
Abstract
Background
Accurate clinical diagnosis and prognosis of retinal degeneration can be aided by the identification of the disease-causing genetic variant. It can confirm the clinical diagnosis as well as inform the clinician of the risk for potential involvement of other organs such as kidneys. It also aids in genetic counseling for affected individuals who want to have a child. Finally, knowledge of disease-causing variants informs laboratory investigators involved in translational research. With the advent of next-generation sequencing, identifying pathogenic mutations is becoming easier, especially the identification of novel pathogenic variants.
Methods
We used whole exome sequencing on a cohort of 69 patients with various forms of retinal degeneration and in whom screens for previously identified disease-causing variants had been inconclusive. All potential pathogenic variants were verified by Sanger sequencing and, when possible, segregation analysis of immediate relatives. Potential variants were identified by using a semi-masked approach in which rare variants in candidate genes were identified without knowledge of the clinical diagnosis (beyond “retinal degeneration”) or inheritance pattern. After the initial list of genes was prioritized, genetic diagnosis and inheritance pattern were taken into account.
Results
We identified the likely pathogenic variants in 64% of the subjects. Seven percent had a single heterozygous mutation identified that would cause recessive disease and 13% had no obviously pathogenic variants and no family members available to perform segregation analysis. Eleven subjects are good candidates for novel gene discovery. Two de novo mutations were identified that resulted in dominant retinal degeneration.
Conclusion
Whole exome sequencing allows for thorough genetic analysis of candidate genes as well as novel gene discovery. It allows for an unbiased analysis of genetic variants to reduce the chance that the pathogenic mutation will be missed due to incomplete or inaccurate family history or analysis at the early stage of a syndromic form of retinal degeneration.
Introduction
Many forms of retinal degeneration result from genetic mutations, including Leber’s congenital amaurosis (LCA), retinitis pigmentosa (RP), Stargardt disease, and cone–rod dystrophy (CORD). Many different genes have been discovered that, when mutated, lead to retinal degeneration. For LCA alone, mutations in 18 different genes have been discovered to be pathogenic, with about 30% of cases having no known genetic cause.Citation1,Citation2 RP can be caused by mutations in any one of over 80 different genes with many cases still having an unknown cause.Citation3–Citation5 While there has been great progress in identifying disease-causing mutations for retinal degenerative diseases, this presents a staggering problem to the clinician: what is the most efficient and cost-effective test for identifying the genetic diagnosis?
Discovering genes that cause retinal degeneration when mutated and the specific mutations that do or do not cause disease is important to advancing the field. If the pathogenic mutation(s) can be identified in every patient with retinal degeneration, we will know how prevalent those mutations are, will be able to provide accurate prognoses, and will be better able to define potential patient populations for clinical trials. With the growing potential of novel strategies using gene editing (such as use of Clustered Regulatory Interspaced Short Palindromic Repeats) to correct specific genetic mutations, identifying not only the gene but also the specific mutations in each patient is becoming even more important. Identifying the pathogenic variants in retinal degeneration benefits basic science as well as clinical research. By identifying the genes that result in retinal degeneration when mutated, we gain a better understanding of how the retina works, which proteins are necessary for the various cellular processes that take place in the retina, and how they interact. By defining the specific mutations that are pathogenic (and which ones are tolerated), we better understand the function of these proteins and domains. This information can also be used to develop gene-based treatments (ie, gene therapy).
The present report reviews our own experience in seeking genetic diagnoses for retinal degeneration patients seen over a 5-year period at ophthalmology clinics at the Perelman School of Medicine, University of Pennsylvania and The Children’s Hospital of Philadelphia as well as other patients who self-referred and agreed to participate in our molecular genetic research study. We carried out preliminary screens of these individuals in an attempt to identify known disease-causing variants. We followed up with whole exome sequencing (WES). Finally, we assessed the efficiency of making the correct genetic diagnosis using these techniques.
Subjects and methods
Recruitment
Individuals seen in the Scheie Eye Institute and The Children’s Hospital of Philadelphia retina clinics who were found to have a degenerative condition (and first-degree relatives) were invited to submit a blood sample for molecular genetics research testing. Study procedures were approved by the University of Pennsylvania Institutional Review Board (#808828) and each individual provided signed informed consent. Parental or guardian permission and signed informed consent as well as signed assent were obtained for subjects younger than 18 years.
Whole exome sequencing
Purified, target-enriched genomic samples from 69 patients with various form of retinal degeneration (including LCA, RP, Stargardt disease, CORD, and achromatopsia) to the Penn Genome Frontiers Institute were evaluated by WES (). Samples had previously been screened using Asper Ophthalmics (Tartu, Estonia) arrayed primer extension (APEX) multigene panels for the relevant disease (using panels that were available from 2009 to 2014) and no disease-causing variants had been identified. Target enrichment was performed with the Agilent SureSelect target enrichment system (Agilent Technologies, Santa Clara, CA, USA), and the sequencing was performed on Illumina HiSeq2000 (Illumina, San Diego, CA, USA).
Table 1 Breakdown of diagnoses included in this study
Mapping and variant identification
Mapping and variant identification was performed using Galaxy.Citation6 The FASTQ files for each patient were mapped to the reference human genome (hg18) using Burrows-Wheeler Aligner for Ilumina. Variants were identified using freebayes.Citation7 Variants were annotated using Annovar.Citation8
Identifying variants of interest
A semi-masked analysis was used to identify potentially pathogenic mutations without knowledge of the type of retinal degeneration. We created a curated list of genes linked to any form of retinal degeneration and used a python script (Python Software Foundation, Fredericksburg, VA, USA) to create a customized list of potential pathogenic variants for each patient. This list consisted of retinal degeneration genes with variants with an allele frequency of, 0.05. Those mutations were then prioritized based on mutation type and known facts about the mutations from the Single Nucleotide Polymorphism Database (dbSNP) (https://www.ncbi.nlm.nih.gov/projects/SNP/) and the Universal Protein Resource (UniProt; http://www.uniprot.org), which are known to be benign, pathogenic, novel variant, etc. We then checked if the variants were consistent with the clinical diagnosis and performed segregation analysis when possible.
We verified the possible pathogenic mutations in the patients and relatives by polymerase chain reaction, Phusion polymerase (New England Biolabs, Ipswich, MA, USA) and custom primers made by Thermo Fisher Scientific (Waltham, MA, USA) followed by Sanger sequencing by the Penn Genomics Analysis Core. The sequences were visualized using SnapGene software (from GSL Biotech, Chicago, IL, USA; available at snapgene.com).
