Abstract
Obstructive sleep apnea (OSA) is a common condition characterized by repetitive episodes of complete (apnea) or partial (hypopnea) obstruction of the upper airway during sleep, resulting in oxygen desaturation and arousal from sleep. Intermittent hypoxia (IH) resulting from OSA may cause structural neuron damage and dysfunction in the central nervous system (CNS). Clinically, it manifests as neurocognitive and behavioral deficits with oxidative stress and inflammatory impairment as its pathophysiological basis, which are mediated by microglia at the cellular level. Microglia are dominant proinflammatory cells in the CNS. They induce CNS oxidative stress and inflammation, mainly through mitochondria, reduced nicotinamide adenine dinucleotide phosphate oxidase, and the release of excitatory toxic neurotransmitters. The balance between neurotoxic versus protective and anti- versus proinflammatory microglial factors might determine the final roles of microglia after IH exposure from OSA. Microglia inflammatory impairments will continue and cascade persistently upon activation, ultimately resulting in clinically significant neuron damage and dysfunction in the CNS. In this review article, we summarize the mechanisms of structural neuron damage in the CNS and its concomitant dysfunction due to IH from OSA, and the potential roles played by microglia in this process.
Introduction
Obstructive sleep apnea (OSA), which is a major public health burden,Citation1 is a common condition characterized by repetitive episodes of complete (apnea) or partial (hypopnea) obstruction of the upper airway during sleep, resulting in oxygen desaturation and arousal from sleep.Citation2 Based on available population-based studies, OSA affects 3%–7% of adult men, 2%–5% of adult women,Citation3–Citation5 and up to 4% of children.Citation6,Citation7 This prevalence is much higher in some specific patient groups, such as those with congestive heart failure (40%),Citation8 end-stage kidney disease (50%),Citation9 and stroke (60%),Citation10 while less than 5% of all OSA patients receive appropriate treatment.Citation11,Citation12 At all ages, it is associated with complications in different organ systems, such as cardiovascular morbidity, hypertension, obesity, dyslipidemia, and insulin resistance.Citation13–Citation16 Moreover, both in children and adults, OSA causes behavioral and neuropsychological deficits in the central nervous system (CNS), including daytime sleepiness, depression,Citation17 impaired memory,Citation18 mood disorders, cognition deficiencies,Citation19 all of which are compatible with impaired hippocampal function,Citation7,Citation20 and there exists a significant correlation between disease severity and CNS functional deficiencies in patients with OSA.Citation7 Cognition deficiencies in OSA patients have typically been found in attention and vigilance, memory and learning, executive functions, and simulated driving. There is evidence showing that sleep parameters can rapidly be normalized with continuous positive airway pressure (CPAP) treatment, but those deficits in cognitive performance often persist.Citation1
Pathophysiologic changes from OSA include intermittent hypoxia (IH), intermittent hypercapnia, intrathoracic pressure changes, sympathetic activation, and sleep fragmentation.Citation21 Because of simultaneous occurrence it is very difficult to differentiate the effects of its two main pathologic traits – IH and sleep fragmentation – in clinical and bench studies of OSA. Many OSA studies utilize IH as the only exposure factor in OSA studies. These approaches simplify the research process and attain most of the academic goals.Citation7 IH and sleep fragmentation can independently and even synergistically lead to neuronal loss in the hippocampus and prefrontal cortex, areas closely associated with memory processes and executive functions.Citation22,Citation23 Veasey et alCitation24 and Polotsky et alCitation25 demonstrated that IH alone can result in increased sleepiness or altered sleep architecture, while Gozal et alCitation26 showed that initially disturbed sleep patterns could be normalized in the course of prolonged IH treatment.
IH can result in deficits in spatial learning and memory in rats that are most likely hippocampal-dependent and persistent, and consequently lead to diminished performance in standard water-maze tests.Citation7,Citation27 The precise roles IH plays in causing hippocampal impairments may involve multiple pathways, including ion-channel alterations,Citation28 glutamate excitotoxicity, oxidative stress, mitochondrial dysfunction, upregulation of proinflammatory mediators, altered regulation of pro- and antiapoptotic gene cascades,Citation29,Citation30 diminished apolipoprotein E,Citation31 and nitric oxide (NO) overproduction.Citation32 It has been reported that IH in the CNS is related to oxygen-reactive species (ROS) production, oxidative stress over-activation, and neuronal apoptosis and/or necrosis. These deleterious effects would contribute to the development of cognitive impairments associated with OSA.Citation33 Microglia are a robust source of oxidative stress in CNS, and microglial mitochondria and reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase are the predominant sources of ROS,Citation27 while intracellular ROS are critical for self-activation of microglia and the overproduction of proinflammatory factors.Citation34
The CNS is composed of two major cell types: neuronal cells and glial cells. Glial cells consist of astrocytes, oligodendrocytes, and microglia. For a long time, they were considered to be a largely uninteresting matrix; “glue” elements that only contributed to feeding and supporting neurons. However, recent evidence has been accumulating that suggests glial cells, in particular microglia, are in fact very important elements in CNS development, repair, and neurotransmission. They are also key elements in CNS vascularization, inflammation, and neuroprotection.Citation35 Microglial cells were first described by Del Rio Hortega et al.Citation36 They are resident immune cells in the CNS and occupy approximately 5% of the total cell population in the brain. During normal CNS conditions, they exhibit a ramified morphology and a constitutive expression of molecules associated with phagocytosis, a state traditionally termed “resting microglia.”Citation36 Recently, many studies have indicated that these so-called resting microglia are not really resting or quiescent, but are rather continually surveying their microenvironment around them with extremely fine protrusions. Microglia are especially prone to be activated in response to brain injuries and inflammatory or immunological stimuli, and undergo dramatic morphological alterations upon activation, changing from resting ramified microglia into activated ameboid microglia. During activation, their surface molecules, such as complement receptors, cytokines, chemokines, ROS, reactive nitrogen species, proinflammatory receptors, and major histocompatibility complex molecules, will be upregulated simultaneously. Meanwhile, a variety of soluble factors, which are proinflammatory in nature and potentially cytotoxic, will be released by activated microglia. Not only acutely contributing to neuronal loss when activated, microglia are also involved in chronic CNS inflammation, such as in Alzheimer’s and Parkinson’s diseases, neuropathic pain, cerebral ischemia, or stroke.Citation36–Citation38 In this review article, we summarize the mechanisms of structural neuron damage in the CNS and their concomitant dysfunction from IH of OSA, and the potential roles played by microglia in this process.
