Abstract
Background:
To compare the safety and efficacy of loteprednol etabonate ophthalmic ointment 0.5% (LE ointment), a new topical ointment formulation, with vehicle for the treatment of inflammation and pain following cataract surgery.
Methods:
Two randomized, multicenter, double-masked, parallel-group, vehicle-controlled studies were conducted. Patients aged ≥18 years with a combined postoperative anterior chamber cells and flare (ACI) ≥ Grade 3 following uncomplicated cataract surgery participated in seven study visits. Patients self-administered either topical LE ointment or vehicle four times daily for 14 days. Efficacy outcomes included the proportion of patients with complete resolution of ACI and the proportion of patients with no (Grade 0) pain at postoperative day 8. Safety outcomes included the incidence of adverse events, ocular symptoms, changes in intraocular pressure and visual acuity, and biomicroscopy and funduscopy findings.
Results:
Data from the two studies were combined. The integrated intent-to-treat population consisted of 805 patients (mean [standard deviation] age 69.0 [9.2] years; 58.0% female and 89.7% white). Significantly more LE ointment-treated patients than vehicle-treated patients had complete resolution of ACI (27.7% versus 12.5%) and no pain (75.5% versus 43.1%) at day 8 (P < 0.0001 for both). Fewer LE ointment-treated patients required rescue medication (27.7% versus 63.8%), and fewer had an ocular adverse event (47.2% versus 78.0%, P < 0.0001) while on study treatment. The most common ocular adverse events with LE ointment were anterior chamber inflammation, photophobia, corneal edema, conjunctival hyperemia, eye pain, and iritis. Mean intraocular pressure decreased in both treatment groups. Four patients had increased intraocular pressure ≥10 mmHg (three LE ointment and one vehicle) prior to rescue medication. Visual acuity and dilated funduscopy results were similar between the treatment groups, with the exception of visual acuity at visits 5 and 6, which favored LE ointment.
Conclusion:
LE ointment was efficacious and well tolerated in the treatment of ocular inflammation and pain following cataract surgery.
Introduction
Topical corticosteroids are useful in a variety of ophthalmic conditions and are generally indicated for treatment of steroid-responsive inflammatory conditions of the palpebral and bulbar conjunctiva, cornea, and anterior segment of the eye.Citation1 Although corticosteroids are widely used as topical agents for ocular inflammation, most possess a risk profile that limits their general utility. A common risk associated with corticosteroid therapy is elevation of intraocular pressure (IOP).Citation2,Citation3 A rise in IOP may result in optic nerve damage and visual field defects. In addition, chronic use of corticosteroids may result in the development of cataracts.Citation4 Therefore, it is recommended that patients limit their exposure to corticosteroids and have their IOP routinely monitored during treatment.
Loteprednol etabonate (LE) is an ester corticosteroid specifically designed to be metabolically labile, subject to predictable hydrolysis, resulting in the formation of inactive metabolites.Citation5,Citation6 Specifically, the labile 17-β-chloromethyl ester function undergoes de-esterification to an inactive carboxylic acid metabolite after exerting its effect or when unbound due to receptor saturation, thereby minimizing the likelihood of toxicity.Citation7 Hence, LE has a lower propensity to induce elevation in IOP even when used in known steroid responders.Citation8 The absence of the ketone group at position C-20 should also reduce the likelihood of molecular interaction with amino acid residues on the ocular lens proteins and formation of steroid-protein adducts, a process which may be involved in cataract formation. The biochemical mechanism of corticosteroid cataractogenesis has been shown to involve reaction of the C-20 steroid ketol moiety with lens protein amino groups to form a Schiff base intermediate, followed by interaction with the C-21 hydroxyl to produce a stable ketoamine via a Heyns rearrangement.Citation9 The metabolism of LE to inactive metabolites and the absence of the ketone group at position C-20 should offer a therapeutic advantage over conventional steroids by reducing the risk for steroid-induced cataracts and elevation of IOP.
LE has been approved by the US Food and Drug Administration and other global health authorities for several clinical indications. In the US, LE ophthalmic suspension 0.5% (Lotemax®, Bausch and Lomb Inc, Rochester, NY) is indicated for the treatment of steroid-responsive inflammatory conditions of the palpebral and bulbar conjunctiva, cornea, and anterior segment of the globe, and postoperative inflammation following ocular surgery.Citation10 In clinical studies, LE ophthalmic suspension 0.5% was shown to be effective in the treatment of inflammation associated with giant papillary conjunctivitis,Citation11,Citation12 seasonal allergic conjunctivitis,Citation13 dry eye,Citation14 anterior uveitis,Citation15 and cataract surgery.Citation16,Citation17 In addition, LE ophthalmic suspension 0.5% has been shown to have a decreased incidence of significant IOP increase (ie, ≥10 mmHg) compared with dexamethasone,Citation18 as well as a smaller mean change from baseline IOP compared with both dexamethasoneCitation18,Citation19 and prednisolone acetate.Citation7
The availability of an ointment formulation in the already well characterized 0.5% LE concentration would allow physicians a choice of dosage forms when treating ocular inflammation. The primary objective of the two Phase III clinical studies described herein was to compare the safety and efficacy of LE ophthalmic ointment 0.5% with that of the vehicle for the treatment of inflammation and pain following cataract surgery. The inflammation and pain associated with cataract surgery is a good model for evaluating treatment of intraocular inflammation. The two studies had identical designs, including study endpoints, choice of control group, study duration, statistical methods, patient population, and dosage. The efficacy and safety data were collected in an identical manner in both studies. Therefore, integrated analyses of the pooled data from the two studies were performed.
