
Abstract
Pompe disease is a rare autosomal recessive hereditary disease caused by genetic defects of acid maltase. This disease could be divided into two forms: infantile and late-onset, which mainly affect cardiac, respiratory, and skeletal muscle systems. Late-onset patients mainly show symptoms of skeletal muscle involvement, but recent reports have found that the central nervous system was also affected in some patients. Herein, we report a case of a female, adolescent-onset Pompe patient, who was diagnosed with complicated intracranial aneurysm in adulthood.
Introduction
Pompe is a rare, autosomal recessive, glycogen storage disease, which is caused by the deficiency or absence of the glycogen-degrading enzyme, acid maltase (acid alpha-glucosidase, GAA).Citation1 Pompe disease can occur at any age, and is classified into infantile and late-onset formsCitation1–Citation3 based on onset age, organ involvement, and disease progression. The late-onset form can be further classified into child, juvenile, and adult forms. The clinical manifestations of the adult form are varied, with a strong heterogeneity. The main clinical manifestation is chronic progressive aggravated girdle muscle weakness. The illness in lower extremity is often worse than in the upper limbs, pelvic girdle muscle is worse than lower proximal muscle. In addition, trunk and abdominal muscles are usually seriously involved at the early stage. But myocardial involvement in this form is usually insignificant. Another prominent clinical manifestation is respiratory muscle involvement, as a result, some patients suffered from disease onset because of acute respiratory failure. However, we found no significant link between respiratory insufficiency and extent of limb weakness. Another special disease pattern whose main clinical manifestation is cerebrovascular involvement could also be a subtype of late-onset Pompe disease.Citation1,Citation4 The symptoms of this subtype include cerebrovascular multiple stenosis, aneurysm, and vertebrobasilar dolichoectasia. Herein, we report a case of a female Pompe patient with complicated giant basilar artery aneurysm.
Case description
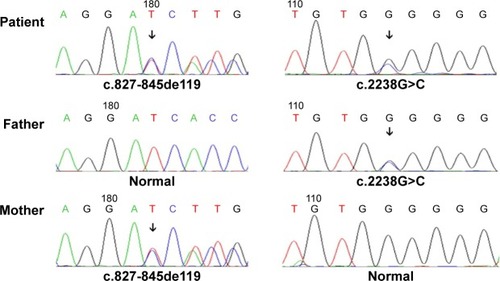
A female patient began to experience weakness in her waist and lower limbs as well as difficulty in climbing stairs since 13 years of age. She sought medical help at the age of 18 after progressive aggravation. She was walking unsteadily, with a waddling gait, and had trouble in taking the bus. She needed to rest after walking for 1.5 km, and was unable to run. The patient had nodular goiter for many years, without taking systematic treatment. Her neurological examination revealed clear mind, fluent speech, no obvious abnormality in the appearance of cranial nerves, and normal muscles in upper limbs. The strength of her iliopsoas was grade 2, gluteus maximus was grade 2–3, quadriceps was grade 4, calf muscles were normal without any obvious sensory disturbance, and no bilateral pyramidal sign was elicited. Muscle biopsy of her left quadriceps under local anesthesia revealed different sizes of muscle fibers with numerous basophilic vacuoles containing amorphous substances, which were seen as purple particles on modified Gomori trichrome staining, and the activity of succinate dehydrogenase, lactic dehydrogenase (LDH) were unevenly distributed. The activity of some fiber cytochrome C oxidase enzyme was enhanced, and amorphous substance was seen in the vacuoles on periodic acid–Schiff staining. The activity of acid phosphatase increased on acid phosphatase staining, which conformed to acid maltase deficiency (). The patient’s hematological examination showed aspartate aminotransferase 107 IU/L, creatine kinase 1,006 U/L, creatine kinase-MB 23 U/L, and LDH 313 U/L. B-scan ultrasound of the abdomen showed hepatomegaly, the thickness of the left and right lobes were 6.4 and 13.5 cm, respectively. The patient also had splenomegaly, and the size of her spleen was 4.4×12.3 cm. No craniocerebral imaging examination