OSA IH may cause structural neuron damage, and CNS dysfunction may be complicated
OSA IH may cause structural neuron damage in the CNS.Citation39 The CNS, especially the cerebral cortex, is particularly susceptible to ischemia.Citation40 Both vascular injuries and disturbances in blood flow may contribute to neural loss. A study measured two serum markers for cerebrovascular disease – soluble CD40 ligand and soluble platelet selectin. Both of these markers were higher in patients with OSA and declined upon effective treatment with nasal CPAP.Citation41 There is evidence suggesting that the hippocampus may become atrophic in patients with OSA. A study reported quantitative regional gray-matter loss in the hippocampus, cerebellum, frontal and parietal cortex, and the anterior cingulate gyrus in OSA.Citation42 In another study, neurocognitive deficits and corresponding brain-morphology changes were reported in adults with severe OSA before and after 3 months of CPAP treatment compared with age-matched healthy controls. The authors found that patients with untreated OSA at baseline compared with controls had significantly reduced gray-matter volumes in the left posterior parietal, right superior frontal, and left parahippocampal gyri; gray-matter volume correlated with errors on executive-function tests. After 3 months of CPAP, the individuals with OSA showed significant improvements involving memory, attention, and executive functioning that paralleled gray-matter volume increases in hippocampal and frontal structures.Citation43 With gray-matter volume reductions up to 18% in some regions of the brain in patients with OSA, the extent of decline increased with the severity of the syndrome. The structural neuron damage in the CNS may be a consequence of OSA or contribute to the genesis or maintenance of OSA.Citation40 OSA IH may contribute to frontal impairment and result in executive dysfunctionCitation23 and memory impairment. This means that the hippocampus, a region associated with memory processing, may be impaired.Citation42 Recent studies have almost confirmed the relationship between structural neuron damage in the CNS and cognitive impairments; and particularly in children, studies have found significant decrements in intelligence quotient (IQ) (15 points), verbal working memory, and verbal fluency.Citation44
Functional magnetic resonance imaging (MRI) with spectroscopy may be used to examine metabolic disturbances in the CNS, mainly because of structural neuronal damage and gliosis. For example, a reduced N-acetylaspartate (NAA)-to-choline ratio was found for the cerebral white matter, where the apnea–hypopnea index (AHI; which is used to evaluate the severity of OSA) correlated negatively with the NAA-to-choline ratio.Citation45 Structural neuronal damage in the CNS with reduced activation responses in some brain regions resulted in cognitive dysfunction,Citation46 while some studies demonstrated overactivation of the bilateral inferior and middle frontal gyri, cingulate gyrus, the temporoparietal junction, the thalamus, and the cerebellum in patients with OSA.Citation47 This kind of activation may be a compensatory mechanism, but may produce another type of CNS impairment. A recent study with functional MRI found reduced prefrontal cortex activation in severe OSA subjects, with impaired verbal memory.Citation48 In addition, they found subjects with OSA had less activation of the prefrontal cortex while performing a working-memory task. A similar deficit was observed in hypoxic and nonhypoxic subjects with OSA, suggesting that hypoxia may not influence the deficit in prefrontal cortical activation during learning. In contrast, hypoxic subjects showed far less activation in the parietal cortex. This suggests that there are regional differences for hypoxia-sensitive neural tissue in the structure and function of the CNS.Citation41 In some functional MRI studies of patient populations, the left middle occipital gyrus, the right middle orbital gyrus, and the caudal pons were less activated in patients with OSA, as brainstem mechanisms of respiratory control are considered to be involved in the pathogenesis of OSA.Citation48 Another MRI study showed medial pontine damage and less activation in symptomatic OSA patients.Citation49 CNS structural injury and functional and metabolic deficits in OSA occur in limbic regions classically associated with negative emotions, such as the amygdala, hippocampus, insular, and cingulate cortices.Citation50 These sites help to mediate emotional behaviors such as fear (amygdala, hippocampus) and dyspnea (insula, cingulate cortex).Citation51 In addition, if OSA subjects show anxiety, or blood pressure changes accompanying emotion, the affected brain regions may include the ventral medial prefrontal, cingulate, parietal and insular cortices, and the uncus of the hippocampal formation, extending to the amygdala.Citation48,Citation52 Using a newer, more sensitive methodology – diffusion tensor imaging with fractional anisotropy – Macey et al identified substantial white-matter lesions in OSA, largely throughout the brain but with more pronounced changes in the cortices, limbic system, pons, and cerebellar tracts.Citation53 A similar study found that severe OSA subjects with sleepiness, depression, and neurobehavioral abnormalities had reduced gray-matter volumes in the hippocampus and frontoparietal cortices.Citation54 With CPAP therapy, cognitive performance normalized in some patients, as did gray-matter lesions. But in other patients, the abnormalities were not reversed with therapy. Consistently, there were persistent reductions in neuron-metabolite measures, suggesting injured, nonviable neurons in particular brain regions, including the frontal cortex and hippocampus. The most important point was that this structural neuronal damage in the CNS was also observed in children.Citation55
Besides functional MRI, other imaging methodologies could also be used to examine structural neuronal damage in the CNS and cognitive dysfunction. For example, positron emission computed tomography (PET-CT) for the brain can provide insight into regional brain metabolism and may also provide insight into abnormalities in specific neurochemical transmission. A study with PET-CT found four of seven subjects had impaired glucose utilization in the frontal and/or temporal cortex, and one additional subject had reduced glucose utilization in the parietal cortex.Citation41
IH may result in CNS neuronal apoptosis and dysfunction