Methods
Two randomized, multicenter, double-masked, parallel-group, vehicle-controlled studies were conducted at 33 centers in the US between 2008 and 2009 (ClinicalTrials.gov. Identifiers: NCT00645671 and NCT00699153). The studies were conducted in accordance with the Declaration of Helsinki (2004) and were approved by the institutional review board of each center or by Schulman Associates Institutional Review Board Inc (Cincinnati, OH). All patients (or a legally authorized representative) gave written informed consent prior to study enrollment.
Patients
Eligible patients were at least 18 years of age, were candidates for routine, uncomplicated cataract surgery (phacoemulsification with posterior chamber intraocular implantation, not combined with any other surgery), had potential postoperative pin-hole Snellen visual acuity of at least 20/200 in the study eye, were not of childbearing potential, or had a negative urine pregnancy test at screening. Furthermore, to be eligible for randomization, patients who had undergone routine, uncomplicated cataract surgery were required to have a combined grade of ≥3 for anterior chamber cells and anterior chamber flare (anterior chamber inflammation [ACI]) at visit 3 (postoperative day 1). Patients were excluded if they were expected to require concurrent ocular therapy (either eye) with nonsteroidal anti-inflammatory drugs, mast cell stabilizers, antihistamines, or decongestants, as well as immunosuppressants, and systemic or ocular corticosteroids during the 18 days following surgery. Limitations of use for each of these therapies were also prespecified prior to cataract surgery. Subjects with known hypersensitivity or contraindication to the study drug or their components were also excluded.
Study procedures
The study period for each patient was approximately four weeks in duration and required seven clinic visits. Eligibility of patients for each study was determined at the screening visit (visit 1) by a clinical assessment of ocular signs and symptoms in both eyes. The screening eye examinations included pinhole visual acuity, biomicroscopy, funduscopy, and IOP measurement. Cataract surgery was performed at visit 2, within 14 days of the screening visit. At visit 3, on postoperative day 1 (18–34 hours after surgery), the eye examinations performed at visit 1 were repeated, with the exception of funduscopy. Anterior chamber cells and flare were assessed and a combined ACI grade determined. Patients with ACI ≥ 3 were considered eligible for the study and were randomized to treatment. Patients with ACI < 3 were considered screen failures. All eligible patients were randomized to receive either study drug (LE ointment or vehicle) in a 1:1 ratio stratified by site. Patient supplies were labeled according to a computer-generated randomization schedule and dispensed sequentially by kit number within a site. Patients self-administered a 0.5 inch (1.3 cm) ribbon of study drug (LE ointment or vehicle) to the lower cul de sac of the study eye four times daily, at approximately four-hour intervals for 14 days, and recorded the times of instillation in a study diary. The initial dose of study drug occurred in the clinic at visit 3. The last treatment administration was the fourth dose on the day before visit 6. The eye examinations performed at visits 1 and 3 were repeated at visit 4 (postoperative day 3), visit 5 (postoperative day 8), visit 6 (postoperative day 15), and visit 7 (postoperative day 18), with the exception of funduscopy. Funduscopy was performed at visits 1 and 6 only.
During the study, any subjects not responding adequately to study treatment could be placed on rescue therapy. The initiation of rescue therapy and the type of medication given to a subject as rescue therapy was at the investigator’s discretion. If the investigator determined that rescue therapy was needed, the use of study medication was stopped and an adverse event of worsening inflammation or persistent inflammation was recorded. The subjects continued participation in the study and were followed through visit 7, completing all study assessments despite having discontinued use of the study medication.
Efficacy and safety outcomes
The primary efficacy endpoints for each study were the proportion of patients with complete resolution of ACI at visit 5, and the proportion of patients with no (Grade 0) pain at visit 5. Secondary efficacy endpoints included assessments at each visit of the proportion of patients with complete resolution of ACI, complete resolution of anterior chamber cells and anterior chamber flare individually, the proportion of patients with no (Grade 0) pain, and the change from baseline in ACI. Anterior chamber cells were rated as 0 = no cells seen, 1 = 1–5 cells, 2 = 6–15 cells, 3 = 16–30 cells, or 4 = 30 cells. Anterior chamber flare was rated as 0 = none, 1 = mild, 2 = moderate, 3 = severe, or 4 = very severe. ACI represented a combination of the grade for cells and flare. Pain was measured on a scale of 0–5, from 0 = no pain to 5 = severe pain.
Safety endpoints included the incidence of adverse events, change from baseline in IOP and visual acuity, and the absence/presence of abnormal slit lamp findings and a variety of subjective symptoms (discharge, dryness, itching, pain, photophobia, and tearing). Ocular symptoms were considered as both safety and tolerability measures. All adverse events were documented, assessed for relationship to study drug, and rated for intensity (mild, moderate, or severe). Slit lamp findings were graded using standard clinical scales. For ocular symptoms, ocular pain was graded as previously described, while photophobia, itching, tearing, dryness, and discharge, were rated on a scale of 0–3, from 0 = absent to 3 = severe. IOP was measured using Goldmann applanation tonometry (or an equivalent technique), following the assessment of ocular signs and symptoms. Dilated fundus examinations were performed at visits 1 and 6 to assess the retina, macula, choroid, and optic nerve for abnormalities.