Results
Prior to embarking on WES studies, APEX screening was carried out on 100 unrelated subjects, screening for ABCA4 variants in individuals diagnosed with Stargardt disease, CORD, and macular degeneration, for variants in a panel of genes known to cause LCA for those diagnosed with LCA, achromatopsia, or nanophthalmos, and for genes associated with autosomal recessive or autosomal dominant RP (ADRP) in those diagnosed with RP, Usher Syndrome, vitreoschisis, or unspecified retinal degeneration ().
Compound heterozygous or homozygous disease-causing variants were identified through APEX screening in 44% of those screened for ABCA4 variants and 31% screened for LCA variants. Heterozygous or compound heterozygous disease-causing variants were identified in ~29% of those screened for RP variants. These disease-causing variants were confirmed by segregation studies in cases where informative family members were available. This left us with 69 individuals in whom either no disease-causing variants or only one potential disease-causing variant (in cases of autosomal recessive RP) had been identified. We identified the pathogenic mutations in 44 of those 69 subjects (64%) using WES. Sixty-six different mutations were found in 26 different genes. Eighteen of these mutations are completely novel and not listed in dbSNP. Eleven more are extremely rare, have no prediction on pathogenicity, and are not in any published study (see Supplementary materials for specific mutations in each form of retinal degeneration).
De novo mutations
De novo mutations pose a particular problem when trying to identify pathogenic mutations. Dominant de novo mutations appear to be recessive when looking at the family history. We found two de novo mutations in our patient cohort. The first was a heterozygous novel frameshift mutation in OTX2, a known mutational hotspot, in a patient with LCA (; JB275). OTX2 is a transcription factor that is essential for development of the brain and retina. All known mutations are autosomal dominant. Knockout of OTX2 is embryonic lethal in mice due to the absence of the forebrain and midbrain.Citation9 OTX2 is essential for photoreceptor differentiation as well as pituitary development. Knockout of OTX2 in mouse retinal cells results in a failure to develop photoreceptors.Citation10 OTX2 mutations can result in syndromic microphthalmia, combined pituitary hormone deficiency, and early-onset retinal dystrophy with or without pituitary dysfunction.Citation11–Citation13 The frameshift mutation was not present in either parent.
Table 2 Pathogenic and probable pathogenic variants in known disease-causing genes identified in subjects in this study
Another subject presented with cone dystrophy beginning at age 15. Neither parent had any form of retinal degeneration, so it was presumed that the genetic cause was recessive. Upon analysis of the WES dataset, we found that the proband was heterozygous for a p.Y99C mutation in GUCA1A (; JB185), a well-characterized dominant mutation leading to cone dystrophy.Citation14,Citation15 Neither parent carried the p.Y99C mutation. Paternity was confirmed using the WES results of the parents and proband, indicating that the p.Y99C mutation was a de novo mutation.
Identification of KIF7 as a candidate gene for cone dystrophy
A 5-year-old boy with cone dystrophy was found to have a compound heterozygous mutation in KIF7 (p.H1115Q and p.Q834R; ; JB307). KIF7 is a cilia gene that plays an important role in hedgehog signaling and microtubule stability.Citation16,Citation17 KIF7 localizes to the primary cilia, specifically the distal tip.Citation18 Mutations in KIF7 cause the cilia to become longer than normal and disorganized.Citation18 Knockout of Kif7 in mice is neonatal lethal.Citation17 Mutations in KIF7 have been shown to cause severe ciliopathies including Joubert syndrome, hydrolethalus syndrome, acrocallosal syndrome, Meckel–Gruber syndrome, and Bardet–Biedl syndrome (BBS).Citation19 Both variants in the patient in this study have been previously identified as hypomorphic variants and have been seen in patients with BBS, Meckel–Gruber syndrome, and hydrolethalus syndrome.Citation20 Typically, when a missense mutation in KIF7 leads to disease, the other allele is a null allele with either a truncation or frameshift mutation.Citation20,Citation21 In this case, both alleles are rare hypomorphic variants (allele frequency in 1,000 genomes are 0.0010 and 0.0058 for p.Q834R and p.H1115Q, respectively). In addition to the compound heterozygous mutations, the subject was heterozygous for mutations in several other cilia genes, including a pathogenic mutation in BBS1. Ciliopathies have been shown to have a range of severities depending on the mutation in the gene.Citation38 For example, CEP290 mutations can result in retinal degeneration alone (specifically LCA), syndromic ciliopathies (Joubert syndrome or Meckel syndrome), and neonatal lethal ciliopathies.Citation38–Citation41 This range appears to be due to the amount of functional CEP290 that remains.Citation22 KIF7 could be similar to CEP290 in that severe mutations like truncations and frameshifts result in more severe ciliopathies, while compound heterozygosity of hypomorphic variants may result in only retinal degeneration. It is unknown why the cone photoreceptors would be affected while the rod photoreceptors are currently preserved.
Additional mutations
Some of the patients had potentially pathogenic mutations in additional genes linked to retinal degeneration. Most of these additional mutations were heterozygous and, therefore, unlikely to be the disease-causing mutation in the patient. However, it is possible that the heterozygous mutations, which can be tolerated in isolation, add to the mutational load and modify the patient’s phenotype.Citation23 Five subjects had an unusually high number of additional mutations ( and ). Some of the mutations are in genes that interact or are in the same pathway, increasing the chances of epistatic effect. It is important to note there is a high likelihood that these are incidental findings that do not contribute to disease phenotype at all. One of the drawbacks of WES is that rare but benign variants are found and can complicate analyses.
Table 3 Additional heterozygous variants of uncertain significance found in this study
Table 4 Multiple compound heterozygous mutations identified in a subject with multiplex RP
No disease-causing variants identified
We were unable to conclusively determine the disease-causing variants in 20 subjects in the study. Five of these had a presumably recessive disease with one pathogenic or likely pathogenic variant identified that is consistent with their disease phenotype (). These patients should be screened for copy number variants in the gene identified or possibly for intronic variants. Of the remaining 20 subjects, 9 do not have family members willing to enroll in the study. Since we did not find any obviously pathogenic variants in the initial screening and are unable to perform segregation analysis for the variants identified, we are unable to determine the pathogenicity of the variants identified. One of these subjects has many known pathogenic, likely pathogenic, and novel variants, but all are inconsistent with the diagnosis of ADRPCitation46–Citation48 and we are unable to perform segregation analysis to narrow down the disease-causing variant(s) (). The remaining 10 subjects should be analyzed for novel genes that could cause retinal degeneration ().