A study found that IH can cause a general compromise of oxidative phosphorylation and consequently poor maintenance of ion gradients of neurons. That is to say, the physiological function of the neurons may be compromised before explicit neuronal apoptosis. Until now, there have been few attempts to examine the direct effects of IH on the excitability of hippocampal neurons and their synaptic transmission in the animal model of OSA. Nevertheless, it has been shown that in the developing nervous system, IH will affect neuronal excitability and its maturation by altering the expression of Na channels and ion transporters. In adult mice, IH could decrease membrane-input resistance and excitability of hippocampal CA1 neurons.Citation56 Exposure to IH during sleep does not induce substantial sleep disturbances, but is associated with impaired spatial learning in the adult rat, as well as with increased apoptosis in the cortex and CA1 region of the hippocampus.Citation57 Apoptosis in the hippocampal CA1 region and the frontoparietal cortex peaked at 1 or 2 days of IH and decreased thereafter,Citation27 with learning impairments and spatial memory deterioration.Citation26,Citation41 Another study found developing rats at postnatal ages 10–25 days exhibit significantly more extensive apoptosis in these neural regions when exposed to 48 hours of IH, indicating an exposure to IH in a developmentally regulated period may enhance susceptibility to OSA IH. In other words, a slowly evolving or weak neural excitotoxicity that may occur as a consequence of impaired cellular energy metabolism, free radical production, and/or modifications in glutamate ion/receptor complexes may finally lead to neuronal apoptosis.Citation57 IH could trigger apoptosis programs in neurons in areas including the hippocampus and cortex, which could lead to cytoarchitectural disorganization.Citation56 Other research indicated that IH could result in cell death in the hippocampus as well as forebrain regions via mechanisms of apoptosis, which may represent the basis for the clinical complications of OSA.Citation58
CNS neuronal impairments and apoptosis from IH may involve other mechanisms. For instance, brain-derived neurotrophic factor (BDNF) is an important neural regulator of CNS function. The expression of BDNF is reduced significantly after IH exposure. However, the levels of some other neurotrophic factors are not decreased, indicating that the effect of IH on BDNF is specific. Meanwhile, these results suggest that the number and frequency of IH could be major factors in determining the effect on BDNF expression, and also argue against the possibility that the decrease in BDNF level is simply due to neuronal number loss.Citation56 Neurotrophic factors such as BDNF can significantly prevent neuronal damage in the CNS caused by oxidative stress, as shown in neurodegenerative diseases,Citation59 or as shown in in vitro cultures against ROS generation and activity directly.Citation60 Thus, it is possible that the lack of BDNF in chronic IH not only contributes to impaired long-term synaptic plasticity but also reduces the neutralizing effect for ROS, and may lead to the increase of neuronal apoptosis induced by ROS. As described above, oxidative stress induced by IH- and ROS-induced apoptosis are two closely related factors that are widely accepted as contributing to CNS damage during IH exposure.Citation56
Oxidative stress from microglia may cause inflammatory damage in CNS during IH exposure
Microglia are major inflammatory cells in the CNS. Recently, some studies examining hippocampal impairments induced by microglia were published.Citation61–Citation64 The role of CNS inflammation and microglia activation in adult neurogenesis has turned out to be very complicated. Recent evidence indicates that microglia can support the different steps in neurogenesis; progenitor proliferation, survival, migration, and differentiation; as well as clearance of dead cells and secretion of neurotrophins.Citation61 Microglia are the primary supervisor of microenvironmental changes in the CNS. Like systemic macrophages, once activated they produce tumor necrosis factor (TNF)-α, which acts in an autocrine and paracrine manner to activate the population of immune cells across the CNS.Citation62 And like peripheral immune cells, microglia also increase the production of inflammatory cytokines to clear the pathogens. This kind of CNS inflammation is generally beneficial; but unfortunately, this beneficial process sometimes gets out of balance and the CNS inflammatory process persists, even when those proinflammatory stimuli are eliminated.Citation63 Neuroinflammation is not an epiphenomenon, nor a mere scavenger of other, putatively more important, pathogenic mechanisms. This inflammation may begin with microglial activation, and may end in both beneficial and harmful effects simultaneously.Citation64
Microglia mediate oxidative stress and inflammation in the CNS mainly through mitochondria, NADPH oxidase, and the release of excitatory toxic neurotransmitters. IH can produce masses of intracellular ROS. Oxidative stress occurs when ROS are produced at a greater rate than they are removed, rendering the activities of antioxidative enzymes and the levels of endogenous antioxidants insufficient, but ultimately resulting in oxidative damage to lipids, proteins, and/or nucleic acids.Citation7,Citation26 Mitochondria and NADPH oxidase are the predominant sources of ROS in the CNS and play crucial roles in IH-induced neuronal impairments.Citation27 Mitochondria are the major cellular source of ROS in most nonphagocytic cells under normal conditions. While commonly regarded as “by-products” of adenosine triphosphate production through oxidative phosphorylation, ROS produced in mitochondria play indispensable roles in various cellular processes, including metabolic regulation, oxygen sensing, and preconditioning, and their production is actively and tightly regulated.Citation65 In primary cortical neurons, the transition phase from hypoxia to normoxia during IH appears to generate more ROS than the transition phase from normoxia to hypoxia or hypoxia alone, all of which generate more ROS than control normoxic conditions. The results of using targeted inhibitors of the major pathways underlying ROS generation in the cell membrane, cytosol, and mitochondria show that the mitochondria emerge as the predominant source of ROS generation during IH in cortical neurons, and that overexpression of manganese superoxide dismutase decreases IH-mediated cortical neuronal impairments and apoptosis, and reduces spatial learning deficits assessed by the Morris water-maze assay.Citation66
