
Abstract
This phase I study aimed at determining the maximum tolerated dose (MTD) and characterizing the safety, tolerability, pharmacokinetics (PKs), and efficacy of pasireotide in patients with advanced neuroendocrine tumors (NETs). Patients were enrolled in two phases: dose-escalation phase (to determine the MTD) at a starting dose of 80 mg pasireotide long-acting release (LAR) i.m. followed by a dose-expansion phase (to evaluate safety and prelimi-nary efficacy). Associations between PK/pharmacodynamic parameters and clinical outcomes were evaluated using linear regression analysis. A total of 29 patients were treated with 80 mg (n=13) and 120 mg (n=16) doses. Most common primary tumor sites included small intestine (44.8%), pancreas (24.1%), and lung (17.2%). No protocol-defined dose-limiting toxicities were observed in the study; however, in post hoc analysis, a higher incidence of bradycardia (heart rate [HR] <40 beats per minute [bpm]) was observed with 120 mg (31.3%) vs 80 mg (0%). Two partial responses (PRs) were observed, both in the 120 mg dose cohort. Pasireotide concentrations correlated with tumor shrinkage, although the association was not statistically significant (P=0.08). Among the biomarkers analyzed, insulin-like growth factor 1 (IGF-1) showed a decreasing trend with increasing pasireotide concentration, while chromogranin A (CgA) and neuron-specific enolase (NSE) levels did not show any dose–response relationship. The most common adverse events in any dose group were hyperglycemia, fatigue, and nausea. MTD was defined at 120 mg for pasireotide LAR in patients with advanced NETs. Although objective radiographic responses were rarely observed with somatostatin analogs, two PRs were observed among 16 patients in the 120 mg cohort. Bradycardia (HR <40 bpm) appears to be a dose-limiting effect; however, the mechanism and clinical significance are uncertain. This study was registered with clinicaltrials.gov (NCT01364415).
Introduction
Somatostatin analogs (SSAs), such as octreotide long-acting release (LAR) and lanreotide autogel, are the standard of care for treatment of symptoms resulting from hormonal secretions in functioning neuroendocrine tumors (NETs).Citation1–Citation4 Although the efficacy of SSAs in symptom control for NETs has been well established,Citation4,Citation5 the role of SSAs in tumor control has been only recently elucidated. Limited data from prospective studies exist on the efficacy and safety of SSAs when combined with targeted agents. Antitumor activity of SSAs in NETs was first demonstrated in the placebo-controlled, double-blind, randomized phase III PROMID study in which octreotide LAR showed a clinically meaningful increase in time to tumor progression compared with placebo in patients with metastatic midgut NETs.Citation6 In the recent phase III CLARINET study, lanreotide autogel, another SSA with a somatostatin receptor type 2 (sst2) affinity profile similar to that of octreotide, demonstrated progression-free survival (PFS) benefit in patients with nonfunctional enteropancreatic NETs and has been recently approved for clinical use in patients with advanced enteropancreatic NETs.Citation7 A literature review conducted by Berardi et alCitation8 on treatment strategy for NETs concluded that SSAs and targeted therapies should be considered as first-line options for the treatment of Grade 1–Grade 2 advanced pancreatic NETs (pNETs).
