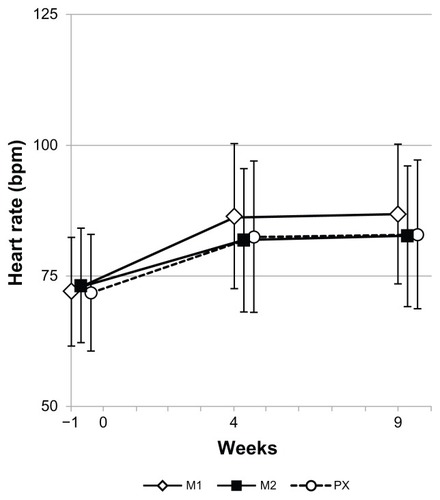
Blood pressure and heart rate data are presented in and , respectively. No noticeable changes in systolic () or diastolic () blood pressure were observed in any of the treatment groups. Increased heart rate was noted in all the three groups and did not differ by type of drug or dose level of milnacipran ().
Figure 5 Systolic (A) and diastolic (B) blood pressure throughout the study.
Abbreviations: M1, milnacipran 100 mg/day group; M2, milnacipran 200 mg/day group; PX, paroxetine group.
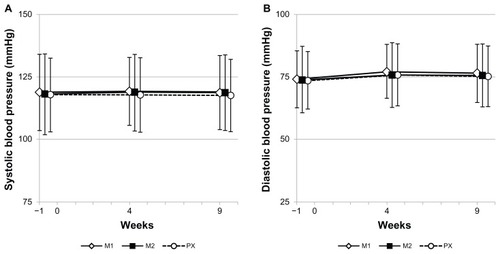
Figure 6 Heart rate throughout the study.
Abbreviations: M1, milnacipran 100 mg/day group; M2, milnacipran 200 mg/day group; PX, paroxetine group.
Discussion
This study was conducted with the aim of verifying the noninferiority of milnacipran (100 mg/day and 200 mg/day) to paroxetine (30 or 40 mg/day) in terms of improvement in depressive symptoms. The results of the study demonstrated that the upper limit of the 95% simultaneous confidence interval of differences in changes in HAM-D total scores between groups M1 and PX and between groups M2 and PX was lower than the noninferiority margin, thus confirming the noninferiority of groups M1 and M2 to group PX. Noninferiority was also confirmed by analysis of full analysis set data, thereby demonstrating the robustness of the above conclusion. The results of the analysis of secondary endpoints also showed that the improvement rates for groups M1 and M2 were comparable with the improvement rate for group PX. The antidepressant effect of paroxetine has already been demonstrated in many placebo-controlled studies.Citation18,Citation19 The present study has thus established that milnacipran has an antidepressant effect comparable with that of paroxetine.
Sechter et al also reported that milnacipran and paroxetine were comparable in antidepressant effect in their study,Citation8 where milnacipran was used at 100 mg/day and paroxetine at 20 mg/day, unlike the dose levels used in the present study. An analysis of reduction in HAM-D total scores in relation to the mean dose level of paroxetine has shown that the antidepressant effect of paroxetine is insufficient at a dose of 10 mg/day and plateaus out when increasing to doses greater than 20 mg/day.Citation20 Thus, taking account the unclear dose-response relationship of paroxetine at doses above 20 mg/day, the results from the present study showing that treatment with milnacipran at 100 mg/day produced an antidepressant effect equivalent to that of paroxetine 30 or 40 mg/day are not considered contradictory to the report of Sechter et al.Citation8
The present study was conducted in a fixed-dose design, on the other hand, and confirmed the noninferiority of milnacipran to paroxetine in patients receiving milnacipran 100 mg/day for 7–8 weeks as well as in those receiving milnacipran 200 mg/day for 5–7 weeks. In view of the different modes of administration, dose, and duration of milnacipran therapy in these two studies, it is difficult to interpret and comment on the possible apparent disparity of the conclusions.
In addition to the confirmation of noninferiority of milnacipran to paroxetine, the present study also assessed the efficacy and safety of treatment with milnacipran 200 mg/day in comparison with the standard dose of 100 mg/day. The results of the efficacy analysis showed comparable efficacy in patients receiving milnacipran 100 mg/day and 200 mg/day. However, it must be borne in mind that the vast majority of patients in the three treatment groups were outpatients with no concurrent psychiatric disorders (). In addition, patients with treatment-refractory depression not improved by use of two or more antidepressants were excluded from the study by the protocol. Many of the patients included in this study responded readily to antidepressant therapy, and it can be concluded that 100 mg/day of milnacipran was a sufficient dose for the treatment of depression in most patients included in this particular study. This may not necessarily be the case in patients with more severe or complex depression.