NADPH oxidase is a multisubunit enzyme complex, and may contribute directly to IH-associated injury. For example, evidence demonstrates that IH results in irreversible wake impairments.Citation27 IH induces oxidative and nitrative stress in regions of wake-active neurons. Mice with NADPH oxidase inhibited or mice with transgenic absence of the NADPH oxidase isoform that is upregulated in response to IH do not develop wake neuron injury or sleepiness. Moreover, mice with absent or inhibited NADPH oxidase develop less inflammatory responses to IH in the CNS, and less nitration and oxidation.Citation67 The activation of NADPH oxidase would then lead to the observed increases in carbonylation, lipid-peroxidation injury, and downstream proinflammatory responses in the CNS after IH exposure.Citation60 NADPH oxidase may be important in conditions in which there is overactivation of glutamate receptors and concomitant release of NO. While NO normally functions as a physiological neuronal mediator, and as a free radical, NO can mediate cellular toxicity by damaging critical metabolic enzymes and by reacting with ROS, such as superoxide, to form an even more potent oxidant – peroxynitrite.Citation27 The other enzyme associated with the release of NO is inducible nitric oxide synthase (iNOS). IH can induce iNOS to produce reactive nitrogen species, lead to nitration of protein and lipid peroxidation, and ultimately result in CNS damage, especially hippocampal injury. IH also can induce iNOS to produce abnormally high levels of glutamate and cause excitotoxicity in hippocampal neurons of the CNS.Citation7,Citation68 It has also been reported that NADPH oxidase and hypoxia inducible factor 1α have bidirectional positive feedback on each other during IH exposure; so that inhibiting or reducing one lowers the IH response of the other and in doing so lessens injury that IH mediates. IH can activate angiotensin 1 and result in NADPH oxidase activation, and so angiotensin 1α antagonists can confer protection across time in cognitive dysfunction and related CNS morphology changes in individuals with OSA.Citation55
While mitochondria and NADPH oxidase are each capable of producing superoxide independently, emerging evidence suggests the existence of cross talk between the two cellular systems in which they appear to be costimulatory.Citation69 The existence of such a cross-talk mechanism between NADPH oxidase and mitochondria as described above would allow the two systems to form a positive-feedback loop that amplifies the original signal regardless of its origin, and result in the increase of cellular oxidative stress and eventual structural neuronal damage in the CNS.Citation27 The causal relationships between enhanced oxidative stress and neuronal cell damage are also supported by various antioxidant/antioxidant enzyme approaches.Citation7 Compared with systemic tissues, the CNS will generate more oxygen free radicals, largely due to the low levels of antioxidant enzymes, the relatively large amount of oxygen consumed, the high lipid content, and the more redox metal ions,Citation70 and this type of IH-induced oxidative stress plays a key role in the pathogenesis of hippocampal injury especially. The unique sensitivity of the CNS to alterations in oxygen homeostasis demonstrates that neural damage and behavioral consequences observed in OSA patients are associated with IH.Citation7 The cumulative evidence supports the view that ROS plays a major intermediary role in the related impairments of IH on hippocampal and other selected CNS structures. Involvement of other ROS-producing systems in OSA-related neuropathology has not been thus far either explored or confirmed. However, such involvement should not be excluded, and definitely warrants additional future investigation.Citation27
The potential role played by microglia in structural neuronal impairments and dysfunction of CNS during IH exposure
A recent study demonstrated that IH-induced neuron apoptosis and CNS dysfunction could in fact be orchestrated largely by microglia-mediated oxidative stress and inflammatory impairments at the cellular level. In some neurodegenerative diseases, such as multiple sclerosis, Alzheimer’s disease, stroke, and Parkinson’s disease, inflammation has been implicated in these diseases, and the involvement of microglia and their important roles have been clearly shown. As previously discussed, IH-induced neuron apoptosis and CNS dysfunction may involve multiple pathways. The most prevalent explanation is oxidative stress, including the resultant inflammation and apoptosis hypotheses. Mitochondria and NADPH oxidase may be the predominant sources of ROS, and microglial mitochondria and NADPH oxidase are a robust source in the CNS. In turn, intracellular ROS are critical for activation of microglia and the enhancement of the production of proinflammatory factors.Citation35 It has been reported that a variety of factors that pose a potential threat to CNS inflammatory substance homeostasis, including bacterial, viral, and fungal structures, abnormal endogenous proteins, complement factors, antibodies, cytokines, inflammatory peptides, and multiple neurotoxins, are sensed by microglial receptors and subsequently induce microglial activation.Citation36
A number of mechanisms by which IH inflammation-induced microglia result in neuron apoptosis and CNS dysfunction have been identified. Phagocytic NADPH oxidase found in microglia can be acutely activated by inflammation.Citation71 NADPH oxidase contributes to microglial activation and microglia-mediated neurotoxicity through two mechanisms. First, activation of NADPH oxidase results in the production of extracellular ROS that are toxic to neurons and the superoxide anion. Second, activation of NADPH oxidase causes an increase in microglial intracellular ROS. This kind of increase in production of ROS is speedy, usually occurring within minutes, and is typically measured in microglia after lipopolysaccharide simulation. After activation, downstream proinflammatory factors (interleukin [IL]-1β, TNF-α, IL-6) that are toxic to neurons will be produced immediately. These factors can function synergistically to produce inflammatory neuronal damage and apoptosis in the CNS. Among these factors released, superoxide is essential for both the amplification and induction of neurotoxicity. Evidence suggests that neuron apoptosis and CNS dysfunction mainly result from microglia-mediated inflammatory changes in the CNS; NADPH oxidase is the key enzyme for producing ROS in the activation of microglia. ROS generated from NADPH oxidase are an essential pathway regulating the microglial expression of neurotoxic proinflammatory factors.Citation62 Another pathway of microglia-induced neuron apoptosis and CNS dysfunction is the release of excitatory toxic neurotransmitters such as glutamate. IH-related inflammation can include iNOS, which is expressed on microglia only during inflammatory conditions. High levels of iNOS expression in microglia cause NO inhibition of neuronal respiration, resulting in neuronal depolarization and glutamate release, with the glutamate released by microglia, which through the activation of neuronal N-methyl-D-aspartic acid receptors can induce neuronal death and CNS dysfunction. Because NO can inhibit cytochrome oxidase in competition with oxygen, hypoxia strongly synergizes with iNOS expression to induce neuronal death. This sensitization to hypoxia is potentially important in stroke, trauma, vascular dementia, Alzheimer’s disease, and brain aging, where both inflammation and hypoxia may coexist.Citation38,Citation62