Figure 1 Final outcome of initial screening for variants in genes associated with retinal degeneration.
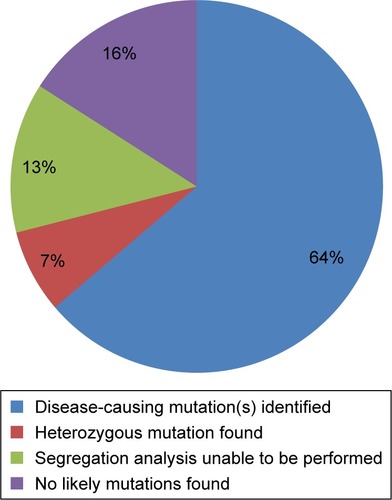
Table 5 Heterozygous variants identified in subjects with recessive disease
Discussion
WES is a powerful tool to help identify new mutations in patients who test negative for the known pathogenic mutations for retinal degeneration. However, the majority of mutations in these patients are in genes already linked to retinal degeneration. Forty-four of the 69 probands in our study, all of whom had had negative screenings using gene panels for their disease, were found to harbor pathogenic mutations in genes already linked to retinal degeneration. Thus, we found that if we failed to find the pathogenic mutation in a patient with retinal degeneration using standard APEX microarrays for their disease, it could be more time- and cost-efficient to use targeted sequencing of all genes linked to retinal degeneration rather than WES. Use of a targeted sequencing approach would also allow good coverage of the area of interest without the expense of deeper coverage of the whole exome. When we restricted our analysis of the WES data to genes linked to retinal degeneration, it allowed us to find novel mutations in those genes as well as mutations that are known to cause a different form of retinal degeneration.
WES is a powerful tool for identifying pathogenic mutations in genes by quickly identifying all variants in a patient’s exome. It sequences the entire protein coding portion of the genome, which is about 2% of the total genome. Since the noncoding portion is not sequenced, it is cheaper than whole genome sequencing (WGS) and allows for greater depth of coverage. The large majority of disease-causing variants are in the exome, so although intronic mutations are missed, it is still usually more cost-effective to use WES and follow-up with WGS or targeted sequencing if WES fails to identify the cause of disease.
Although the size of the dataset generated by WES is smaller than WGS, sorting through the large amount of data generated to find the relevant variants is still a challenge. Filtering the dataset based on inheritance pattern, disease phenotype, and other predetermined criteria can not only make the dataset more manageable, but also risks filtering out pathogenic variants. De novo mutations can result in a dominant mutation in a patient whose disease is presumed to be recessive. Dominant mutations can also be revealed by findings of non-paternity (and concomitant incorrect family history). Diagnosis is based on qualitative assessment and psychophysical testing, and many forms of retinal degeneration have significant phenotypic overlap with each other. A patient can be diagnosed with a nonsyndromic form of retinal degeneration when they have a syndromic form if they have not yet developed extraocular symptoms. In these cases, it is easy to filter out pathogenic variants if the filters are too strict.
Limiting our initial screening to the genes that are known to cause one particular form of retinal degeneration after the subject has already come back negative from the typical first-pass screening (such as APEX array or single nucleotide polymorphism chip) would have been unnecessarily restrictive. It would have placed too much confidence in the accuracy of the clinical diagnosis when there is significant overlap between different forms of inherited retinal degeneration, especially given the fact that the subjects are known to be negative for the typical disease-causing mutations associated with that diagnosis. Further, at both early and late stages of retinal degeneration, it may be difficult to distinguish one form of disease from another. The possibility of de novo mutations and inaccurate family history makes it impossible to determine the inheritance pattern with complete certainty even in the best case scenario. There are also multiple genes that are linked to multiple forms of retinal degeneration, with the mechanism by which they can differentially affect rods and cones still not understood. It is entirely possible that a gene in which mutations are known to cause only RP or LCA currently could in fact also have mutations that lead to cone dystrophy. When one adds in the possibility of digenic cause and epistatic effects, it becomes even clearer that limiting analysis by the form of retinal degeneration is overly simplistic and can lead to missed pathogenic mutations.
Using a broader filter (retinal degeneration in general instead of the specific diagnosis) does come with an increased analytical burdened with more variants to sort through and can run the risk of becoming distracted by irrelevant variants. A mutation in rhodopsin is unlikely to cause cone dystrophy, but irrelevant variants can be quickly filtered out by a knowledgeable analyst and mutations in more relevant genes are prioritized. Since the list of genes to be screened is still specialized, our knowledge of the genes and the diseases that they are usually associated with can be used to prioritize the list of candidate genes. This would not be the case if the list used consisted of simply genes expressed in the retina where there are many genes whose function is unknown and, thus, with unknown disease involvement.
So far, most of the mutations we have found that lead to retinal degeneration follow typical Mendelian genetics. However, many patients with retinal degeneration do not have a known cause. It is probable that at least some of these cases will be found to be due to the additive or epistatic effects of mutations in more than one gene. Especially in the case of a presumed recessive inheritance pattern, the possibility that the cause is polygenic rather than monogenic cannot be discounted. In light of that possibility, screening all genes with mutations known to cause or contribute to retinal degeneration is important. The fact that these genes code for proteins vital to the proper functioning of the retina has already been established. A mutation or variant in a gene that reduces the ability to function (but not so much that it is sufficient to cause disease in the general population) might very well add to the mutational load that predisposes someone to retinal degeneration.
By using a semi-masked analysis in our screening, we were able to easily identify two patients with dominant de novo mutations as well as several with an inaccurate or incomplete incoming diagnosis. Many of the subjects in our study self-reported the initial diagnosis, which decreases the initial accuracy. However, there is some inherent unreliability in the initial diagnosis for early-stage retinal degeneration, particularly if this is made by a nonspecialist. Some forms of retinal degeneration, like RP, could be a part of a syndromic form of retinal degeneration like BBS. By not taking into account the type of retinal degeneration until after the possible mutations have been identified, we eliminate the possibility of filtering out obviously pathogenic variants before we even begin the analysis. Elicitation of a detailed family and clinical history as well as a focused clinical examination would significantly narrow down the search for the disease-causing gene during first-pass screening and is also likely most economical in avoiding the cost and time associated with WES and subsequent bioinformatics analyses (Stone et al,Citation25 who demonstrated in a study of 1,000 different families that the disease-causing gene/variant could be identified in 57.6% of families without using WES). Identifying the disease-causing gene during first-pass screening would also eliminate the possibility of evaluating irrelevant seemingly pathogenic variations (such as those identified in families evaluated by Stone et alCitation25).