Statistical analyses
Approximately 400 patients were planned to be enrolled in each study (200 patients per treatment group), to yield approximately 180 patients per treatment group completing the study based on a sample size of 180 patients per treatment group providing 99% power to detect a difference between the LE ointment and vehicle treatment groups in the proportion of patients with complete resolution of ACI as well as in the proportion of patients with no (Grade 0) pain at visit 5 (postoperative day 8). With more than 300 patients/eyes in the integrated safety analysis, there was at least a 95% chance of observing adverse events with an incidence of 1% or greater.
Results from the two studies were combined for integrated analyses of all endpoints. Efficacy endpoints were evaluated in all randomized patients (the intent-to-treat [ITT] population). The analyses of the primary efficacy endpoints tested the difference in the proportion of patients with complete resolution of ACI and the difference in the proportion of patients with no (Grade 0) pain between treatments at visit 5 using the Pearson χ2 test and Cochran-Mantel-Haenszel test stratified by center at the α = 0.05 level.Citation20 Missing data and data from patients placed on rescue medication prior to visit 5 were imputed as failures. For analysis of the secondary efficacy endpoints, the differences in the proportion of patients with complete resolution of ACI and the proportion of patients with no (Grade 0) pain were independently tested at each visit using the Pearson χ2 test. Additionally, anterior chamber cells and anterior chamber flare were analyzed separately at each visit, as was the change from baseline in ACI. Again, differences between treatments were tested using the Pearson χ2 test and Cochran-Mantel-Haenszel test stratified by center, and patients placed on rescue medication were imputed as failures. For consistency of presentation, results for the Pearson χ2 analysis are reported, and any differences in statistical test results are noted.
The safety analysis included all patients who received at least one dose of study drug. The proportions of patients reporting adverse events, mean changes in IOP, and proportion of patients with change in IOP ≥ 10 mmHg, visual acuity, biomicroscopy, and funduscopy findings were tabulated for each treatment group at each visit. To avoid the confounding effect of concomitant rescue medication, safety data collected prior to rescue medication were analyzed. Between-group comparisons of the proportions of patients experiencing adverse events were evaluated using the Fisher’s exact test.Citation20
Ocular symptoms were compared between treatment groups both as absence versus presence of a symptom (Pearson χ2 test) and as change from baseline severity (Wilcoxon rank sum test). For these analyses, patients who used rescue medication prior to the visit being summarized were excluded.
All analyses were conducted using the Statistical Analysis System, version 9.1 (SAS Institute Inc, Cary, NC).
Results
Study population
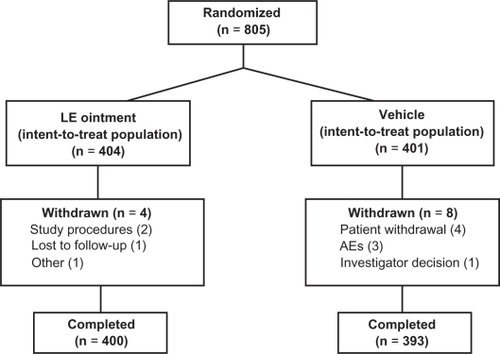
Patients were recruited from 33 US sites. A total of 805 patients were randomized to treatment (404 patients to LE ointment and 401 patients to vehicle) and were included in the ITT population (); all 805 patients received at least one dose of study drug and were included in the safety population. Six patients randomized to vehicle received LE ointment and five patients randomized to LE ointment received vehicle, resulting in a safety population of 405 and 400 patients in the LE ointment and vehicle treatment groups, respectively. Almost all randomized patients completed the study, ie, 400 (99.0%) patients randomized to LE ointment and 393 (98.0%) patients randomized to vehicle. Twelve patients (1.5%) were discontinued from the study, including four LE ointment and eight vehicle. Reasons for discontinuation included patient withdrawal (four vehicle), adverse events (three vehicle), failure to follow study procedures (two LE ointment), investigator decision (one vehicle), loss to follow-up (one LE ointment), and “other reasons” (one LE ointment).
Figure 1 Flow of patients through the study.
Patient demographic characteristics for the ITT population are shown in . Demographics were similar for the LE ointment and vehicle treatment groups. The majority of patients were white (89.7%) and female (58.0%). The mean (standard deviation [SD]) age was 69.0 (9.16) years, with an overall age range of 21 to 94 years. Ocular and nonocular medical histories were generally similar between treatments, and indicative of a study population nearing the seventh decade of life. Based on patient diaries, more than 90% of patients in the LE ointment and vehicle treatment groups were at least 80% compliant with treatment administration.