Compared with other organs, the inflammation in CNS depends more on the nuclear factor (NF)-κB signal-transduction pathway.Citation62 A recent study suggests that proteases known to modify the extracellular matrix may be a critical factor through which damaged neurons activate microglia to produce extracellular superoxide.Citation35 Stressed or damaged neurons send signals to resident microglia, and the latter produce specific chemokines to cause recruitment of bone marrow-derived microglia. A large number of endogenous ligands for Toll-like receptors express on the surface of microglia and then trigger NF-κB signaling. This may lead to a subtle inflammatory response and consequently chemotaxis of microglia. Therefore, neurons, especially those in danger, can interact with microglia and stimulate their proliferation and recruitment. This process will influence several signals and impact on stressed, injured, or infected neurons.Citation62 Imaging of microglia, such as functional PET or MRI, may be able to help in the early assessment of CNS inflammation, monitor the severity and progression of CNS disease, and evaluate the effectiveness of CNS therapies.Citation36 In neurodegenerative disease patients and animal models, PET will find microglial activation and detect the specific position of disease with the specific ligand of benzodiazepine receptor.Citation35 And it was reported that similar results could be obtained by using oxide nanoparticles such as ultrasmall superparamagnetic iron oxide (USPIO) as cell-specific contrast agents for MRI.Citation72
CNS microglia activation is a phenotypically and functionally bidirectional process, which is dependent on stimulus type, stimulus intensity and context.Citation73 Microglia form the first line of defense for the CNS parenchyma, but uncontrolled activation of microglia may directly or indirectly toxic to CNS neurons by releasing various inflammatory cytokines.Citation74 From the defensive insight, microglia may have neuroprotective and anti-inflammatory potential. Similar to peripheral macrophages, microglia can be induced into a classical (pro-inflammatory) activation state, but are also capable of entering an alternative (anti-inflammatory) activation state involved in tissue repair and extracellular matrix remodeling. Moreover, some other studies demonstrated that activated microglia phagocytose not only damaged cell debris but also neighboring intact cells.Citation75–Citation77 This further supports their active participation in self-repair and self-update cycles. The balance between neurotoxic versus protective and anti-versus pro-inflammatory microglial factors might determine the role of microglia in a given disease or condition.Citation36 Neurotoxic substances are frequently used to produce animal models of acute injuries or diseases and they may activate microglia either directly or indirectly by their ability to cause neuronal death and demyelination. Resident microglia and those derived from the bone marrow play important roles in the stimulation of myelin repair, removal of toxic proteins from the CNS and the prevention of neurodegeneration. CNS inflammatory process is driven by activated microglia and the induction of proinflammatory molecules and related signaling pathways, leading to both synaptic and neuronal damage as well as further microglia activation. There is a fine regulation in this process to avoid an exaggerated response by microglial cells. Inflammation inhibitory factors (regulatory factors) include anti-inflammatory cytokines (IL-10, transforming growth factor [TGF]-β), inhibitory proteins (nuclear transcription factor inhibitory protein [I]-κBα, mitogen-activated protein kinase phosphatase [MKP]-1, suppressors of cytokine signaling [SOCS]) and the release of glucocorticoids (GCs) by the adrenal gland. In CNS inflammatory process, GCs are probably the most powerful endogenous inhibitors of the innate immune reaction.Citation62 Learning how to harness the positive side of microglia, as well as to suppress their negative side represents a major scientific and clinical problem which is need to be resolved.Citation38
CNS microglia consistently generate ROS when activated by environmental factors (lipopolysaccharide, diethyl phthalate, rotenone, paraquat), endogenous protein toxins (β-amyloid peptide, α-synuclein), and neuronal injury. On the other hand, the production of pro-inflammatory factors such as NO, TNF-α or phenyl glycidyl ether from microglia can further activate other microglia and produce ROS.Citation78 Activation of microglial phagocytic NADPH oxidase alone causes no neuronal death, but when iNOS is overexpressed, they will result in extensive neuronal death via peroxynitrite production. At present, it is assumed that the activation of microglia in response to injury, illness, aging or other causes begins a cascade of events. It manifests that proinflammatory cytokines are overexpressed by the activated microglia, and lead to large quantities of neuronal damage and death through all kinds of channels. Dying or damaged neurons have the potential to activate microglia, regardless of how the neurons are damaged (environmental toxin, endogenous disease protein, or reactive microgliosis) or neurodegenerative disease. depicts the relationships between neuronal damage and microglial activation and characterizes how damaged neurons will activate microglia to initiate a self-propelling cycle of neuron death. Reactive microgliosis will release more pro-inflammatory factors in a self-cycling, self-sustaining and self-amplifying fashion. Over a period of years, this slow, smoldering inflammatory process in the CNS destroys sufficient numbers of neurons to cause the clinical signs of CNS diseases.Citation35
Figure 1 The potential mechanisms of microglia-induced structural neuron damage during IH exposure from OSA.