One cautionary finding from our study is that there were several patients who had areas of low coverage in genes of interest. One patient with Stargardt disease had a gap in coverage in ABCA4 that masked the pathogenic mutation. Such gaps need to be filled in with Sanger sequencing. A gap in coverage can complicate analysis if not detected as it would essentially show no mutation in the area with no or low coverage.
We believe that all subjects should undergo appropriate psychophysical testing in order to refine the clinical diagnosis and then undergo a panel mutation screening for that diagnosis before being enrolled in a study for novel gene detection. This should be followed by targeted exome sequencing of all genes linked to retinal degeneration. In our study, 64% of the subjects were eliminated from further analysis after screening only genes linked to retinal degeneration. Subjects with a recessive disease in which only one pathogenic mutation is found (7% of the patients in this study, 20% of those without a genetic diagnosis) should be screened for copy number variants and intronic variants in that gene. After eliminating those enrolled in the study with no relatives, this would leave only 16% for a more in-depth analysis using WES.
The subjects with an unusually high mutational load merit further study as well. We tend to view sporadic cases of early-onset retinal degeneration as being due to an autosomal recessive mode of inheritance, or occasionally a de novo mutation. Modifying alleles are sometimes identified which increase the severity of the disease phenotype, like the p.A229T variant in RPGRIP1L.Citation24 However, the consequence of multiple heterozygous variants that can be pathogenic if there is a second mutation in the same gene has not been studied. Screening the unaffected relatives of the three subjects with an unusually high mutational load could help illuminate how many mutations can be tolerated. Studying the progression of retinal degeneration in these subjects can help show whether the additional mutations result in a more severe phenotype or have no obvious effect.
Conclusion
WES is a very effective approach for identifying novel mutations and narrowing down candidate genes for further analysis, especially when paired with targeted analysis based on known retinal pathways. If WES does not yield a molecular genetic diagnosis or a likely candidate for further analysis, WGS can be used to identify noncoding (intronic) mutations. While analysis of the parents and immediate family of the proband is not always required for identification of pathogenic variants, it does increase the likelihood of identifying pathogenic variants and could be needed if the mutations are not obviously pathogenic.
Acknowledgments
This study was supported by the Pennsylvania Department of Health, Foundation Fighting Blindness-sponsored CHOP-Penn Pediatric Center for Retinal Degenerations, National Eye Institute/NIH grant R24EY019861, NIH Vision Training Grant 5T32EY007035-37, Research to Prevent Blindness, the Brenda and Matthew Shapiro Stewardship, the Robert and Susan Heidenberg Investigative Research Fund for Ocular Gene Therapy, the Paul and Evanina Mackall Foundation Trust, the Scheie Eye Institute, the Center for Advanced Retinal and Ocular Therapeutics, and the FM Kirby Foundation. We thank the participants and their families for participating in this study and Jeannette Bennicelli, Grace Han, Jessica IF Morgan, Angela Luo, Michael Colucciello, Georgia Spies, Nicholas Phelps, and Ayodele Maja for technical assistance.
Supplementary materials
Syndromic disease
Three siblings in one family were diagnosed with retinitis pigmentosa (RP). Whole exome sequencing (WES) revealed two variants in BBS4 in the proband (; JB319). The first mutation was a frameshift mutation likely resulting in nonsense mediated decay. The second mutation was p.R295G. While the arginine to glycine mutation is novel, an arginine to proline mutation at the same location has been previously reported to be pathogenic, making it very likely that the glycine substitution would be pathogenic as well.Citation1 Mutations in BBS4 have been shown to cause Bardet–Biedl syndrome (BBS), but not nonsyndromic RP. Segregation analysis confirmed that all three siblings were compound heterozygous for the BBS4 mutations and clinical analysis showed that their symptoms (RP, obesity, and learning difficulties) were consistent with mild BBS. Renal dysfunction, polydactyly, and male hypogonadism are also commonly seen in BBS, but were absent in this family.Citation2
Another subject had Usher syndrome, a condition characterized by hearing loss and RP. Clinical testing failed to reveal any known mutations leading to Usher syndrome. WES revealed two novel frameshift mutations in GPR98 (; JB324), a gene known to cause Usher syndrome when mutated.Citation3 Another subject with Usher syndrome was found to have two mutations in USH2A (, JB252) that had been already classified as pathogenic.
Stargardt, cone dystrophy, and achromatopsia
Stargardt disease is a childhood-onset macular degeneration and is most commonly caused by mutations in ABCA4. Characteristic yellow flecks are typically seen under the macula during a fundus exam. Five subjects presented with Stargardt disease and were found to have mutations in ABCA4 (; JB260, JB333, JB358, JB16, JB320). Six of the mutations had been previously characterized and determined to be pathogenic, while three novel frameshift mutations were found.
Autosomal dominant Stargardt disease is caused by mutations in PROM1. Interestingly, the dominant mutations seem to cause primarily cone degeneration, while the recessive mutations can cause either rod or cone degeneration. The dominant mutations in PROM1 are typically gain-of-function missense mutations, while the recessive mutations are loss-of-function mutations including frameshift mutations, splice site mutations, and truncations. Two loss-of-function variants were identified in patients with either Stargardt disease or cone dystrophy (; JB189, JB241). A second variant was not identified in either case. Since a dominant inheritance pattern was not established in these families, the pathogenicity of these variants is currently undetermined.
One subject diagnosed with Stargardt disease was found to have a heterozygous p.V242M variant in CRX (; JB195). This mutation is reported to be pathogenic and cause autosomal dominant cone–rod dystrophy.Citation4 CRX is a transcription factor that is required for the development and maintenance of cone photoreceptors.Citation5 While many CRX mutations have been shown to reduce the ability of CRX to activate the rhodopsin promoter in vitro, the p.V242M mutation did not appear to affect the ability of CRX to activate the rhodopsin promoter, casting some doubt on the pathogenicity.Citation6 It is possible that the p.V242M variant is pathogenic but has a different functional effect. The variant did not segregate with disease in the family in this study. Both the mother and half-sister are heterozygous for the variant, but do not have retinal degeneration. It is possible that the p.V242M variant contributes to mutational load or digenic cause for disease, but based on the lack of segregation and lack of demonstrated functional effect, the variant is probably benign.
