
Abstract
The aim of this study was to investigate the prognostic and diagnostic value of genes with promoter methylation in hepatocellular carcinoma (HCC) patients. On the basis of The Cancer Genome Atlas data, we identified genes with differentially methylated promoters in HCC tissues and adjacent non-tumor tissues, using the linear models for microarray data approach. Cox proportional hazard regression analysis was applied to access the prognostic value of identified differentially methylated genes. The diagnostic value of the genes was evaluated through receiver operating characteristic. Pathway analyses were performed to illustrate biological functions of the identified genes. Compared to adjacent tissues, 77 genes with hypermethylated promoters and 2,412 genes with hypomethylated promoters were identified in HCC. The promoter hypomethylations of RNA5SP38, IL21, SDC4P, and MIR4439 were found to be associated with HCC patient survival (P=0.035, 0.040, 0.004, and 0.024, respectively). Hypomethylated SDC4P was associated with a better prognosis (hazard ratio, 0.482; 95% confidence interval [CI], −0.147–1.110; P=0.007). The combination of the promoter hypomethylations with RNA5SP38, IL21, and SDC4P showed an area under receiver operating characteristic curves of 0.975 (95% CI, 0.962–0.989; P=4.811E-25). Several pathways, including olfactory transduction, cytokine–cytokine receptor interaction, natural killer cell–mediated cytotoxicity, as well as inflammation mediated by chemokine and cytokine signaling pathway, were annotated with the hypomethylated promoter genes. SDC4P promoter hypomethylation may be a potential prognosis biomarker. A panel of promoter methylations in RNA5SP38, IL21, and SDC4P was proven a novel approach to diagnosis HCC. The pathway analysis defined the extensive functional role of DNA hypomethylation in cancer.
Introduction
Hepatocellular carcinoma (HCC) is a major health problem worldwide, which causes ~600,000 deaths every year.Citation1 Liver transplantation, surgical resection, target therapy, and chemotherapy are currently available therapeutic strategies for HCC.Citation2 However, the prognosis of HCC remains extremely dismal, with long-term 5-year survival rate ranging from 17% to 53%.Citation3,Citation4 HCC patients are commonly diagnosed at advanced stage, which may also contribute to the poor prognosis.Citation5 Understanding the molecular mechanisms in HCC could help identify new therapy target and find effective diagnostic and prognostic biomarkers for HCC.
Accumulated evidences have demonstrated the vital roles of DNA methylation in many biological activities, especially in cancer initiation and progression.Citation6–Citation8 Regional hypermethylation and global hypomethylation are two common kinds of aberrant methylation in cancers.Citation9 Some of the tumor-specific DNA methylations have been proposed to be potential diagnostic or prognostic biomarkers.Citation10,Citation11 Aberrant DNA methylations have been frequently found in HCC, which contributed to carcinogenesis by transcriptional silencing of tumor-suppressor genes (TSGs).Citation12,Citation13 Recent studies have attempted to find promising epigenetic aberrations to evaluate prognosis.Citation14,Citation15 Remarkably, DNA hypomethylation genes exhibit upregulated expression level in tumors and have effective impact upon tumor cell growth and metastasis.Citation16 This group of genes is supposed to constitute targets of epigenetic therapy. However, most studies were conducted to investigate the association between a specific gene promoter methylation with cancer survival, rather than screening the association between genome-wide methylation and cancer survival.Citation17,Citation18 Few concordant gene methylation patterns were observed across the individual studies. Thus, we hypothesized that the study conducted in the manner of assessing global differential promoter methylation of genes and clinical features may provide in-depth results.
The Cancer Genome Atlas (TCGA) database contains a collection of genomic alterations, DNA methylation, RNA, proteomic expression, and clinicopathological data profiles, which could help explore the molecular characteristics of HCC comprehensively (Cancer Genome Atlas Research N 2013). With the aim to identify a methylation profile informative for HCC clinical features, we stringently conducted a stepwise study taking advantage of the data from TCGA project to 1) ascertain the differential promoter methylation expression profiles between HCC tumors and non-cancerous tissues, 2) identify the methylation associated with prognosis potential from the differential expression profiles, 3) find out methylation with reliable diagnosis potential, and 4) understand the biological pathways of the differential methylation profiles.