Table 1 Demographics, intent-to-treat population
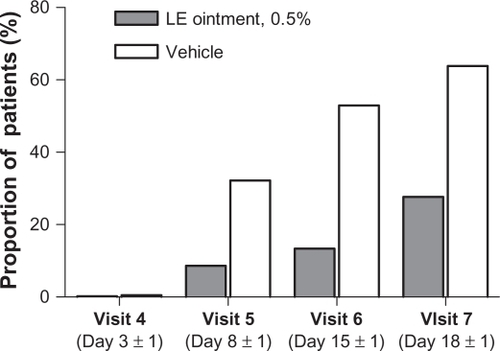
A total of 112 (27.7%) patients randomized to LE ointment and 256 (63.8%) patients randomized to vehicle required rescue therapy (nonsteroidal anti-inflammatory drugs and/or corticosteroids) during the study. An additional 18 and four patients in the LE ointment and vehicle treatment groups, respectively, received rescue therapy at study exit. shows the proportion of subjects who required rescue medication use prior to each visit. Prior to visit 5, rescue therapy was initiated for 35 (8.7%) patients in the LE ointment group versus 129 (32.2%) patients in the vehicle group. The use of rescue medication decreased overall exposure to assigned treatments, especially in the vehicle group. Mean (SD) treatment exposure was 12.9 (3.43) days for patients randomized to LE ointment, with a median exposure of 14 days, and 9.0 (5.12) days for patients randomized to vehicle, with a median exposure of 8.0 days.
Figure 2 Proportion of patients with rescue medication use prior to each visit.
Efficacy outcomes
Significantly more patients randomized to LE ointment had complete resolution of ACI, and no (Grade 0) pain at visit 5 () compared with patients randomized to vehicle. Complete resolution of ACI was observed for 112 (27.7%) patients in the LE ointment group compared with 50 (12.5%) patients in the vehicle group (P < 0.0001). Additionally, 305 (75.5%) LE ointment patients compared with 173 (43.1%) vehicle patients had no (Grade 0) pain (P < 0.0001). When the primary analyses were repeated using the per protocol population, the results were similar to that of the ITT population, (P < 0.0001 for ACI and for pain). Furthermore, subgroup analysis for patients stratified by age (<65 years, 65–75 years, and >75 years) returned similar results (P ≤ 0.0245 for ACI and P ≤ 0.0001 for pain). As indicated above, six patients randomized to vehicle received LE ointment and five patients randomized to LE ointment received vehicle. Outcomes for the primary analysis were also similar when the ITT population was analyzed as treated, with complete resolution of ACI observed for 115 (28.4%) versus 47 (11.8%) of LE ointment and vehicle-treated patients, respectively, and no (Grade 0) pain observed in 308 (76.0%) versus 170 (42.5%) of LE ointment and vehicle-treated patients, respectively (P < 0.0001 for both).
Table 2 Resolution of anterior chamber cells and flare and pain at visit 5: integrated intent-to-treat population
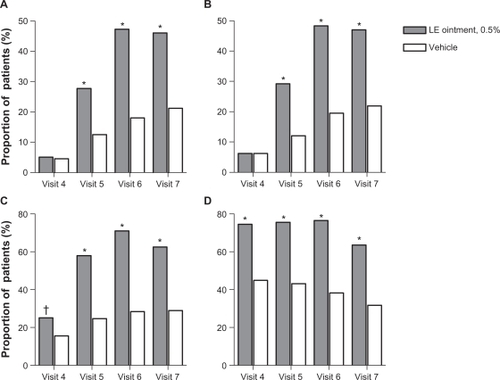
Results of the secondary outcome measures were in agreement with the primary outcome measures. Significantly more patients randomized to LE ointment compared with patients randomized to vehicle had complete resolution of ACI and anterior chamber cells at visits 5–7; and anterior chamber flare as well as no (Grade 0) pain at visits 4–7 (). Baseline mean (SD) ACI severity was 3.7 (0.75) and 3.7 (0.82) in the LE ointment and vehicle treatment groups, respectively. Mean change from baseline ACI showed an improvement in both groups, with a mean (SD) change of −1.1 (1.14), −2.2 (1.41), −2.6 (1.48), and −2.6 (1.52) for the LE ointment group and a mean change of −0.5 (1.45), −0.7 (1.81), −1.0 (1.95), and −1.1 (1.98) for the vehicle group at visits 4–7, respectively. Mean changes were consistently and significantly lower in the LE ointment treatment group at each of these visits (P < 0.0001).
Figure 3 Proportion of patients with complete resolution of anterior chamber inflammation A), complete resolution of anterior chamber cells B), complete resolution of anterior chamber flare C), and no (Grade 0) pain D) at each study visit.
Abbreviation: LE, loteprednol etabonate.