The mean change in HAM-D total score on milnacipran100 mg/day in this study (−12.9) is by no means inferior to the changes in HAM-D total scores observed with various antidepressants in clinical trials recently conducted in Japan.Citation21–Citation23
No deaths were reported in this study, and only one patient in group M1 experienced a serious adverse event (headache). The incidence of adverse events was 71.6% with milnacipran 100 mg/day, 68.8% with milnacipran 200 mg/day, and 69.3% for paroxetine 30–40 mg/day, with no significant difference between the groups. Adverse events that occurred with relatively high frequency in patients treated with milnacipran, including nausea, constipation, headache, and tachycardia, were the same as those seen previously.Citation24 A comparison of the incidence rates of different types of adverse events among the three treatment groups revealed no adverse events with an incidence noticeably different between the groups. The above results indicate that there was no difference in tolerability between milnacipran 100 mg/day and milnacipran 200 mg/day or between either dose strength of milnacipran and paroxetine.
Overall, there were very few post-treatment emergent events (about 3%, ). Events following abrupt discontinuation of drugs that block serotonin reuptake are thought to result from rapid reduction of plasma and/or cerebral serotonin levels, and paroxetine has been found to have a greater effect than milnacipran.Citation25 In the present study, there is a suggestion that patients withdrawn stepwise from milnacipran 200 mg/day (via 100 mg/day) experienced a greater number of post-treatment emergent events than those on either 100 mg/day milnacipran or paroxetine. However, the numbers are small and it is impossible to do any meaningful analysis.
In conclusion, the noninferiority of milnacipran to paroxetine has been confirmed in the present double-blind, controlled study of Japanese patients with major depressive disorder. The present study is the first to verify the noninferiority of milnacipran to a standard antidepressant using a fixed-dose scheme. Our results also show that the antidepressant effect of milnacipran 100 mg/day and that of milnacipran 200 mg/day were essentially comparable with each other and with the antidepressant effect of paroxetine. However, before generalizing these conclusions, it is important to consider the characteristics of this population. It is possible that the higher dose of milnacipran may be more effective in patients with more severe or harder to treat depression.
Acknowledgments
We are grateful for the cooperation of the following clinical investigators who participated in the present study: H Ishikawa, H Maeda, W Miura, M Shimode, K Kaneta, N Hasegawa, M Shimizu, Y Chiba, M Nakayama, K Nakajima, H Tao, T Ikeda, R Matsubara, S Kashiwakura, T Hayashishita, F Okada, K Kawamura, M Tsukamoto, J Shiraki, K Kawaharada, M Kurita, K Tsunoda, K Ohtomo, Y Mori, T Oda, H Kanome, S Itagaki, H Enguchi, M Sanga, I Tadama, H Kusanagi, Y Miura, M Kimura, T Nakayama, H Sakamoto, T Ohtsubo, J Aoki, E Atarashi, Y Watanabe, N Harikae, K Sugimoto, M Shirai, K Tada, M Nankai, J Kobayashi, T Murata, H Muraoka, H Shibata, Y Aoki, I Asai, Y Miyasaka, H Ozawa, Y Itakura, H Harai, O Furukawa, K Tanaka, I Arii, Y Ikeuchi, S Gon, Y Matsumoto, S Nakayama, H Hayakawa, Y Kaneda, K Yoshikawa, J Sanada, N Imatoh, M Kimura, J Hatakeyama, I Kanagami, Y Ariyoshi, H Kaku, M Matsunaga, N Matsuguchi, S Sakamoto, T Okano. We also thank Mike Briley for his help in the preparation of the manuscript.
Disclosure
KK has received honoraria from Asahi-kasei Pharma, Astellas Pharma, Dainihon Sumitomo Pharma, Eli Lilly Japan, Esai, GlaxoSmithKline, Mochida Pharma, MSD, Otsuka Pharma, Pfizer, and Shionogi. He is an advisory board member of Abbott Japan, and has served as a consultant for Asahi-kasei Pharma. TK has received honoraria from Astellas Pharma, Eli Lilly Japan, Meiji Seika Pharma, Mitsubishi Tanabe Pharma, MSD, Otsuka Pharma, and Pfizer, and has received research grants from Astellas Pharma, Dainippon Sumitomo Pharma, GlaxoSmithKline, and Pfizer. He is also an advisory board member of GlaxoSmithKline, and has served as a consultant for Asahi-kasei Pharma. SH and EN are employed by Asahikasei Pharma.