Another subject presented with severe retinal degeneration that was classified as either achromatopsia or Leber’s congenital amaurosis (LCA; ; JB301). The proband had two novel mutations in GNAT2, a frameshift mutation and a missense mutation. GNAT2 encodes the alpha subunit of cone transducing, which is essential for phototransduction in cones.Citation7 Truncations are the most common pathogenic mutations in GNAT2, but pathogenic missense mutations in GNAT2 have been seen in both patients and mice.Citation8,Citation9 Complicating the interpretation of the sequencing results for this subject, she had a p.A249E heterozygous mutation in GDF6 that has been reported to be autosomal dominant with incomplete penetrance.Citation10–Citation12 This variant has been reported to lead to LCA, microphthalmia, and Klippel–Feil syndrome. Notably, this subject does not have any skeletal defects that are sometimes reported to be associated with the p.A249E variant. The mother, who does not exhibit any symptoms of retinal degeneration, is positive for the missense variant in GDF6. Therefore, we determined that the GNAT2 mutations are the most likely genetic cause in this subject, with the GDF6 mutation potentially acting as a modifying allele and increasing the severity or playing no role in retinal degeneration in this subject.
Another subject had an incoming diagnosis of LCA that was changed to cone dystrophy upon examination. The diagnosis was further modified to achromatopsia after WES revealed two heterozygous mutations in CNGB3, one of which was novel (; JB426).
Leber’s congenital amaurosis
LCA is a severe retinal degeneration which is symptomatic before the first year of age.Citation13 The retinal degeneration eventually includes cone loss as well. It is one of the most severe forms of hereditary blindness. In this study, we found mutations in five different genes that resulted in LCA (). Nine of the mutations (in ABCA4, CNGB3, KCNJ13, and PROM1) were novel.
CEP290 mutations are the most common cause of LCA. CEP290 is essential for cilia development and maintenance.Citation14,Citation15 Mutations in CEP290 cause many different ciliopathies including Joubert syndrome, Bardet–Biedl syndrome, Meckel syndrome, and Senior–Loken syndrome.Citation15,Citation16 We found a novel splice site mutation in CEP290 in one patient with LCA (; JB165). The second mutation was the deep intronic splice mutation.Citation17 This case illustrates one of the limitations of WES for finding pathogenic mutations. WES did not reveal the deep intronic mutation. Since the CEP290 c.2991+1655A>G mutation is the most common mutation leading to LCA, the patient had been screened for it previously. If the mutation had not already been identified and specifically looked for, we would not have identified the second mutation or been able to determine if the CEP290 splice site mutation was actually disease causing in this case or if the patient was simply a carrier for the mutation.
Mutations in PRPH2 tend to be autosomal dominant due to haploinsufficiency.Citation18 Typically, these mutations cause either RP or macular degeneration.Citation19,Citation20 Homozygous or compound heterozygous mutations are much rarer and cause LCA or early-onset RP when they do occur.Citation21 The parents do not always have any functional vision loss, but will usually show signs of mild retinal degeneration when examined. One subject in this study was homozygous for a novel truncating mutation in PRPH2 (; JB310).
One subject was initially diagnosed with LCA. WES revealed two mutations in PDE6A (; JB28), which have been shown to result in RP. Upon re-evaluation, the diagnosis was changed to early-onset RP, which has a later age of onset and typically less severe progression.
Retinitis pigmentosa
RP is a progressive form of retinal degeneration that primarily affects rod photoreceptors. It has a later onset than LCA with a typical age of diagnosis of 35 years. There is a wide range for the age of onset with some patients diagnosed during childhood and others being diagnosed in their 60s or even later.Citation22,Citation23 RP can be x-linked, autosomal dominant, or autosomal recessive. Over 80 genes have been identified that can cause RP, and the genetic cause is unknown in almost half of all patients diagnosed.Citation24 In this study, 10 different genes were found to cause RP with two novel mutations (). All of the genes have been previously shown to cause RP.
Autosomal dominant RP accounts for 15%–40% of all cases of RP.Citation25,Citation26 So far, more than 20 genes have been identified that cause autosomal dominant RP.Citation25,Citation26 These include CA4, PRPH2, and PRPF31. Dominant mutations tend to be pathogenic either due to heterozygous insufficiency or they cause a toxic gain of function. Mutations in all three of these genes seem to cause disease at least in part due to haploinsufficiency as a null allele is sufficient to cause disease. The novel mutation we found in PRPH2 is an early stop codon, so we did not follow-up with functional testing. The mutations in CA4 and PRPF31 are missense mutations that are near-known pathogenic missense mutations in the genes. CA4 mutations have been shown to impair the ability of the cells to regulate intracellular pH.Citation27 The p.Q254P mutation in CA4 (; JB42) is very rare and currently classified as a variant with uncertain significance on dbSNP (rs150432787). We were unable to perform a segregation analysis due to the lack of DNA from family members. Interestingly, this patient was heterozygous for several mutations that are known to cause recessive forms of retinal degeneration, including USH2A, ABCA4, and CDH23, as well as novel mutations in several other genes linked to retinal degeneration, including BBS12, ROM1, and GPR98. This is an unusually high mutational load ().