Tolerability of the study medications was judged from assessment of ocular symptoms at baseline and at each visit. At baseline (visit 3), under 5% of subjects had symptoms of ocular discharge, dryness, or itching. The proportion of subjects with these symptoms and their change from baseline at visits 4–7 were similar between treatment groups, with the exception of ocular discharge at post-treatment visit 7, which favored vehicle (4.8% versus 0.7%, P = 0.0306), and dryness at visit 5, which favored LE ointment (13.7% versus 20.6%, P = 0.0213). Of the subjects who had discharge at visit 7, all had mild discharge, and all but one either had discharge that was resolved previously and reported again or had discharge reported for the first time at visit 7. The proportions of patients in the LE ointment group and vehicle group with ocular pain, photophobia, and tearing at baseline were 44.1% versus 46.6%, 57.9% versus 55.9%, and 37.1% versus 35.7%, respectively. There were fewer subjects in the LE ointment group compared with the vehicle group having pain at visit 4 (24.9% versus 54.5%) and visit 5 (16.4% versus 35.2%), and ocular pain either improved or did not change from baseline for 92.0% versus 66.2% of subjects, respectively, at visit 4 and for 91.0% versus 78.7% of subjects, respectively, at visit 5 (P < 0.0001 for all). Likewise, there were fewer subjects in the LE ointment group compared with the vehicle group with photophobia at visit 4 (45.6% versus 64.9%) and visit 5 (40.0% versus 58.8%) and tearing at visit 4 (22.9% versus 34.6%) and visit 5 (16.4% versus 25.1%, P < 0.01 for all). Photophobia either improved or did not change from baseline for 88.5% versus 70.7% of subjects at visit 4 and for 86.3% versus 71.2% of subjects at visit 5 (P < 0.0001 for both), while tearing improved or did not change from baseline in 91.8% versus 82.8% of subjects at visit 4 (P = 0.003) and for 92.1% versus 89.9% at visit 5 (P = 0.0287) in the LE ointment and vehicle groups, respectively. At visit 6, there was a significant difference in the proportion of eyes with stable or improved photophobia (90.8% versus 84.2%, respectively, P = 0.0157) but no difference in the presence of photophobia. Visit 6 tearing rate was significantly lower in the LE ointment group when controlling for center (P = 0.1216/0.0482; Pearson/Cochran-Mantel-Haenszel).
Safety outcomes
Ocular adverse events were consistent with ocular signs after cataract surgery. presents a listing of ocular adverse events that occurred in ≥3% of study eyes prior to rescue medication. There was a lower incidence of ocular adverse events in the LE ointment treatment group versus the vehicle treatment group, and the percentage of patients who had one or more ocular adverse events was significantly less in the LE ointment group (47.2%) than in the vehicle group (78.0%, P < 0.0001). The most prevalent ocular adverse events with LE ointment were ACI, photophobia, corneal edema, conjunctival hyperemia, eye pain, and iritis. Ocular adverse events that occurred at statistically different rates between treatment groups included ACI, conjunctival hyperemia, eye pain, iritis, ciliary hyperemia, increased lacrimation, and eye pruritus. In each case, these ocular adverse events occurred significantly less frequently in eyes randomized to LE ointment (P ≤ 0.0336). Most ocular adverse events were considered unrelated or unlikely to be related to study treatment (72.5% [227/313] and 63.3% [368/581] for the LE ointment and vehicle treatment groups, respectively). Most ocular adverse events considered definitely related to study treatment were of mild or moderate severity (82.6% [19/23] across treatment groups). Four of the adverse events were definitely related to treatment and included ACI (two vehicle), eye pain (LE ointment), and eye irritation (LE ointment).
Table 3 Ocular adverse events occurring in ≥3% study eyes in any treatment group prior to rescue medication
The protocol required the recording of an adverse event of ACI prior to initiation of rescue therapy. To differentiate between adverse events related to increased ACI and those cases where inflammation was stable or improved, a supplemental analysis was performed in which any adverse event associated with rescue medication without increased inflammation relative to visit 3 (baseline) was removed from the listing. Patients with stable ACI of maximum severity (Grade 8) at the rescue visit, however, remained in the analysis. Results from this additional analysis showed that ACI adverse event rates in the study eye prior to rescue medication use decreased from the original rates of 27.2% (110 patients) versus 50.0% (200 patients) for LE ointment versus vehicle treatment groups to 12.8% (52 patients) and 27.5% (110 patients), respectively (P < 0.0001 for both).
Nonocular adverse events were less commonly reported. The percentage of patients who had one or more nonocular adverse events did not differ between the LE ointment treatment group and the vehicle treatment group (5.2% [21/405] and 4.5% [18/400], respectively, P = 0.743). The only non-ocular adverse event reported at a rate of at least 1% was headache (1.5% [6/405] and 1.3% [5/400], respectively, P > 0.9999).
There were no deaths during the study period. Three patients in the vehicle treatment group withdrew from the study due to a serious adverse event. All three serious adverse events were considered by the investigator to be unrelated to the study drug. One patient developed endophthalmitis, which resolved through use of antibiotics, and was considered by the investigator to be related to the study procedure (ie, cataract surgery). The other two were discontinued due to nonocular serious adverse events. Other serious adverse events were all nonocular, with the exception of one report of cystoid macular edema in the LE ointment group. Three additional ocular serious adverse events were reported at study exit, and included mild cystoid macular edema present in three vehicle-treated patients.
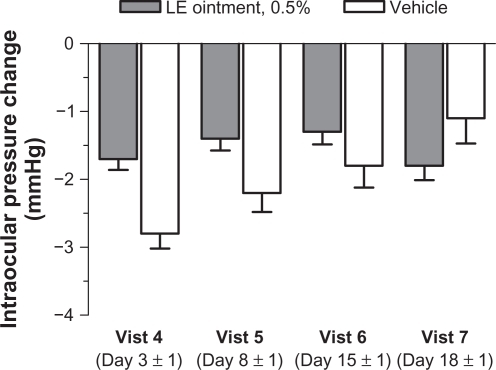
Mean baseline (postoperative day 1) IOP for study eyes was similar between treatment groups. At subsequent visits, mean IOP was consistently lower than baseline for both treatment groups (). Over the course of the study, four patients (three LE ointment and one vehicle) had IOP elevations ≥10 mmHg from baseline prior to rescue medication, with a maximum IOP of 32 mmHg for an LE ointment-treated subject at visit 5. The difference between treatment groups in the percentage of study eyes with an increase in IOP ≥10 mmHg from baseline was not significant at any post-baseline visit.