Materials and methods
Patients and samples from TCGA
All data for HCC patients were retrieved from TCGA data portal up to June 1, 2016. The data of the patients who have suffered from other malignancies or received neo-adjuvant therapy were not included. The full clinical information including sex, age, race, vital status, tumor grade, tumor pathologic stage, lymph node pathologic stage, metastasis stage, the American Joint Committee on Cancer (AJCC) pathologic stage, and methylation values (level 1 data, Illumina Infinium Human Methylation 450K) were then downloaded. Adjacent tissues were away from the tumor margin at least 2 cm. Because the data were obtained from TCGA, further approval by an ethics committee was not required. This study meets the publication guidelines provided by TCGA (http://cancergenome.nih.gov/publicationguidelines).
Illumina infinium human methylation 450K analysis
The differential methylated genes in the HCC tissues (Cohort T) and adjacent non-tumor tissues (Cohort N) were investigated. Our study calculated TCGA Illumina Human Methylation 450 array of HCC using RnBeads version 0.99.19 in the R software 3.1.2, where the methylation signal data were extracted and processed. In processing and filtering, a probe was filtered out when the last five bases in its target sequence overlapped with single-nucleotide polymorphism and removed CpG sites with >10% missing values in all samples. Methylation measures with a detection P-value of >0.01 were removed. Both sites and samples were filtered using a greedy approach. In addition, CpG sites on the sex chromosomes were removed to avoid sex-specific methylation bias. Background subtraction with method “methylumi.noob” and normalization with method “bmiq” were performed, respectively. Surrogate variable analysis was performed to exclude covariates against the variable of tissue types (tumor and adjacent non-tumor tissues). The linear models for microarray data (LIMMA) package were used to identify differential methylated genes between HCC and adjacent tissues.
Biological functions analysis
To access main biological functions that were regulated by DNA methylation, we utilized differential methylated promoter genes for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) analyses based on the Database for Annotation, Visualization, and Integrated Discovery (DAVID). GO can organize genes into hierarchical categories and show the gene regulatory network, biological process, and molecular functions.Citation19
Statistical analysis
The differences of clinical characteristics as mentioned above were evaluated using chi-square tests in GraphPad Prism 5.0 software. The univariate Cox proportional hazards regression analysis was firstly performed to elucidate the association between differential promoter methylated genes and overall survival (OS). Kaplan–Meier survival analysis and univariate/multivariate Cox proportional hazards regression analyses were sequentially carried out in IBM Statistical Package for the Social Sciences (SPSS) Statistics Version 22.0. The predicted values from the logistic regression model were employed to construct receiver operating characteristic (ROC) curves and then to calculate the area under the ROC curves (AUC) through “pROC” package of R soft. P-values <0.05 were considered statistically significant.
Results
Patient characteristics
To explore the HCC whole-genome promoter DNA methylomes, we analyzed 327 HCC tumor tissues (Cohort T) and 46 adjacent non-tumor tissues (Cohort N) using the TCGA Illumina Human Methylation 450 microarray. All 327 patients were pathologically diagnosed with HCC. After quality control and filtering, DNA methylation analysis on 323 HCC and 45 adjacent tissues was carried out. These samples were collected between 1995 and 2013. The detailed clinical information is presented in . The mean age and standard deviation for all 323 patients were 59.040±12.885 years. Among the 323 participants (Cohort T), a total of 45 patients with adjacent non-tumor tissues were included in Cohort N. As summarized in , no significant difference was observed in the distribution of sex (P=0.239), tumor grade (P=0.113), tumor pathologic stage (P=0.523), node pathologic stage (P=0.355), metastasis pathologic stage (P=0.727), and AJCC pathologic stage (P=0.052) between the two cohorts.