was performed at that time. The activity of acid maltase could not be tested. The patient had no siblings, her parents were healthy, and there was no one with similar diseases in her clan. At the age of 24, she had difficulty in breathing and lying down, so she was admitted to the respiratory department, where she could breathe with the help of a ventilator. But when the ventilator was removed, she had difficulty in spontaneous breathing after lying flat for 3 minutes or lying on one side for 10 minutes, so she started using a noninvasive positive pressure ventilator. At the age of 25, she developed scoliosis, but did not use orthotics or receive surgical treatment. At the age of 28, she was admitted to our hospital again due to paroxysmal headache and dizziness, which were relieved after symptomatic treatment. Further examination revealed clear mind, fluent speech, lateral curvature and deformity in her spine, and unsteady walking. On neurological examination, the cranial nerves were found to be intact. A motor examination revealed the strength of her neck extensor was grade 3, upper limbs was grade 4, and the proximal and remote portions of both lower limbs was grade 3 and 5, respectively. Gowers’ sign was positive. The GAA activity measured by peripheral blood lymphocytes enzyme activity method was 4.17 nmol/L h/mg (normal value >14 nmol/L h/mg). Her hematological examination revealed creatine kinase 488 U/L, aspartate aminotransferase 40 U/L, alanine aminotransferase 59 U/L, creatine kinase-MB 9.7 NG/L, and LDH 275 U/L. B-ultrasound of the upper abdomen showed that her liver, gallbladder, pancreas, and spleen were normal. Echocardiography showed tricuspid regurgitation. Pulmonary function test showed severe restrictive pulmonary dysfunction. Electrocardiography was normal. Spinal X-ray showed thoracic scoliosis and deformity. Head and neck computed tomographic angiography showed a cystic aneurysm in basilar artery (~3.0×2.8 cm in size), and expanded P1 segment in left posterior cerebral artery (). DNA was extracted from peripheral blood lymphocytes. Each exon and flanking sequence of the GAA gene were amplified via polymerase chain reaction. We identified a compound heterozygous mutations involving exon 4 (c.827-845del19) and exon 16 (c.2238G>C) in the patient. The patient’s mother was heterozygous for c.827-845del19 mutation in exon 4. The patient’s father was heterozygous for c.2238G>C mutation in exon 16 (). One month after the diagnosis of intracranial aneurysm, the patient died of a sudden loss of consciousness. The patient’s family refused craniocerebral computed tomography examination and autopsy of her body. Despite this, it was believed that the rupture of the basilar artery aneurysm caused the patient’s death. The study was approved by the Ethics Committee of Shandong University’s Qilu Hospital, and written informed consents were obtained from the patient and her guardians.
Figure 1 Hematoxylin–eosin staining showed the different sizes of muscle fiber, and lots of basophilic cavitation (arrow) contains amorphous materials in muscle fiber (A). (B) is normal. Acid phosphatase staining showed the increase of acid phosphatase activity, which caused the vacuolation turn ragged-red (arrow), and enzyme activity of the vacuoloid unchanged muscle fiber got a slight increase, which was stellate distributed (C). (D) is normal. (×400).
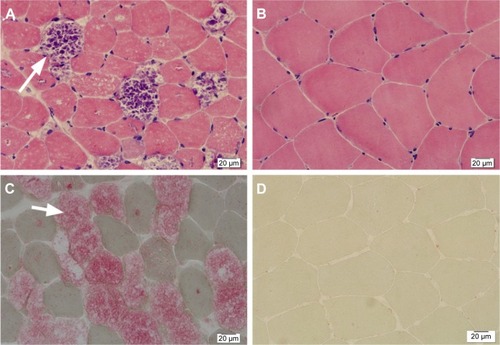
Figure 2 Craniocerebral CT scan (D) and CTA (A–C) show spherical, high-density shadow to the right of the basilar artery. The size was about 3.0 cm×2.8 cm. P1 of the left posterior cerebral artery was dilated, and the vertebral artery showed a shift to the left due to compression.
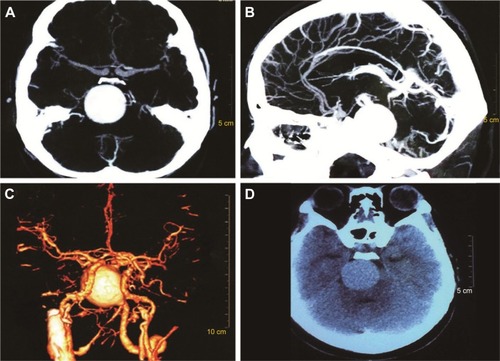
Figure 3 Gene analysis revealed heterozygous mutations involving exon 4 (c.827-845del19) and exon 16 (c.2238G>C) in the patient. The patient’s mother was heterozygous for c.827-845del19 mutation in exon 4. The patient’s father was heterozygous for c.2238G>C mutation in exon 16.