Figure 4 Mean (± standard error of the mean) change from baseline in intraocular pressure (mmHg) by visit.
The distribution of slit lamp signs reported at baseline (postoperative day 1) was similar between treatment groups. At visits 4–7, the proportion of patients with an increase in severity from baseline for each ocular sign were consistently either similar between treatments or significantly different between treatments in favor of LE ointment. There were no instances where there was a significant difference in favor of the vehicle (data not shown).
Dilated funduscopy and visual acuity results were comparable across treatment groups and visits with few exceptions. No fundus pathology was noted for the majority of eyes (>80%) at baseline and there was only one treatment-emergent finding at visit 6 of a mild retina hemorrhage in an eye treated with LE ointment that was normal at screening. At visits 5 and 6, significantly fewer eyes treated with LE ointment had a ≥2 line drop in visual acuity from baseline compared with vehicle (1.9% [7/367] versus 7.2% [19/264], P = 0.0010 at visit 5) and (0.6% [2/348] versus 2.8% [5/180], P = 0.0359 at visit 6).
Discussion
LE ointment 0.5% administered four times daily for 14 days postoperatively was shown to be superior to vehicle in the treatment of inflammation and pain following cataract surgery in this integrated analysis of two randomized, multicenter, double-masked, parallel-group, vehicle-controlled studies. A significantly greater proportion of patients randomized to LE ointment had complete resolution of ACI and no (Grade 0) pain at postoperative day 8 compared with patients randomized to vehicle (P < 0.0001 for both). Also, fewer patients in the LE ointment group than in the vehicle group required rescue medication during the course of the study. Secondary analyses were consistent with the primary analysis. These included analyses at individual visits for complete resolution of ACI, for cells and flare considered separately, for the change from baseline ACI severity, and for no pain. Resolution of ACI was significantly improved (P < 0.001) at visit 4 (postoperative day 3), while cells were not significantly different until visit 5, suggesting that a dosing period of more than three days may be required for postoperative resolution of cells.
Treatment with LE ointment was well tolerated and safe. Symptoms of ocular pain, photophobia, and tearing between the treatment groups and their change from baseline significantly favored LE ointment at most visits, while symptoms of ocular discharge, dryness, and itching were consistently similar across the treatment groups, with few exceptions. Ocular adverse events were consistent with ocular signs after cataract surgery and occurred less frequently, overall, in the LE ointment group compared with the vehicle group. The most common ocular adverse events in patients randomized to LE ointment were ACI, photophobia, corneal edema, conjunctival hyperemia, eye pain, and iritis. Of these, ACI, conjunctival hyperemia, eye pain, and iritis occurred significantly less frequently in the LE ointment compared with the vehicle group (P < 0.05). Headache was the only nonocular adverse event, and was reported by more than 1% of patients.
Mean IOP for all study eyes in both treatment groups was consistently lower than baseline at all post-treatment visits. Prior to rescue medication, there were only four patients who had an IOP increase of ≥10 mmHg from baseline over the course of the study (three LE ointment and one vehicle). These findings are consistent with the low incidence of clinically significant IOP elevations observed in studies of LE 0.5% ophthalmic suspension in postoperative inflammation following cataract surgery,Citation16,Citation17 as well as other indications,Citation19,Citation21 and are attributed to the rapid de-esterification of LE to inactive metabolites.Citation5,Citation6 Dilated funduscopy results were comparable across treatment groups, while significantly fewer eyes treated with LE ointment had a ≥2 line drop in visual acuity from the day 1 postoperative visit compared with vehicle-treated eyes at visits 5 and 6.