Table 1 Characteristics of patients with hepatocellular cell carcinoma from the TCGA data portal
Differential methylated genes within promoter between tumor and adjacent non-tumor tissues
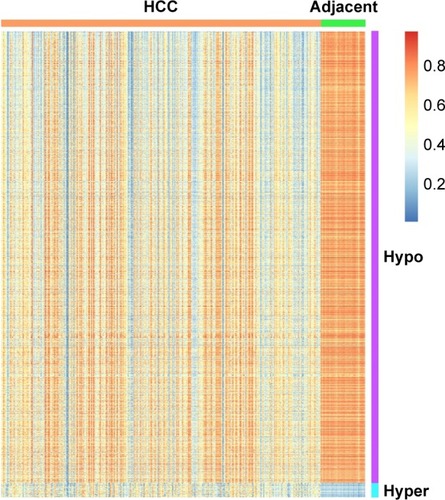
The study utilized LIMMA approach to identify the statistically different DNA methylation status between HCC and adjacent tissues, based on the remaining 445,428 probes. Promoter methylations of 2,489 genes were found to be significantly different with false discovery rate (FDR) <0.05 and |delta beta| >0.2 bases on 323 cancer tissues and 45 control tissues. Compared to adjacent tissue, 77 genes were hyper-methylated and 2,412 genes were hypomethylated within promoters in HCC, showing a surprisingly outstanding gene promoter hypomethylation in Table S1 and . A differential methylated pattern was presented between HCC and adjacent tissues in . Besides, the methylation profile in the 45 patients with both cancer tissues and adjacent non-tumor tissues was evaluated and is presented in Table S2, and the results were almost overlapped with those based on 323 cancer tissues and 45 control tissues. However, given the 323 cancer tissues had contained 45 cancer tissues, as well as to include more samples for survival and diagnostic analyses, only the results from 323 cancer tissues were chosen for further analyses.
Figure 1 Volcano plot showing the distribution of genes from the promoter-level test assessed by methylation differences and adjusted P-values.
Abbreviation: FDR, false discovery rate.
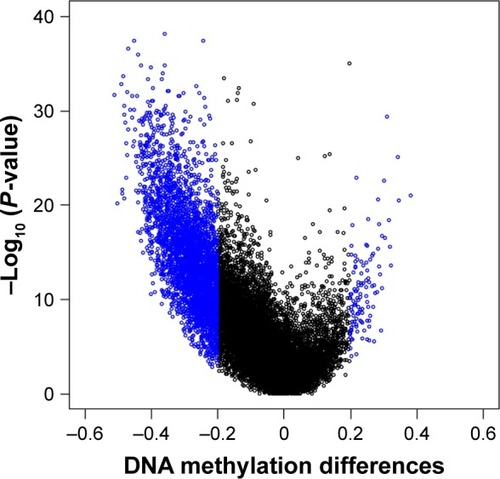
Figure 2 Heatmap of differentially methylated genes within promoters between HCC cancer tissues and adjacent tissues.
Abbreviations: FDR, false discovery rate; HCC, hepatocellular carcinoma.
Genes with methylated promoter associated with HCC patient survival
Follow-up data on survival were available for the 323 TCGA HCC patients, and 78 patients died by the end of follow-up. To identify the potential promoter methylations of genes with prognostic characteristics, a total of 864 differential methylated genes within promoter (FDR <0.05, |delta beta| >0.3) were subjected to univariate Cox proportional hazard regression analysis. Then four promoter hypomethylations of genes: RNA 5S ribosomal pseudogene 38 (RNA5SP38), interleukin-21 (IL21), syndecan 4 pseudogene (SDC4P), and microRNA 4439 (MIR4439) were found to be associated with HCC patient survival (P=0.035, 0.040, 0.004, and 0.024, respectively). Multivariate Cox proportional hazard regression analysis was further performed to determine whether the four gene promoter methylations could be independent prognostic factors (). Under the control of potential confounding factors, including sex, node stage, and metastasis stage, we concluded that RNA5SP38, IL21, and SDC4P promoter methylations would be individually independent prognostic variables (P=0.008, <0.001, and 0.035, respectively), but not MIR4439 promoter methylation (P=0.419).