Discussion and conclusion
Pompe is a rare, autosomal recessive, inherited, systemic disease that can affect various tissues and organs of the body, such as respiratory, cardiac, skeletal, and smooth muscles. The clinical manifestations and prognosis are related to the age of onset. Typically, earlier onset entails lesser residual enzyme activity, severe clinical manifestation, faster progression, and worse prognosis. Late-onset patients have more clinical manifestations, including fatigue intolerance, difficulty in climbing stairs, inability to squat, run and jump, chest tightness on exertion to suppress asthma, acute respiratory failure, and other skeletal and respiratory muscle symptoms. The late-onset patients are classified into three categories.Citation5 Common symptom patterns include: 1) classic limb girdle and diaphragmatic weakness, 2) rigid spine syndrome, scoliosis, and low body mass, and 3) several cardiocerebrovascular patterns, manifesting as tortuous and dilated vertebral basilar artery or carotid artery vascular system, and multiple stenoses. So far, less than ten patients with intracranial aneurysms () have been reported worldwide, predominantly due to vascular posterior circulation involvement. Peric et alCitation6 reported subarachnoid hemorrhage with middle cerebral artery aneurysm in male patients, with no significant sex differences. In addition, basilar artery aneurysm complicated with thrombosis of the basilar pons and cerebellum on acute infarction has been reported.Citation7 In 2010, Sacconi et alCitation8 reported six patients, who underwent magnetic resonance imaging (MRI) examination, of which four patients were found to have abnormal intracranial vascular morphology, including basilar artery dilatation and internal carotid artery dilatation. Interestingly, all six patients had gastrointestinal symptoms, including diarrhea and constipation. The exact etiology of cerebral vascular involvement is not known, but a possible cause is cell structural damage induced by glycogen accumulation in smooth muscle cells, which leads to changes in vascular morphology. Earlier, its pathogenesis was thought to be due to lack or defects of acid maltase activity that leads to lysosomal accumulation of glycogen in the body due to inability to degrade glycogen in time, thereby causing muscle cell destruction. However, enzyme replacement therapy and further research on animal models in recent years found that its pathogenesis is not simply due to glycogen accumulation. Autopsy of Pompe patients who died after sudden comaCitation9–Citation11 revealed that the cause of death was basilar artery aneurysm, and pathology examination showed glycogen accumulation and extensive vacuolization to different degrees in the gastrointestinal tract, bladder, artery endothelial smooth muscle cells, and capillaries by positive periodic acid–Schiff staining. Electron microscopy revealed the size of artery vacuolar degeneration in the tube wall, accumulation of excessive glycogen in muscle cells, causing cell degeneration, necrosis, and interference in the synthesis of extracellular matrix, damage of elasticity, and integrity of the pipe wall.Citation8 The intracranial artery elastic layer was relatively weak as compared to other parts of the vessel, and more vulnerable to the formation of aneurysm expansion and vascular disease.Citation12 Diarrhea, constipation, and other gastrointestinal symptoms, along with gastrointestinal smooth muscle vacuolization degeneration were also observed.Citation13 Additionally, in-depth in vitro and in vivo studies along with pathological staining experiments and electron microscopy revealed vacuoles containing numerous molecules related to autophagy.Citation14,Citation15 Studies on autophagy and Pompe disease have recently intensified, and increasing evidence shows that autophagy plays a very important role in the pathogenesis of Pompe. Further research is required to understand if any difference exists in the intracranial aneurysm associated with Pompe autophagy in patients without stroke. Except in the present case where the patient had a history of nodular goiter associated with intracranial aneurysm, there is currently no clear relationship between nodular goiter and Pompe disease. However, we have seen a case with the onset of acute respiratory failure along with hypothyroidism, which needs further study. The GAA gene is located on chromosome 17q25.2-q25.3, is ~28 kb in length and contains 20 exons. Mutations in the gene vary widely including missense, nonsense, frame shift, and insertions and deletions. More than ~400 mutations (www.pompecenter.nl) up to June 2015 have been found in the gene encoding GAA, leading to total or partial deficiency of lysosomal GAA. The level of residual enzymatic activity is known to correlate with the location of mutations, age of disease onset, and severity of disease, although a definite genotype–phenotype correlation cannot be made. Intracranial aneurysm as a symptom of neuromuscular disease is rare; however, it has been previously described, not only in patients with Pompe disease but also in those with Marfan and Loeys-Dietz syndrome. Diagnosing adult-onset Pompe disease is sometimes challenging due to its various clinical manifestation. Young and middle-aged patients, especially with aneurysm, presenting with headaches, dizziness, unexplained cerebral vascular morphology changes, and skeletal muscle involvement, should be tested for Pompe disease. This should be most stringently followed for patients with sudden cerebral ischemia or stroke (especially posterior circulation) symptoms, accompanied with cerebral vascular disease. Additionally, all Pompe patients should undergo routine brain imaging examination.