References
- MykytynKBraunTCarmiRIdentification of the gene that, when mutated, causes the human obesity syndrome BBS4Nat Genet200128218819111381270
- PriyaSNampoothiriSSenPSripriyaSBardet-biedl syndrome: genetics, molecular pathophysiology, and disease managementIndian J Ophthalmol201664962062727853007
- WestonMDLuijendijkMWHumphreyKDMollerCKimberlingWJMutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of usher syndrome type IIAm J Hum Genet200474235736614740321
- SwainPKChenSWangQLMutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degenerationNeuron1997196132913369427255
- ChenSWangQLNieZCrx, a novel otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genesNeuron1997195101710309390516
- ChenSWangQLXuSFunctional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophyHum Mol Genet200211887388411971869
- NishiguchiKMSandbergMAGorjiNBersonELDryjaTPCone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseasesHum Mutat200525324825815712225
- JoblingAIVesseyKAWaughMMillsSAFletcherELA naturally occurring mouse model of achromatopsia: characterization of the mutation in cone transducin and subsequent retinal phenotypeInvest Ophthalmol Vis Sci20135453350335923580486
- KuniyoshiKMuraki-OdaSUeyamaHNovel mutations in the gene for alpha-subunit of retinal cone cyclic nucleotide-gated channels in a japanese patient with congenital achromatopsiaJpn J Ophthalmol201660318719727040408
- Asai-CoakwellMFrenchCRYeMIncomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypesHum Mol Genet20091861110112119129173
- Asai-CoakwellMMarchLDaiXHContribution of growth differentiation factor 6-dependent cell survival to early-onset retinal dystrophiesHum Mol Genet20132271432144223307924
- TassabehjiMFangZMHiltonENMutations in GDF6 are associated with vertebral segmentation defects in klippel-feil syndromeHum Mutat20082981017102718425797
- CremersFPvan den HurkJAden HollanderAIMolecular genetics of leber congenital amaurosisHum Mol Genet200211101169117612015276
- DrivasTGHolzbaurELBennettJDisruption of CEP290 microtubule/ membrane-binding domains causes retinal degenerationJ Clin Invest2013123104525453924051377
- D’AngeloAFrancoBThe dynamic cilium in human diseasesPathogenetics200921319439065
- DrivasTGWojnoAPTuckerBAStoneEMBennettJBasal exon skipping and genetic pleiotropy: a predictive model of disease pathogenesisSci Transl Med20157291291ra97
- den HollanderAIKoenekoopRKYzerSMutations in the CEP290 (NPHP6) gene are a frequent cause of leber congenital amaurosisAm J Hum Genet200679355656116909394
- ConleySMStuckMWBurnettJLInsights into the mechanisms of macular degeneration associated with the R172W mutation in RDSHum Mol Genet201423123102311424463884
- MeinsMGruningGBlankenagelAHeterozygous ‘null allele’ mutation in the human peripherin/RDS geneHum Mol Genet1993212218121828111389
- WroblewskiJJWellsJA3rdEcksteinAMacular dystrophy associated with mutations at codon 172 in the human retinal degeneration slow geneOphthalmology1994101112228302543
- WangXWangHSunVComprehensive molecular diagnosis of 179 leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencingJ Med Genet2013501067468823847139
- StylesCJDhillonBWrightAFThe diagnosis of autosomal dominant late-onset retinal degeneration in two sistersEye (Lond)200317453053212802359
- TsujikawaMWadaYSukegawaMAge at onset curves of retinitis pigmentosaArch Ophthalmol2008126333734018332312
- NashBMWrightDCGriggJRBennettsBJamiesonRVRetinal dystrophies, genomic applications in diagnosis and prospects for therapyTransl Pediatr20154213916326835369
- HartongDTBersonELDryjaTPRetinitis pigmentosaLancet200636895491795180917113430
- FahimATDaigerSPWeleberRGNonsyndromic retinitis pigmen-tosa overviewPagonRAAdamMPArdingerHHGeneReviews(R)Seattle, WAUniversity of Washington Seattle1993
- YangZAlvarezBVChakarovaCMutant carbonic anhydrase 4 impairs pH regulation and causes retinal photoreceptor degenerationHum Mol Genet200514225526515563508
Disclosure
The authors report no conflicts of interest in this work.
References
- Chacon-CamachoOFZentenoJCReview and update on the molecular basis of leber congenital amaurosisWorld J Clin Cases20153211212425685757
- den HollanderAIRoepmanRKoenekoopRKCremersFPLeber congenital amaurosis: genes, proteins and disease mechanismsProg Retin Eye Res200827439141918632300
- HartongDTBersonELDryjaTPRetinitis pigmentosaLancet200636895491795180917113430
- FahimATDaigerSPWeleberRGNonsyndromic retinitis pigmentosa overviewPagonRAAdamMPArdingerHHGeneReviews(R)Seattle, WAUniversity of Washington Seattle1993
- Bravo-GilNGonzalez-Del PozoMMartin-SanchezMUnravelling the genetic basis of simplex retinitis pigmentosa casesSci Rep201774193728157192
- AfganEBakerDvan den BeekMThe galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 updateNucleic Acids Res201644W1W3W1027137889
- GarrisonEMarthGHaplotype-based variant detection from short-read sequencingarXiv preprint Epub2012720
- WangKLiMHakonarsonHANNOVAR: functional annotation of genetic variants from high-throughput sequencing dataNucleic Acids Res20103816e16420601685
- AcamporaDMazanSLallemandYForebrain and midbrain regions are deleted in Otx2-/- mutants due to a defective anterior neuroectoderm specification during gastrulationDevelopment199512110327932907588062
- NishidaAFurukawaAKoikeCOtx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland developmentNat Neurosci20036121255126314625556
- TajimaTOhtakeAHoshinoMOTX2 loss of function mutation causes anophthalmia and combined pituitary hormone deficiency with a small anterior and ectopic posterior pituitaryJ Clin Endocrinol Metab200994131431918854396
- HendersonRHWilliamsonKAKennedyJSA rare de novo nonsense mutation in OTX2 causes early onset retinal dystrophy and pituitary dysfunctionMol Vis2009152442244719956411
- DatekiSKosakaKHasegawaKHeterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotypeJ Clin Endocrinol Metab201095275676419965921
- PayneAMDownesSMBessantDAA mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1Hum Mol Genet1998722732779425234
- LauraRPDizhoorAMHurleyJBThe membrane guanylyl cyclase, retinal guanylyl cyclase-1, is activated through its intracellular domainJ Biol Chem19962712011646116518662612
- DafingerCLiebauMCElsayedSMMutations in KIF7 link joubert syndrome with sonic hedgehog signaling and microtubule dynamicsJ Clin Invest201112172662266721633164