Table 2 Univariate and multivariate analysis of prognostic variables for cancer-specific survival in patients with hepatocellular cell carcinoma
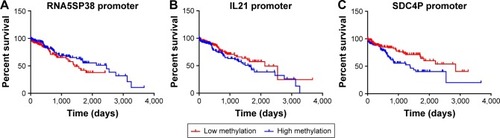
The median levels of RNA5SP38, IL21, and SDC4P promoter methylations were cutoff, respectively. Kaplan–Meier survival curves stratified by high and low levels of RNA5SP38, IL21, and SDC4P promoter methylation are shown in . Log-rank test showed that lower methylated levels of SDC4P promoter have a significantly favorable prognosis for HCC patients (hazard ratio =0.482, 95% confidence interval [CI] =−0.147–1.110, P=0.007), while not in RNA5SP38 (P=0.108) and IL21 (P=0.127) promoter methylations. HCC patients with low methylated levels of SDC4P promoter survived longer than those with high methylated levels of SDC4P promoter.
Figure 3 Kaplan–Meier plot of overall survival.
Abbreviations: HCC, hepatocellular carcinoma; IL21, interleukin-21; RNA5SP38, RNA 5S ribosomal pseudogene 38; SDC4P, syndecan 4 pseudogene.
Diagnostic value of promoter methylations of RNA5SP38, IL21, and SDC4P
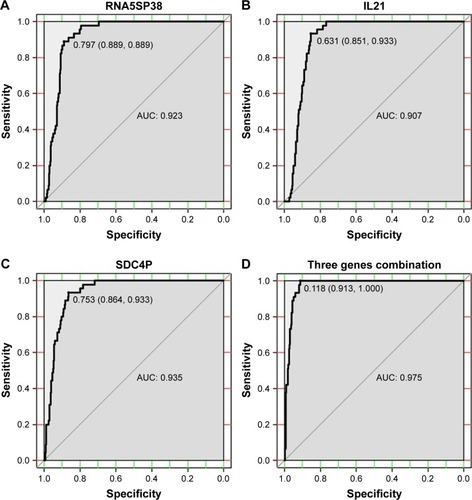
ROC analysis was applied to calculate the significance of the promoter methylations to RNA5SP38, IL21, and SDC4P as potential biomarkers to differentiate HCC tissues from adjacent tissues (). The promoter methylation of SDC4P (AUC =0.935, 95% CI =0.909–0.961, P=3.211E-21) demonstrated a higher diagnostic capability than promoter methylations of RNA5SP38 (AUC =0.923, 95% CI =0.895–0.951, P=3.585E-20) and promoter methylation of IL21 (AUC =0.907, 95% CI =0.877–0.937, P=9.248E-19). Subsequently, the logistic regression approach was applied for the combination of these three genes with promoter methylations, and the ROC curve revealed a much better diagnostic accuracy than when they were calculated individually. The optimal cutoff value was set at a maximal sum of sensitivity and specificity. The result showed an AUC of 0.975 (95% CI =0.962–0.989, P=4.811E-25), and the diagnosis sensitivity and specificity of the combination of these three promoter methylations were found to be 91.3% and 100.0%, respectively.
Figure 4 The areas under the ROC curves for the three gene promoter methylations and their combination that differentiate between HCCs and adjacent tissues.
Abbreviations: AUC, area under the curve; HCC, hepatocellular carcinoma; IL21, interleukin-21; RNA5SP38, RNA 5S ribosomal pseudogene 38; ROC, receiver operating characteristic; SDC4P, syndecan 4 pseudogene.
KEGG pathway and GO analysis
To avoid bias from promoter hypermethylation genes, only the substantial promoter hypomethylation genes were subjected to KEGG and GO analysis. To identify pathways and biological functions that enriched by genes with differential hypomethylation promoter, we analyzed GO and KEGG pathways through the 2,412 genes and displayed the GO results in Table S3. In the KEGG pathway, the genes showed dominant enrichments in the pathways of olfactory transduction (P=5.740E-188), cytokine–cytokine receptor interaction (P=1.962E-02), natural killer cell–mediated cytotoxicity (P=8.702E-03), and inflammation mediated by chemokine and cytokine signaling pathway (P=4.300E-04), which are shown in . Besides, GO analysis revealed the biological processes, cellular components, and molecular functions of differentially hypomethylated genes within promoter.