The availability of an ointment formulation for LE allows physicians an additional choice in dosage forms when treating patients with ocular inflammation. Ointment formulations increase the ocular contact time of applied drugs approximately two-fold in blinking eyes and four-fold in the nonblinking (patched) eyes as compared with saline vehicle.Citation22 They are generally used to increase drug absorption for night-time therapy or for treating children because they do not wash out readily with tearing. Disadvantages include transient blurred vision and more difficult administration.Citation22 Fluorometholone ointment 0.1% is currently the only approved ophthalmic t opical corticosteroid ointment in the US, and is indicated for the treatment of steroid-responsive inflammation of the palpebral and bulbar conjunctiva, cornea, and anterior segment of the globe.Citation23 There has been only one clinical study comparing the safety and efficacy of any LE ophthalmic formulation with a fluorometholone ophthalmic formulation. Stewart compared the efficacy and safety of fluorometholone acetate 0.1% suspension with LE ophthalmic suspension 0.5%, both administered four times a day for 14 days in a small randomized, double-masked, parallel-group study (n = 30) of patients with postoperative inflammation following cataract surgery.Citation24 No statistical differences were found between treatments in resolution of flare, anterior chamber cells, or conjunctival hyperemia, and there were no significant adverse events in either group. Concentrations of active ingredients in the formulations compared in the Stewart study were the same as those in their respective ointment formulations; however, both formulations contained benzalkonium chloride 0.01% as a preservative.Citation10,Citation25 LE ointment is preservative-free, while fluorometholone ointment is preserved with 0.0008% phenylmercuric acetate,Citation23 a preservative subject to degradation to mercury and which has been associated with time- and dose-dependent ocular cytotoxicityCitation26 and periorbital contact dermatitis.Citation27
Both Howes et al and Samudre et al studied the anti-inflammatory activity of LE ophthalmic suspension 0.5% in comparison with that of other topical ophthalmic steroids, including fluorometholone acetate 0.1% suspension, in a rabbit model of acute anterior uveitis.Citation28,Citation29 While there was no difference between fluorometholone acetate and LE in reducing anterior chamber cells and iris hyperemia in the study by Howes et al, LE was less effective in reducing conjunctival injection, anterior chamber flare, and protein.Citation28 In contrast, Samudre et al reported that LE 0.5% was associated with the most glucocorticoid receptor migration compared with other ophthalmic steroids, including fluorometholone, and was highly efficacious using several independent measures of steroid anti-inflammatory efficacy.Citation29 In addition, confocal microscopy indicated that only LE reverted abnormal corneal endothelial cell shape back to normal.Citation29 These results suggest that there may be some differences in efficacy between LE and fluorometholone that warrant further study. Fluorometholone itself is a C-20 ketone corticosteroid, and there have been case reports of fluorometholone-induced cataract formation in the literature.Citation30,Citation31 As indicated earlier, LE is a C-20 ester corticosteroid, and therefore does not form Schiff base intermediates with lens protein, a likely first step in cateracterogenesis.Citation9 Finally, Cantrill et al reported a mean increase in IOP from baseline of 6.1 ± 1.4 mmHg with fluorometholone acetate 0.1% suspension in steroid-responsive subjects (n = 6) treated four times daily for two to six weeks;Citation32 while Bartlett reported a mean IOP rise of 4.1 mmHg in steroid-responsive subjects treated with LE ophthalmic suspension 0.5% (n = 14) also treated four times daily but for a full six weeks, a change from baseline that was not statistically significant.Citation7 These findings suggest that there may also be differences in safety with these two steroids.
In conclusion, the integrated results of these studies indicate that LE ointment is effective and well tolerated in the treatment of postoperative inflammation and pain following ocular surgery. The availability of an ointment formulation of the already well characterized LE 0.5% concentration allows physicians a choice of dosage forms in treating ocular inflammation following ocular surgery. The availability of a preservative-free ointment formulation of LE may provide a safety advantage over fluorometholone ointment, although further randomized comparative clinical studies are warranted.
Acknowledgements
This study was designed and conducted by Bausch and Lomb, Inc. The authors thank the following principal investigators who participated in the studies: R Arleo, Ithaca, NY; P Arnold, Fort Collins, CO; D Brown, Fort Myers, FL; R Chu, Bloomington, MN; L Cibik, Monroeville, PA; A Cottingham, San Antonio, TX; T Croley, Ocala, FL; R DeBarge, Fort Oglethorpe, GA; A Fishman, Pembroke Pines, FL; W Flynn, San Antonio, TX; W Fried, Gurnee, IL; J Gira, Des Peres, MO; M Graham, Orlando, FL; P Hartman, Rochester, NY; G Henderson, Brandon, FL; J Hunkeler, Overland Park, KS; K Kansupada, Belmont, NC; M Korenfeld, Washington, MO; S Lane, Stillwater, MN; D Lorenz, Henderson, NV; T Macejko, Fairfield, OH; J Macy, Los Angeles, CA; J McDonald, Fayetteville, AR; J Peace, Inglewood, CA; B Perez, Tampa, FL; T Peters, Portsmouth, NH; H Reiser, Kingston, PA; M Rotberg, Charlotte, NC; K Sall, Artesia, CA; S Smith, Fort Myers, FL; R Smyth-Medina, Mission Hills, CA; W Colby Stewart, Houston, TX; L Taustine, Louisville, KY; M Tepedino, High Point, NC; S Trocme, Cleveland, OH; F Tyson, Cape Coral, FL; S Updegraff, St Petersburg, FL; T Walters, Austin, TX. The authors also wish to thank Dan Wilson, PhD, and Heleen H DeCory, PhD, both from Bausch and Lomb Inc, for writing and editorial assistance.
Disclosure
TL Comstock, MR Paterno, A Singh, and T Erb are employees of Bausch and Lomb Inc. Elizabeth Davis is a consultant for Bausch and Lomb Inc.