Table 1 Literature review of eight Pompe patients with complicated intracranial aneurysm
Disclosure
The authors report no conflicts of interest in this work.
References
- Di RoccoMBuzziDTaroMGlycogen storage disease type II: clinical overviewActa Myol2007261424417915568
- FukudaTRobertsAPlotzPHRabenNAcid alpha-glucosidase deficiency (Pompe disease)Curr Neurol Neurosci Rep200771717717217857
- KishnaniPSSteinerRDBaliDPompe disease diagnosis and management guidelineGenet Med20068526728816702877
- HirschornRReuserAGlycogen storage disease type II: acid alpha glucosidase (acid maltase) deficiencyScriverThe Metabolic and Molecular Bases of Inherited DiseaseNew YorkMcGraw-Hill200133893420
- SchullerAWenningerSStrigl-PillNSchoserBToward deconstructing the phenotype of late-onset Pompe diseaseAm J Med Genet C Semin Med Genet2012160C1808822253010
- PericSFumicKBilicKReuserARakocevic StojanovicVRupture of the middle cerebral artery aneurysm as a presenting symptom of late-onset Pompe disease in an adult with a novel GAA gene mutationActa Neurol Belg2014114216516624338761
- RefaiDLevRCrossDTShimonyJSLeonardJRThrombotic complications of a basilar artery aneurysm in a young adult with Pompe diseaseSurg Neurol200870551852018207222
- SacconiSBocquetJDChanaletSTanantVSalviatiLDesnuelleCAbnormalities of cerebral arteries are frequent in patients with late-onset Pompe diseaseJ Neurol2010257101730173320559845
- MatsuokaYSendaYHirayamaMMatsuiTTakahashiALate-onset acid maltase deficiency associated with intracranial aneurysmJ Neurol198823563713733139844
- KretzschmarHAWagnerHHubnerGDanekAWittTNMehraeinPAneurysms and vacuolar degeneration of cerebral arteries in late-onset acid maltase deficiencyJ Neurol Sci1990982–31691832243227
- MiyamotoYEtohYJohRNodaKOhyaIMorimatsuMAdult-onset acid maltase deficiency in siblingsActa Pathol Jpn1985356153315423937437
- MakosMMMcCombRDHartMNBennettDRAlpha-glucosidase deficiency and basilar artery aneurysm: report of a sibshipAnn Neurol19872256296333322184
- BijvoetAGVan HirtumHVermeyMPathological features of glycogen storage disease type II highlighted in the knockout mouse modelJ Pathol1999189341642410547605
- RabenNTakikitaSPittisMGDeconstructing Pompe disease by analyzing single muscle fibers: to see a world in a grain of sandAutophagy20073654655217592248
- NascimbeniACFaninMMasieroEAngeliniCSandriMThe role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII)Cell Death Differ201219101698170822595755
- BraunsdorfWEFusiform aneurysm of basilar artery and ectatic internal carotid arteries associated with glycogenosis type2 (Pompe’s disease)Neurosurgery19872157487493480453
- LaforetPPetiotPNicolinoMDilative arteriopathy and basilar artery dolichoectasia complicating late-onset Pompe diseaseNeurology200870222063206618505979