Table 3 KEGG and PANTHER pathways enriched in differentially methylated genes within promoters
Discussion
TCGA dataset provides a powerful approach to investigate the association between epigenetic alterations and clinical outcomes, paving way to elucidate new diagnostic and prognostic markers.Citation20 Herein, we gained 2,412 hypomethylated and 77 hypermethylated promoter genes between tumor and adjacent non-tumor tissues. Stefanska et al reported that nearly 2,000 genes within promoter hypomethylation were found in another HCC epigenetic study.Citation21 These evidences suggested that the broad scope of DNA hypomethylation influenced HCC profoundly and potential biomarkers might be detected from hypomethylated genes. Some hyperm-ethylated promoter genes identified in the current study have been reported in recently published studies, such as TBX15, RASSF10, AKR1B1, CDKL2, and ZNF154.Citation10,Citation13 Consistent with a previous study, S100A8 was presented a lower methylation level in HCC tissues compared to adjacent tissues.Citation22 Hypermethylation of TSGs was considered as one of the hallmarks in cancer initiation and progression, while the effects of these alterations were mostly proven to depress TSGs expression in several kinds of malignancies.Citation23,Citation24 On the contrary, promoter hypomethylation in genes was observed related to the activation of oncogenes and pro-metastatic genes in HCC.Citation21,Citation25
This study has found four promoter hypomethylations of RNA5SP38, IL21, SDC4P, and MIR4439 that may be associated with HCC patient survival. RNA5SP38, IL21, and SDC4P promoter methylations would be individually independent prognostic variables. Noteworthy, RNA5SP38 and SDC4P were pseudogenes and found to be associated with HCC survival for the first time. A great number of pseudogenes have been discovered to play an important role in cancer through relating activity to parental genes or functioning independently.Citation26 It was becoming clear that certain pseudogenes were also transcribed and maintained functions.Citation27 The association between pseudogene methylation and cancer progression should be considered exploratory.Citation28 In a pan-cancer analysis of pseudogene, it was reported that the pseudogene expression subtypes not only significantly related to patient survival, but also enabled to stratify patients in combination with clinical variables in kidney cancer.Citation29 Yu et al have illustrated that pseudogene methylation could downregulate the corresponding gene expression, which might contribute to clear-cell renal cell carcinoma progression.Citation30 Another noteworthy gene was IL21, which was discovered in 2000 and has been known as an important regulator of immune responses. It harbored pleiotropic functions dominantly at modulating the activity of T cells, B cells, and natural killer cells.Citation31 IL21 contributed to antitumor effects by direct and immune-mediated cytotoxicity.Citation32 Combined with other factors, IL21 had potent to predict the outcome of ovarian cancer patients.Citation33 Our study has suggested the potential prognostic role of promoter hypomethylation of RNA5SP38, IL21, and SDC4P in HCC patients for the first time. Furthermore, the study revealed that hypomethylation of SDC4P was associated with a better outcome, suggesting that SDC4P might be a potential TSG for HCC.
Our study demonstrated the significant diagnostic value of the combination of RNA5SP38, IL21, and SDC4P. It was not surprising that a panel of genes with promoter hypomethylation displayed better diagnosis accuracy than an individual methylated alteration. Aberrant methylations occurred far more frequently than genetic mutations in cancers.Citation34 Thus, identification of specific DNA methylation signatures gained robust potential to generate diagnostic markers for detection of HCC.Citation35 It was notable that our study provided this diagnostic finding based on the TCGA data, most of which were of early stage HCC patients (stage I + stage II, accounting for 71.5% HCC patients). To some extent, this result may indicate a novel diagnostic approach for the early HCC detection. Future clinically verified experiments are in great demand.
The signaling pathway analysis showed that cytokine–cytokine receptor interaction, natural killer cell–mediated cytotoxicity, and inflammation mediated by chemokine and cytokine signaling pathway were correlated with HCC. GO annotation yielded multiple signaling pathways in the biological process, cellular component, and molecular function. They linked to DNA replication, cell cycle, immune response, and defense response. All these data together indicated that multiple pathways, which were influenced by hypomethylation phenomenon, were involved in comprehensive cell developmental processes coordinately. The immune system was essential in body defense system, and the dysfunction of immune system might be correlated with cancer initiation.Citation36 Interestingly, a large number of identified genes were enriched in olfactory transduction pathway in KEGG analysis. Likewise, similar pathways (olfactory receptor activity, sensory perception of chemical stimulus, and sensory perception) were obtained by GO analysis. Previous reports have shown widespread ectopic olfactory receptor expression in non-olfactory tissues.Citation37,Citation38 A study reported that olfactory receptor activation increased intracellular Ca2+ and reduced cell proliferation in Huh7 HCC cell line.Citation39 Olfactory receptor activation was also observed to inhibit human leukemia cell proliferation, evoke apoptosis, and regulate differentiation.Citation40 Although the association between the olfactory transduction and HCC remained unclear, the current evidences suggested the potential anti-tumor behavior of olfactory transduction.