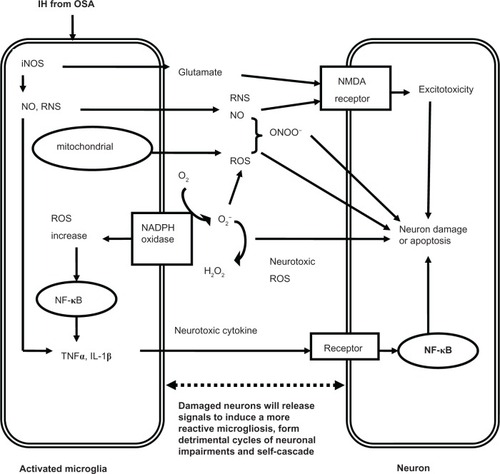
At present, little is known about the mechanisms of structural neuron damage in the CNS and the concomitant dysfunction during IH exposure from OSA, and the potential roles played by microglia in this process. Questions left unanswered include: Which type of patients with OSA are at risk for CNS inflammatory damage? Can we prevent the progression of CNS inflammation from OSA IH? What CNS injuries in OSA patients are reversible? Are there markers, such as evoked potentials or electromyography, able to predict CNS inflammatory injuries and dysfunction in OSA patients? We need to piece together these comprehensive phenotypes (imaging, electrophysiology, and behavioral testing) to answer these actual questions and uncover these mechanisms.Citation55 From this article, we may conclude some preliminary pathophysiological pathways through which IH from OSA can cause structural neuron damage in the CNS:
IH resulting from OSA may cause structural neuron damage and dysfunction in the CNS (especially in the hippocampus). Clinically, it manifests as neurocognitive and behavioral deficits.
At the cellular level, impairments and dysfunction of the CNS, especially in the hippocampus, may be orchestrated largely by microglia-mediated oxidative stress and inflammation. Microglia are dominant pro-inflammatory cells in CNS. They induce CNS oxidative stress and inflammation mainly through mitochondria, NADPH oxidase and the release of excitatory toxic neurotransmitters.
The inflammatory process mediated by microglia is a double-edged sword. The balance between neurotoxic versus protective and anti- versus pro-inflammatory microglial factors might determine the final roles played by microglia after IH exposure from OSA.
Microglia inflammatory impairments will continue and cascade persistently upon activation, ultimately resulting in clinically significant neuron damage and dysfunction in the CNS.
Therefore, the crucial roles of microglia-mediated inflammation in CNS damage needs further exploration. Drugs and other individual treatments which can selectively alter microglial function in such a way that they can promote neuroprotection, decrease the release of pro-inflammatory mediators, control infectious pathogens and suppress deleterious inflammatory impairment to the CNS simultaneously, may access more therapeutic objectives.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81270144, 30800507, 81170071).