References
- MochizukiDTsujitaRYamadaSNeurochemical and behavioral characterization of milnacipran a serotonin and noradrenaline reuptake inhibitor in ratsPsychopharmacology200216232333212122491
- StahlSMGradyMMMoretCBrileyMSNRIs: their pharmacology, clinical efficacy, and tolerability in comparison with other class of antidepressantsCNS Spectr20051073274716142213
- LecrubierYPletanYSollesATournouxAMagneVClinical efficacy of milnacipran: placebo-controlled trialsInt Clin Psychopharmacol199611 Suppl 429338923124
- AnsseauMvon FrenckellRGérardMAInterest of a loading dose of milnacipran in endogenous depressive inpatients. Comparison with the standard regimen and with fluvoxamineEur Neuropsychopharmacol199111131211821700
- Lopez-IborJGuelfiJDPletanYTournouxAProstJFMilnacipran and selective serotonin reuptake inhibitors in major depressionClin Psychopharmacol199611 Suppl 44146
- GuelfiJDAnsseauMCorrubleEA double-blind comparison of the efficacy and safety of milnacipran and fluoxetine in depressed inpatientsInt Clin Psychopharmacol1998131211289690979
- ClercGMilnacipran/Fluvoxamine Study GroupAntidepressant eff icacy and tolerability of milnacipran, a dual serotonin and noradrenaline reuptake inhibitor: a comparison with fluvoxamineInt Clin Psychopharmacol20011614515111354236
- SechterDVandelPWeillerEA comparative study of milnacipran and paroxetine in outpatients with major depressionJ Affect Disord20048323323615555719
- OliéJPGourionDMontagneARostinMPoirierMFMilnacipran and venlafaxine at flexible doses (up to 200 mg/day) in the outpatient treatment of adults with moderate-to-severe major depressive disorder: a 24-week randomized, double-blind exploratory studyNeuropsychiatr Dis Treat20106717920396639
- von FrenckellRAnsseauMSerreCSutetPPooling two controlled comparisons of milnacipran (F2207) and amitriptyline in endogenous inpatients. A new approach in dose ranging studiesInt Clin Psychopharmacol1990549562185303
- KasperSPletanYSollesATournouxAComparative studies with milnacipran and tricyclic antidepressants in the treatment of patients with major depression: a summary of clinical trial resultsInt Clin Psychopharmacol199611 Suppl 4S35S39
- Van AmerongenAPFerreyGTournouxAA randomised, double-blind comparison of milnacipran and imipramine in the treatment of depressionJ Affect Disord200272213112204314
- Lopez-IborJJConesaASpanish Milnacipran/Imipramine Study GroupA comparative study of milnacipran and imipramine in the treatment of major depressive disorderCurr Med Res Opin20042085586015200743
- KanemotoKMatsubaraMYamashitaKTaraoYInadaESekineTControlled comparison of two different doses of milnacipran in major depressive outpatientsInt Clin Psychopharmacol20041934334615486520
- OkumuraKFurukawaTARemission rates with milnacipran 100 mg/day and 150 mg/day in the long-term treatment of major depressionClin Drug Investig200626135142
- HayashiMMimuraMOtsuboTKamijimaKEffect of high-dose milnacipran in patients with depressionNeuropsychiatr Dis Treat2007369970219300602
- McEntegartDThe pursuit of balance using stratified and dynamic randomization techniques: an overviewDrug Inf J200337293308
- ClaghornJLKievARickelsKSmithWTDunbarGCParoxetine versus placebo: a double-blind comparison in depressed patientsJ Clin Psychiatry1992534344381487471
- FeighnerJPCohnJBFabreLFJrA study comparing paroxetine placebo and imipramine in depressed patientsJ Affect Disord19932871798354771
- JennerPNParoxetine: an overview of dosage, tolerability, and safetyInt Clin Psychopharmacol19926 Suppl 4S69S80
- KinoshitaTA double-blind, placebo-controlled study of a new antidepressant, mirtazapine, in depressed patientsJpn J Clin Psychopharmacol200912289306
- HiguchiTMurasakiMKamijimaKClinical evaluation of duloxetine in the treatment of major depressive disorder – placebo-and paroxetine-controlled double-blind comparative studyJpn J Clin Psychopharmacol20091216131634
- HirayasuYA dose-response and non-inferiority study evaluating the efficacy and safety of escitalopram in patients with major depressive disorder: a placebo- and paroxetine-controlled, double-blind, comparative studyJpn J Clin Psychopharmacol201114883899
- PuechAMontgomerySAProstJFSollesABrileyMMilnacipran, a new serotonin and noradrenaline reuptake inhibitor: an overview of its antidepressant activity and clinical tolerabilityInt Clin Psychopharmacol199712991089219045
- VandelPSechterDWeillerEPost-treatment emergent adverse events in depressed patients following treatment with milnacipran and paroxetineHum Psychopharmacol20041958558615570574