With regard to limitations of this study, we were unable to conduct stratified analysis based on hepatitis B virus (HBV) infection status using the available TCGA data. HBV infection constituted the major etiology in Asian HCC patients, whereas fatty liver disease and high alcohol consumption were dominant in the white race.Citation41,Citation42 Our study might have missed some significant differential methylated promoter genes.
Conclusion
In summary, our data suggested that SDC4P promoter hypomethylation may be a potential prognostic biomarker. A panel of promoter methylation in RNA5SP38, IL21, and SDC4P was proven a novel approach to diagnosis HCC. The pathway analysis defined the wide functional role of DNA hypomethylation in cancer. A future study to validate the potential prognostic and diagnostic biomarkers in HCC patients is necessary.
Acknowledgments
The authors acknowledge the great contributions of The Cancer Genome Atlas database.
Disclosure
The authors report no conflicts of interest in this work.
References
- TorreLABrayFSiegelRLFerlayJLortet-TieulentJJemalAGlobal cancer statistics, 2012CA Cancer J Clin20156528710825651787
- FornerALlovetJMBruixJHepatocellular carcinomaLancet2012397982212451255
- ByamJRenzJMillisJMLiver transplantation for hepatocellular carcinomaHepatobiliary Surg Nutr201321223024570911
- SinghalAJayaramanMDhanasekaranDNKohliVMolecular and serum markers in hepatocellular carcinoma: predictive tools for prognosis and recurrenceCrit Rev Oncol Hematol201282211614021680198
- ChengALKangYKChenZEfficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trialLancet Oncol2009101253419095497
- KongHKParkSJKimYSEpigenetic activation of LY6K predicts the presence of metastasis and poor prognosis in breast carcinomaOncotarget2016734556775568927494879
- PaskaAVHudlerPAberrant methylation patterns in cancer: a clinical viewBiochem Med (Zagreb)201525216117626110029
- WuWRSunHZhangRMethylation-associated silencing of miR-200b facilitates human hepatocellular carcinoma progression by directly targeting BMI1Oncotarget2016714186841869326919246
- RuanPShenJSantellaRMZhouSWangSNEpiC: a network-assisted algorithm for epigenetic studies using mean and variance combined signalsNucleic Acids Res20164416e13427302130
- WangFFengYLiPRASSF10 is an epigenetically inactivated tumor suppressor and independent prognostic factor in hepatocellular carcinomaOncotarget2016744279429726701853
- ZhuXShanLWangFHypermethylation of BRCA1 gene: implication for prognostic biomarker and therapeutic target in sporadic primary triple-negative breast cancerBreast Cancer Res Treat2015150347948625783183
- LiQFLiQYGaoARShiQFCorrelation between promoter methylation in the GSTP1 gene and hepatocellular carcinoma development: a meta-analysisGenet Mol Res20151426762677226125884
- YamadaNYasuiKDohiOGenome-wide DNA methylation analysis in hepatocellular carcinomaOncol Rep20163542228223626883180
- ZhangXYouQZhangXChenXSOCS3 methylation predicts a poor prognosis in HBV infection-related hepatocellular carcinomaInt J Mol Sci2015169226622267526393582
- LvZZouHPengKThe suppressive role and aberrent promoter methylation of BTG3 in the progression of hepatocellular carcinomaPLoS One2013810e7743724146994
- StefanskaBCheishviliDSudermanMGenome-wide study of hypomethylated and induced genes in patients with liver cancer unravels novel anticancer targetsClin Cancer Res201420123118313224763612
- QiuXHuangYZhouYZhengFAberrant methylation of TRIM58 in hepatocellular carcinoma and its potential clinical implicationOncol Rep201636281181827373520
- SunFKSunQFanYCMethylation of tissue factor pathway inhibitor 2 as a prognostic biomarker for hepatocellular carcinoma after hepatectomyJ Gastroenterol Hepatol201631248449226313014