Disclosure
None of the authors in the study group has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- StaatsRStollPZinglerDVirchowJCLommatzschMRegulation of brain-derived neurotrophic factor (BDNF) during sleep apnoea treatmentThorax20056068869216061712
- SaunamäkiTHimanenSLPoloOJehkonenMExecutive dysfunction in patients with obstructive sleep apnea syndromeEur Neurol20096223724219672077
- FengJChenBYPrevalence and incidence of hypertension in obstructive sleep apnea patients and the relationship between obstructive sleep apnea and its confoundersChin Med J (Engl)20091221464146819567173
- YoungTPeppardPEGottliebDJEpidemiology of obstructive sleep apnea: a population health perspectiveAm J Respir Crit Care Med20021651217123911991871
- National Heart, Lung, and Blood Institute Working Group on Sleep ApneaSleep apnea: is your patient at risk?Am Fam Physician1996532472538546051
- LumengJCChervinRDEpidemiology of pediatric obstructive sleep apneaProc Am Thorac Soc2008524225218250218
- FengJWuQZhangDChenBYHippocampal impairments are associated with intermittent hypoxia of obstructive sleep apneaChin Med J (Engl)201212569670122490498
- SinDDFitzgeraldFParkerJDNewtonGFlorasJSBradleyTDRisk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failureAm J Respir Crit Care Med19991601101110610508793
- WetterTCBöhmGBArticle reviewed: Improvement of sleep apnea in patients with chronic renal failure who undergo nocturnal hemodialysisSleep Med2001246346514592400
- YaggiHKConcatoJKernanWNLichtmanJHBrassLMMohseninVObstructive sleep apnea as a risk factor for stroke and deathN Engl J Med20053532034204116282178
- YoungTPaltaMDempseyJSkatrudJWeberSBadrSThe occurrence of sleep-disordered breathing among middle-aged adultsN Engl J Med1993328123012358464434
- YoungTEvansLFinnLPaltaMEstimation of the clinically diagnosed proportion of sleep apnea syndrome in middle-aged men and womenSleep1997207057069406321
- LavieLOxidative stress – a unifying paradigm in obstructive sleep apnea and comorbiditiesProg Cardiovasc Dis20095130331219110132
- LévyPBonsignoreMREckelJSleep, sleep-disordered breathing and metabolic consequencesEur Respir J20093424326019567607
- SateiaMJNeuropsychological impairment and quality of life in obstructive sleep apneaClin Chest Med20032424925912800782
- CarotenutoMSantoroNGrandoneAThe insulin gene variable number of tandemrepeats (INS VNTR) genotype and sleep disordered breathing in childhood obesityJ Endocrinol Invest20093275275519574727
- CarotenutoMBruniOSantoroNDel GiudiceEMPerroneLPascottoAWaist circumference predicts the occurrence of sleep-disordered breathing in obese children and adolescents: a questionnaire-based studySleep Med2006735736116713341
- CarotenutoMEspositoMParisiLDepressive symptoms and childhood sleep apnea syndromeNeuropsychiatr Dis Treat2012836937322977304
- DaulatzaiMADeath by a thousand cuts in Alzheimer’s disease: hypoxia-the prodromeNeurotox Res201324221624323400634
- CarotenutoMGimiglianoFFiordelisiGRubertoMEspositoMPositional abnormalities during sleep in children affected by obstructive sleep apnea syndrome: the putative role of kinetic muscular chainsMed Hypotheses20138130630823660129
- SuzukiYJJainVParkAMDayRMOxidative stress and oxidant signaling in obstructive sleep apnea and associated cardiovascular diseasesFree Radic Biol Med2006401683169216678006
- PrilipkoOHuynhNSchwartzSTask positive and default mode networks during a parametric working memory task in obstructive sleep apnea patients and healthy controlsSleep201134293A301A21358846
- SforzaERocheFSleep apnea syndrome and cognitionFront Neurol201238722661967
- VeaseySCDavisCWFenikPLong-term intermittent hypoxia in mice: protracted hypersomnolence with oxidative injury to sleep-wake brain regionsSleep20042719420115124711
- PolotskyVYRubinAEBalbirAIntermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouseSleep Med2006771616309961
- GozalDDanielJMDohanichGPBehavioral and anatomical correlates of chronic episodic hypoxia during sleep in the ratJ Neurosci2001212442245011264318
- WangYZhangSXGozalDReactive oxygen species and the brain in sleep apneaRespir Physiol Neurobiol201017430731620833273
- BakerTLFullerDDZabkaAGMitchellGSRespiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapniaRespir Physiol2001129253511738644
- GoldbartARowBWKheirandishLIntermittent hypoxic exposure during light phase induces changes in cAMP response element binding protein activity in the rat CA1 hippocampal region: water maze performance correlatesNeuroscience200312258559014622901
- GozalDRowBWGozalETemporal aspects of spatial task performance during intermittent hypoxia in the rat: evidence for neurogenesisEur J Neurosci2003182335234214622195
- O’HaraRThe role of hypoxia in apolipoprotein E4-associated cognitive impairment: implications for neurodegeneration and sleep-disordered breathingSleep2005281355135716335323
- ZhanGFenikPPraticoDVeaseySCInducible nitric oxide synthase in long-term intermittent hypoxia: hypersomnolence and brain injuryAm J Respir Crit Care Med20051711414142015750040
- AlmendrosIFarréRPlanasAMTissue oxygenation in brain, muscle, and fat in a rat model of sleep apnea: differential effect of obstructive apneas and intermittent hypoxiaSleep2011341127113321804675
- BadoerEMicroglia: activation in acute and chronic inflammatory states and in response to cardiovascular dysfunctionInt J Biochem Cell Biol2010421580158520638485
- BlockMLHongJSMicroglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanismProg Neurobiol200576779816081203
- VennetiSWileyCAKoflerJImaging microglial activation during neuroinflammation and Alzheimer’s diseaseJ Neuroimmune Pharmacol2009422724319052878
- BedardKKrauseKHThe NOX family of ROS-generating NADPH oxidases: physiology and pathophysiologyPhysiol Rev20078724531317237347
- RockRBPetersonPKMicroglia as a pharmacological target in infectious and inflammatory diseases of the brainJ Neuroimmune Pharmacol2006111712618040778