References
- JaanusSDLesherGAAnti-inflammatory drugsBartlettJDJaanusSDClinical Ocular Pharmacology3rd edBoston, MAButterworth-Heinemann1995
- BeckerBIntraocular pressure response to topical corticosteroidsInvest Ophthalmol19654219820514283013
- ArmalyMFEffects of corticosteroids on intraocular pressure and fluid dynamics. I. The effect of dexamethasone in the normal eyeArch Ophthalmol1963701048249114078870
- BurdeRMBeckerBCorticosteroid-induced glaucoma and cataracts in contact lens wearersJAMA197021312207520785468759
- BodorNLoftssonTWuWMMetabolism, distribution, and transdermal permeation of a soft corticosteroid, loteprednol etabonatePharm Res1992910127512781448425
- DruzgalaPWuWMBodorNOcular absorption and distribution of loteprednol etabonate, a soft steroid, in rabbit eyesCurr Eye Res199110109339371959381
- BartlettJDHorwitzBLaibovitzRHowesJFIntraocular pressure response to loteprednol etabonate in known steroid respondersJ Ocul Pharmacol1993921571658345288
- NovackGDHowesJCrockettRSSherwoodMBChange in intraocular pressure during long-term use of loteprednol etabonateJ Glaucoma1998742662699713785
- ManabeSBucalaRCeramiANonenzymatic addition of glucocorticoids to lens proteins in steroid-induced cataractsJ Clin Invest1984745180318106501572
- Lotemax [Package insert]Rochester, NYBausch and Lomb Inc2006
- AsbellPHowesJA double-masked, placebo-controlled evaluation of the efficacy and safety of loteprednol etabonate in the treatment of giant papillary conjunctivitisCLAO J199723131369001768
- FriedlanderMHHowesJAA double-masked, placebo-controlled evaluation of the efficacy and safety of loteprednol etabonate in the treatment of giant papillary conjunctivitis. Loteprednol Etabonate Giant Papillary Conjunctivitis Study Group 1Am J Ophthalmol199712344554649124242
- DellSJShulmanDGLowryGMHowesJA controlled evaluation of the efficacy and safety of loteprednol etabonate in the prophylactic treatment of seasonal allergic conjunctivitis. Loteprednol Allergic Conjunctivitis Study GroupAm J Ophthalmol199712367917979535623
- PflugfelderSCMaskinSLAndersonBNovackGDHartKA randomized, double-masked, placebo-controlled, multicenter comparison of loteprednol etabonate ophthalmic suspension, 0.5%, and placebo for treatment of keratoconjunctivitis sicca in patients with delayed tear clearanceAm J Ophthalmol2004138344445715364229
- Loteprednol Etabonate US Uveitis Study GroupControlled evaluation of loteprednol etabonate and prednisolone acetate in the treatment of acute anterior uveitisAm J Ophthalmol1999127553754410334346
- StewartRHorwitzBHowesJNovakGDHartKDouble-masked, placebo-controlled evaluation of loteprednol etabonate 0.5% for postoperative inflammation. Loteprednol Etabonate Postoperative Inflammation Study Group 1J Cataract Refract Surg19982410148014899818338
- Loteprednol Etabonate Postoperative Inflammation Study Group 2A double-masked, placebo-controlled evaluation of loteprednol etabonate 0.5% for postoperative inflammationOphthalmology19981059178017869754192
- HollandEJBartlettJDPaternoMRUsnerDWComstockTLEffects of loteprednol/tobramycin versus dexamethasone/tobramycin on intraocular pressure in healthy volunteersCornea2008271505518245967
- WhiteEMMacyJIBatemanKMComstockTLComparison of the safety and efficacy of loteprednol 0.5%/tobramycin 0.3% with dexamethasone 0.1%/tobramycin 0.3% in the treatment of blepharo-keratoconjunctivitisCurr Med Res Opin200824128729618062846
- AgrestiACategorical Data AnalysisNew York, NYJohn Wiley and Sons1990
- PavesioCEDeCoryHHTreatment of ocular inflammatory conditions with loteprednol etabonateBr J Ophthalmol200892445545918245274
- FiscellaRGOphthalmic drug formulationsBartlettJDJaanusSDClinical Ocular Pharmacology5th edSt. Louis, MOButterworth-Heinemann2008
- Fluorometholone [Package insert]Irvine, CAAllergan Inc2001
- StewartRSControlled evaluation of fluorometholone acetate and loteprednol etabonate in the treatment of postoperative inflammation following cataract surgeryInvest Ophthalmol Vis Sci200445Suppl 1U159
- Flarex [Package insert]Fort Worth, TXAlcon Laboratories Inc1999
- TakahashiNCytotoxicity of mercurial preservative in cell cultureOphthalmic Res1982141163697070758
- HerbstRAUterWPirkerCGeierJFroschPJAllergic and non-allergic periorbital dermatitis: Patch test results of the Information Network of the Departments of Dermatology during a 5-year periodContact Dermatitis2004511131915291826
- HowesJFBaruHVeredMNeumannRLoteprednol etabonate: Comparison with other steroids in two models of intraocular inflammationJ Ocular Pharmacol1994101289293
- SamudreSSLattanzioFAWilliamsPBSheppardJDComparison of topical steroids for acute anterior uveitisJ Ocular Pharmacol Ther2004206533547
- BilgihanKGurelikGAkataFHasanreisogluBFluorometholone-induced cataract after photorefractive keratectomyOphthalmologica199721163943969380362
- CostagliolaCCati-GiovannelliBPriccirrilloADelfinoMCataracts associated with long-term topical steroidsBr J Dermatol198912034724732713268
- CantrillHLPalmbergPFZinkHAWaltmanSRPodosSMBeckerBComparison of in vitro potency of corticosteroids with ability to raise intraocular pressureAm J Ophthalmol1975796101210171173539