- AshburnerMBallCABlakeJAGene ontology: tool for the unification of biology. The gene ontology consortiumNat Genet2000251252910802651
- BarbanoRPalumboOPasculliBA miRNA signature for defining aggressive phenotype and prognosis in gliomasPLoS One2014910e10895025279461
- StefanskaBHuangJBhattacharyyaBDefinition of the landscape of promoter DNA hypomethylation in liver cancerCancer Res201171175891590321747116
- LiuKZhangYZhangCMethylation of S100A8 is a promising diagnosis and prognostic marker in hepatocellular carcinomaOncotarget2016735567985681027462864
- LiWLiXWangWNOR1 is an HSF1- and NRF1-regulated putative tumor suppressor inactivated by promoter hypermethylation in nasopharyngeal carcinomaCarcinogenesis20113291305131421803736
- ZhangCPengYYangFQinRLiuWZhangCPCDH8 is frequently inactivated by promoter hypermethylation in liver cancer: diagnostic and clinical significanceJ Cancer20167444645226918058
- StefanskaBBouzelmatAHuangJDiscovery and validation of DNA hypomethylation biomarkers for liver cancer using HRM-specific probesPLoS One201388e6843923950870
- GranderDJohnssonPPseudogene-expressed RNAs: emerging roles in gene regulation and diseaseCurr Top Microbiol Immuol2016394111126
- Kalyana-SundaramSKumar-SinhaCShankarSExpressed pseudogenes in the transcriptional landscape of human cancersCell201214971622163422726445
- ByunDSChoKRyuBKFrequent monoallelic deletion of PTEN and its reciprocal association with PIK3CA amplification in gastric carcinomaInt J Cancer2003104331832712569555
- HanLYuanYZhengSThe pan-cancer analysis of pseudogene expression reveals biologically and clinically relevant tumour subtypesNat Commun20145396324999802
- YuGYaoWGumireddyKPseudogene PTENP1 functions as a competing endogenous RNA to suppress clear-cell renal cell carcinoma progressionMol Cancer Ther201413123086309725249556
- LeonardWJSpolskiRInterleukin-21: a modulator of lymphoid proliferation, apoptosis and differentiationNat Rev Immunol20055968869816138102
- BhattSMatthewsJParvinSDirect and immune-mediated cytotoxicity of interleukin-21 contributes to antitumor effects in mantle cell lymphomaBlood2015126131555156426194763
- ChenYLChouCYChangMCIL17a and IL21 combined with surgical status predict the outcome of ovarian cancer patientsEndocr Relat Cancer201522570371126150382
- KulisMEstellerMDNA methylation and cancerAdv Genet201070275620920744
- HaradaKBabaYIshimotoTLINE-1 methylation level and patient prognosis in a database of 208 hepatocellular carcinomasAnn Surg Oncol20152241280128725319577
- LiuPJiangWRenHZhangHHaoJExploring the molecular mechanism and biomakers of liver cancer based on gene expression microarrayPathol Oncol Res20152141077108325907256
- WieseHGelisLWieseSQuantitative phosphoproteomics reveals the protein tyrosine kinase Pyk2 as a central effector of olfactory receptor signaling in prostate cancer cellsBiochim Biophys Acta20151854663264025219547
- FeldmesserEOlenderTKhenMWidespread ectopic expression of olfactory receptor genesBMC Genomics2006712116716209
- MassbergDSimonAHaussingerDYanaiIOphirRLancetDMonoterpene (−)-citronellal affects hepatocarcinoma cell signaling via an olfactory receptorArch Biochem Biophy2015566100109
- ManteniotisSWojcikSBrauhoffPFunctional characterization of the ectopically expressed olfactory receptor 2AT4 in human myelogenous leukemiaCell Death Discov201621507027551494
- KimBKHanKHAhnSHPrevention of hepatocellular carcinoma in patients with chronic hepatitis B virus infectionOncology201181 Suppl 1414922212935
- McGlynnKALondonWTThe global epidemiology of hepatocellular carcinoma: present and futureClin Liver Dis201115222324321689610