- RaeCBartlettDJYangQDynamic changes in brain bioenergetics during obstructive sleep apneaJ Cereb lood Flow Metab20092914211428
- MaceyPMHendersonLAMaceyKEBrain morphology associated with obstructive sleep apneaAm J Respir Crit Care Med20021661382138712421746
- DempseyJAVeaseySCMorganBJO’DonnellCPPathophysiology of sleep apneaPhysiol Rev2010904711220086074
- JacksonMLHowardMEBarnesMCognition and daytime functioning in sleep-related breathing disordersProg Brain Res2011190536821531244
- Grigg-DambergerMRallsFCognitive dysfunction and obstructive sleep apnea: from cradle to tombCurr Opin Pulm Med20121858058722990657
- HalbowerACDegaonkarMBarkerPBChildhood obstructive sleep apnea associates with neuropsychological deficits and neuronal brain injuryPLoS Med20063e30116933960
- KambaMInoueYHigamiSSutoYOgawaTChenWCerebral metabolic impairment in patients with obstructive sleep apnoea: an independent association of obstructive sleep apnoea with white matter changeJ Neurol Neurosurg Psychiatry20017133433911511706
- PetrellaJRWangLKrishnanSCortical deactivation in mild cognitive impairment: high-field-strength functional MR imagingRadiology200724522423517885190
- AyalonLAncoli-IsraelSKlemfussZShalautaMDDrummondSPIncreased brain activation during verbal learning in obstructive sleep apneaNeuroimage2006311817182516626972
- CastronovoVCanessaNStrambiLFBrain activation changes before and after CPAP treatment in obstructive sleep apneaSleep2009321161117219750921
- CrossRLKumarRMaceyPMNeural alterations and depressive symptoms in obstructive sleep apnea patientsSleep2008311103110918714782
- HaslerGFrommSAlvarezRPLuckenbaughDADrevetsWCGrillonCCerebral blood flow in immediate and sustained anxietyJ Neurosci2007276313631917554005
- EvansKCBanzettRBAdamsLMcKayLFrackowiakRSCorfieldDRBOLD fMRI identifies limbic, paralimbic, and cerebellar activation during air hungerJ Neurophysiol2002881500151112205170
- MaceyKEMaceyPMWooMAInspiratory loading elicits aberrant fMRI signal changes in obstructive sleep apneaRespir Physiol Neurobiol2006151446015993658
- MaceyPMKumarRWooMAValladaresEMYan-GoFLHarperRMBrain structural changes in obstructive sleep apneaSleep20083196797718652092
- CanessaNCastronovoVCappaSFObstructive sleep apnea: brain structural changes and neurocognitive function before and after treatmentAm J Respir Crit Care Med20111831419142621037021
- VeaseySCPiecing together phenotypes of brain injury and dysfunction in obstructive sleep apneaFront Neurol2012313923087666
- XieHYungWHChronic intermittent hypoxia-induced deficits in synaptic plasticity and neurocognitive functions: a role for brain-derived neurotrophic factorActa Pharmacol Sin20123351022212429
- RowBWKheirandishLNevilleJJGozalDImpaired spatial learning and hyperactivity in developing rats exposed to intermittent hypoxiaPediatr Res20025244945312193683
- ZhangJHFungSJXiMSampognaSChaseMHRecurrent apnea induces neuronal apoptosis in the guinea pig forebrainExp Neurol200921629029419124019
- NumakawaTMatsumotoTNumakawaYRichardsMYamawakiSKunugiHProtective action of neurotrophic factors and estrogen against oxidative stress-mediated neurodegenerationJ Toxicol2011201140519421776259
- BoutaharNReynaudELassabliereFBorgJBrain-derived neurotrophic factor inhibits cell cycle reentry but not endoplasmic reticulum stress in cultured neurons following oxidative or excitotoxic stressJ Neurosci Res2010882263227120209632
- KhanSANanduriJYuanGNADPH oxidase 2 mediates intermittent hypoxia-induced mitochondrial complex I inhibition: relevance to blood pressure changes in ratsAntioxid Redox Signal20111453354220618070
- GlezerISimardARRivestSNeuroprotective role of the innate immune system by microgliaNeuroscience200714786788317459594
- Van EldikLJThompsonWLRalay RanaivoHBehannaHAMartin WattersonDGlia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approachesInt Rev Neurobiol20078227729617678967
- RogersJMastroeniDLeonardBJoyceJGroverANeuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder?Int Rev Neurobiol20078223524617678964
- MatsuzakiSSzwedaPASzwedaLIHumphriesKMRegulated production of free radicals by the mitochondrial electron transport chain: cardiac ischemic preconditioningAdv Drug Deliv Rev2009611324133119716389
- ShanXChiLKeYManganese superoxide dismutase protects mouse cortical neurons from chronic intermittent hypoxia-mediated oxidative damageNeurobiol Dis20072820621517719231
- ZhuYFenikPZhanGSelective loss of catecholaminergic wake active neurons in a murine sleep apnea modelJ Neurosci200727100601007117855620
- FungSJXiMCZhangJHApnea promotes glutamate-induced excitotoxicity in hippocampal neuronsBrain Res20071179425017888415
- DaiberARedox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen speciesBiochim Biophys Acta2010179789790620122895
- FloydRAHensleyKOxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseasesNeurobiol Aging20022379580712392783
- BrownGCMechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidaseBiochem Soc Trans2007351119112117956292
- JanderSSchroeterMSalehAImaging inflammation in acute brain ischemiaStroke20073864264517261707
- HanischUKKettenmannHMicroglia: active sensor and versatile effector cells in the normal and pathologic brainNat Neurosci2007101387139417965659
- KimYSJohTHMicroglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s diseaseExp Mol Med20063833334716953112
- GaoHMZhouHZhangFWilsonBCKamWHongJSHMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegenerationJ Neurosci20113131081109221248133
- ZhangDHuXQianLMicroglial MAC1 receptor and PI3K are essential in mediating β-amyloid peptide-induced microglial activation and subsequent neurotoxicityJ Neuroinflammation201181321232086
- WuHMTzengNSQianLNovel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activationNeuropsychopharmacology200934102344235719536110
- BlockMLWuXPeiZNanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidaseFASEB J2004181618162015319363