- Endoh-YamagamiSEvangelistaMWilsonDThe mammalian Cos2 homolog Kif7 plays an essential role in modulating hh signal transduction during developmentCurr Biol200919151320132619592253
- HeMSubramanianRBangsFThe kinesin-4 protein Kif7 regulates mammalian hedgehog signalling by organizing the cilium tip compartmentNat Cell Biol201416766367224952464
- BarakehDFaqeihEAnaziSThe many faces of KIF7Hum Genome Var201521500627081521
- PutouxAThomasSCoeneKLKIF7 mutations cause fetal hydrolethalus and acrocallosal syndromesNat Genet201143660160621552264
- PutouxANampoothiriSLaurentNNovel KIF7 mutations extend the phenotypic spectrum of acrocallosal syndromeJ Med Genet2012491171372023125460
- DrivasTGWojnoAPTuckerBAStoneEMBennettJBasal exon skipping and genetic pleiotropy: a predictive model of disease pathogenesisSci Transl Med20157291291ra97
- Sanchez-AlcudiaRCortonMAvila-FernandezAContribution of mutation load to the intrafamilial genetic heterogeneity in a large cohort of spanish retinal dystrophies familiesInvest Ophthalmol Vis Sci201455117562757125342620
- KhannaHDavisEEMurga-ZamalloaCAA common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathiesNat Genet200941673974519430481
- StoneEMAndorfJLWhitmoreSSClinically focused molecular investigation of 1000 consecutive families with inherited retinal diseaseOphthalmol2017124913141331
- MykytynKBraunTCarmiRIdentification of the gene that, when mutated, causes the human obesity syndrome BBS4Nat Genet200128218819111381270
- PriyaSNampoothiriSSenPSripriyaSBardetbiedl syndrome: genetics, molecular pathophysiology, and disease managementIndian J Ophthalmol201664962062727853007
- WestonMDLuijendijkMWHumphreyKDMollerCKimberlingWJMutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of usher syndrome type IIAm J Hum Genet200474235736614740321
- SwainPKChenSWangQLMutations in the cone-rod homeo-box gene are associated with the cone-rod dystrophy photoreceptor degenerationNeuron1997196132913369427255
- ChenSWangQLNieZCrx, a novel otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genesNeuron1997195101710309390516
- ChenSWangQLXuSFunctional analysis of cone-rod homeo-box (CRX) mutations associated with retinal dystrophyHum Mol Genet200211887388411971869
- NishiguchiKMSandbergMAGorjiNBersonELDryjaTPCone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseasesHum Mutat200525324825815712225
- JoblingAIVesseyKAWaughMMillsSAFletcherELA naturally occurring mouse model of achromatopsia: characterization of the mutation in cone transducin and subsequent retinal phenotypeInvest Ophthalmol Vis Sci20135453350335923580486
- KuniyoshiKMuraki-OdaSUeyamaHNovel mutations in the gene for alpha-subunit of retinal cone cyclic nucleotide-gated channels in a japanese patient with congenital achromatopsiaJpn J Ophthalmol201660318719727040408
- Asai-CoakwellMFrenchCRYeMIncomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypesHum Mol Genet20091861110112119129173
- Asai-CoakwellMMarchLDaiXHContribution of growth differentiation factor 6-dependent cell survival to early-onset retinal dystrophiesHum Mol Genet20132271432144223307924
- TassabehjiMFangZMHiltonENMutations in GDF6 are associated with vertebral segmentation defects in klippel-feil syndromeHum Mutat20082981017102718425797
- CremersFPvan den HurkJAden HollanderAIMolecular genetics of leber congenital amaurosisHum Mol Genet200211101169117612015276
- DrivasTGHolzbaurELBennettJDisruption of CEP290 microtubule/ membrane-binding domains causes retinal degenerationJ Clin Invest2013123104525453924051377
- D’AngeloAFrancoBThe dynamic cilium in human diseasesPathogenetics200921319439065
- den HollanderAIKoenekoopRKYzerSMutations in the CEP290 (NPHP6) gene are a frequent cause of leber congenital amaurosisAm J Hum Genet200679355656116909394
- ConleySMStuckMWBurnettJLInsights into the mechanisms of macular degeneration associated with the R172W mutation in RDSHum Mol Genet201423123102311424463884
- MeinsMGruningGBlankenagelAHeterozygous ‘null allele’ mutation in the human peripherin/RDS geneHum Mol Genet1993212218121828111389
- WroblewskiJJWellsJA3rdEcksteinAMacular dystrophy associated with mutations at codon 172 in the human retinal degeneration slow geneOphthalmology1994101112228302543
- WangXWangHSunVComprehensive molecular diagnosis of 179 leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencingJ Med Genet2013501067468823847139
- StylesCJDhillonBWrightAFThe diagnosis of autosomal dominant late-onset retinal degeneration in two sistersEye (Lond)200317453053212802359
- TsujikawaMWadaYSukegawaMAge at onset curves of retinitis pigmentosaArch Ophthalmol2008126333734018332312
- NashBMWrightDCGriggJRBennettsBJamiesonRVRetinal dystrophies, genomic applications in diagnosis and prospects for therapyTransl Pediatr20154213916326835369
- YangZAlvarezBVChakarovaCMutant carbonic anhydrase 4 impairs pH regulation and causes retinal photoreceptor degenerationHum Mol Genet200514225526515563508
- ZernantJCollisonFTLeeWGenetic and clinical analysis of ABCA4-associated disease in african american patientsHum Mutat201435101187119425066811
- FujinamiKZernantJChanaRKABCA4 gene screening by next-generation sequencing in a british cohortInvest Ophthalmol Vis Sci201354106662667423982839
- MaugeriAKleveringBJRohrschneiderKMutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophyAm J Hum Genet200067496096610958761
- KitiratschkyVBGrauTBerndAABCA4 gene analysis in patients with autosomal recessive cone and cone rod dystrophiesEur J Hum Genet200816781281918285826
- BriggsCERucinskiDRosenfeldPJHiroseTBersonELDryjaTPMutations in ABCR (ABCA4) in patients with stargardt macular degeneration or cone-rod degenerationInvest Ophthalmol Vis Sci200142102229223611527935
- SohockiMMBowneSJSullivanLSMutations in a new photoreceptor-pineal gene on 17p cause leber congenital amaurosisNat Genet2000241798310615133
- CideciyanAVRachelRAAlemanTSCone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathyHum Mol Genet20112071411142321245082
- SarpongASchottmannGRutherKProtracted course of juvenile ceroid lipofuscinosis associated with a novel CLN3 mutation (p.Y199X)Clin Genet2009761384519489875
- MunroePBMitchisonHMO’RaweAMSpectrum of mutations in the batten disease gene, CLN3Am J Hum Genet19976123103169311735
- KohlSVarsanyiBAntunesGACNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsiaEur J Hum Genet200513330230815657609
- SeongMWKimSYYuYSHwangJMKimJYParkSSMolecular characterization of leber congenital amaurosis in koreansMol Vis2008141429143618682808
- Abu-SafiehLAlrashedMAnaziSAutozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genesGenome Res201323223624723105016
- den HollanderAIten BrinkJBde KokYJMutations in a human homologue of drosophila crumbs cause retinitis pigmentosa (RP12)Nat Genet199923221722110508521
- Bandah-RozenfeldDMizrahi-MeissonnierLFarhyCHomozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosaAm J Hum Genet201087338239120705279
- EisenbergerTNeuhausCKhanAOIncreasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophiesPLoS One2013811e7849624265693
- BandahDMerinSAshhabMBaninESharonDThe spectrum of retinal diseases caused by NR2E3 mutations in israeli and palestinian patientsArch Ophthalmol2009127329730219273793
- HaiderNBJacobsonSGCideciyanAVMutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fateNat Genet200024212713110655056
- SharonDSandbergMACarusoRCBersonELDryjaTPShared mutations in NR2E3 in enhanced S-cone syndrome, goldmann-favre syndrome, and many cases of clumped pigmentary retinal degenerationArch Ophthalmol200312191316132312963616
- CoppietersFLeroyBPBeysenDRecurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosaAm J Hum Genet200781114715717564971
- DryjaTPRucinskiDEChenSHBersonELFrequency of mutations in the gene encoding the alpha subunit of rod cGMP-phosphodiesterase in autosomal recessive retinitis pigmentosaInvest Ophthalmol Vis Sci19994081859186510393062
- PayneAVithanaEKhaliqSRP1 protein truncating mutations predominate at the RP1 adRP locusInvest Ophthalmol Vis Sci200041134069407311095597
- TiwariABahrABahrLNext generation sequencing based identification of disease-associated mutations in swiss patients with retinal dystrophiesSci Rep201662875527353947
- SimovichMJMillerBEzzeldinHFour novel mutations in the RPE65 gene in patients with leber congenital amaurosisHum Mutat2001182164
- YzerSvan den BornLISchuilJA Tyr368His RPE65 founder mutation is associated with variable expression and progression of early onset retinal dystrophy in 10 families of a genetically isolated populationJ Med Genet200340970971312960219
- WestonMDEudyJDFujitaSGenomic structure and identification of novel mutations in usherin, the gene responsible for usher syndrome type IIaAm J Hum Genet20006641199121010729113
- DreyerBBroxVTranebjaergLSpectrum of USH2A mutations in scandinavian patients with usher syndrome type IIHum Mutat2008293451
- McGeeTLSeyedahmadiBJSweeneyMODryjaTPBersonELNovel mutations in the long isoform of the USH2A gene in patients with usher syndrome type II or non-syndromic retinitis pigmentosaJ Med Genet201047749950620507924
- AllerEJaijoTBeneytoMIdentification of 14 novel mutations in the long isoform of USH2A in spanish patients with usher syndrome type IIJ Med Genet20064311e5517085681
- Le Quesne StabejPSaihanZRangeshNComprehensive sequence analysis of nine usher syndrome genes in the UK national collaborative usher studyJ Med Genet2012491273622135276
- EudyJDWestonMDYaoSMutation of a gene encoding a protein with extracellular matrix motifs in usher syndrome type IIaScience19982805370175317579624053
- FriedmanJSChangBKannabiranCPremature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degenerationAm J Hum Genet20067961059107017186464
- SlavotinekAMSearbyCAl-GazaliLMutation analysis of the MKKS gene in McKusick-kaufman syndrome and selected bardetbiedl syndrome patientsHum Genet2002110656156712107442
- SrourMSchwartzentruberJHamdanFFMutations in C5ORF42 cause joubert syndrome in the french canadian populationAm J Hum Genet201290469370022425360
- HuangXFHuangFWuKCGenotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencingGenet Med201517427127825356976
- KroesHYvan ZonPHFransen van de PutteDDNA analysis of AHI1, NPHP1 and CYCLIN D1 in joubert syndrome patients from the netherlandsEur J Med Genet2008511243418054307
- DreyerBTranebjaergLRosenbergTWestonMDKimberlingWJNilssenOIdentification of novel USH2A mutations: implications for the structure of USH2A proteinEur J Hum Genet20008750050610909849
- HuangXFXiangPChenJTargeted exome sequencing identified novel USH2A mutations in usher syndrome familiesPLoS One201385e6383223737954
- YatsenkoANShroyerNFLewisRALupskiJRLate-onset stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4)Hum Genet2001108434635511379881
- BreuerDKYasharBMFilippovaEA comprehensive mutation analysis of RP2 and RPGR in a north american cohort of families with X-linked retinitis pigmentosaAm J Hum Genet20027061545155411992260
- SharonDBrunsGAMcGeeTLSandbergMABersonELDryjaTPX-linked retinitis pigmentosa: mutation spectrum of the RPGR and RP2 genes and correlation with visual functionInvest Ophthalmol Vis Sci20004192712272110937588
- Riveiro-AlvarezRAguirre-LambanJLopez-MartinezMAFrequency of ABCA4 mutations in 278 spanish controls: an insight into the prevalence of autosomal recessive stargardt diseaseBr J Ophthalmol200993101359136418977788
- OuyangXMYanDDuLLCharacterization of usher syndrome type I gene mutations in an usher syndrome patient populationHum Genet2005116429229915660226
- MykytynKNishimuraDYSearbyCCEvaluation of complex inheritance involving the most common bardetbiedl syndrome locus (BBS1)Am J Hum Genet200372242943712524598
- RoepmanRBernoud-HubacNSchickDEThe retinitis pigmentosa GTPase regulator (RPGR) interacts with novel transport-like proteins in the outer segments of rod photoreceptorsHum Mol Genet20009142095210510958648
- GerberSPerraultIHaneinSComplete exon-intron structure of the RPGR-interacting protein (RPGRIP1) gene allows the identification of mutations underlying leber congenital amaurosisEur J Hum Genet20019856157111528500
- BealesPLBadanoJLRossAJGenetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-mendelian bardet-biedl syndromeAm J Hum Genet20037251187119912677556
- LangmannTDi GioiaSARauINonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosaAm J Hum Genet201087337638120705278
- OshimaAJaijoTAllerEMutation profile of the CDH23 gene in 56 probands with usher syndrome type IHum Mutat2008296E37E4618429043
- SeyedahmadiBJRivoltaCKeeneJABersonELDryjaTPComprehensive screening of the USH2A gene in usher syndrome type II and non-syndromic recessive retinitis pigmentosaExp Eye Res200479216717315325563