SSAs act via interaction with sst of which five subtypes (sst1–sst5) with clinical activity have been described in gastroenteropancreatic NETs (GEP-NETs).Citation9 Octreotide and lanreotide exert their activity primarily via binding to sst2Citation9–Citation11. However, tumor cells may become resistant, leading to symptomatic and/or radiographic progression. Potential mechanisms of resistance include internalization of sst2, downregulation of sst2, and overexpression of other sst.Citation9,Citation12–Citation15
Pasireotide, a second-generation multireceptor-targeted SSA, has a broader binding profile and higher binding affinity for sst1–3 and sst5 than those of octreotide and lanreotide ().Citation16,Citation17 Pasireotide is available as short-acting pasireotide for subcutaneous (SC) administration with twice-daily administration schedule and the LAR formulation for intramuscular (IM) injection administered once every 28 days with similar pharmacokinetics (PKs)/pharmacodynamics and safety profile.Citation18 In an exploratory analysis from a phase III study in patients with advanced carcinoid syndrome refractory to octreotide LAR, pasireotide LAR 60 mg showed encouraging antitumor activity compared with octreotide LAR 30 mg.Citation19 Median (95% CI) PFS was 11.8 months (11.0–not reached) with pasireotide LAR vs 6.8 months (5.6–not reached) with octreotide LAR (hazard ratio, 0.46; 95% CI, 0.20–0.98; two-sided P=0.045). Tumor control rate at month 6 was 62.7% with pasireotide and 46.2% with octreotide (odds ratio, 1.96; 95% CI, 0.89–4.32; P=0.09). A phase II study of first-line standard-dose pasireotide LAR (60 mg every 4 weeks) in a more heterogeneous cohort of metastatic NETs demonstrated a median PFS of 11 months.Citation20
Figure 1 Postulated mechanism of action of pasireotide.
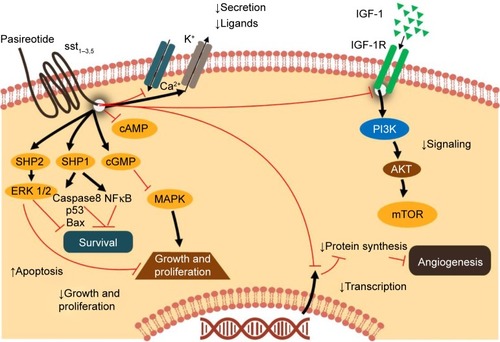
The phase II COOPERATE-2 study was conducted to assess the efficacy and safety of pasireotide (LAR; 60 mg/28 days, intramuscularly) in combination with everolimus (10 mg/day, orally) in patients with advanced, well-differentiated, progressive pNETs. The study failed to show the benefit of combining pasireotide LAR 60 mg with everolimus in terms of prolongation of PFS.Citation21 Additionally, the maximum tolerated dose (MTD) of pasireotide has been unknown, and clinical outcomes associated with high doses of this drug have not been explored. Therefore, this phase I study (NCT01364415) was designed to determine the MTD of pasireotide LAR and characterize the safety, tolerability, and antitumor efficacy trends in patients with advanced NETs with a starting dose of 80 mg/28 days. The study further evaluated the PKs of pasireotide LAR and its effects on biochemical and tumor biomarkers specific for NETs in this patient population.
Methods
Study design
This phase I, multi-center, open-label, dose-escalation study investigated the safety and tolerability, PKs/pharmacodynamics, and preliminary efficacy of pasireotide LAR in patients with advanced NETs. The study consisted of two phases: a dose-escalation phase (n=3–6 patients in each cohort) and a dose-expansion phase (n≥12 patients). The starting dose of pasireotide LAR in the dose-escalation phase was 80 mg/28 days, which was administered by IM injection. Additional doses were selected with the assistance of the two-parameter Bayesian logistic regression model (BLRM) using the dose escalation with overdose control (EWOC) principle in multiples of 20 mg. Intra-patient dose escalation was not allowed in the study; however, dose de-escalation was permitted. The primary objective of the study was to determine the MTD of pasireotide LAR in patients with advanced NETs. A key secondary objective was to characterize the safety and tolerability of pasireotide LAR in this patient population. Other end points included assessment of PKs/pharmacodynamics and preliminary evaluation of the antitumor activity of high-dose pasireotide LAR in NETs.
Patients could discontinue the study treatment prematurely due to adverse events (AEs), abnormal laboratory values or test procedure results, protocol deviation, consent withdrawal, loss to follow-up, administrative problems, death, or disease progression.
Study population
Adults (aged ≥18 years) with histologically confirmed advanced (unresectable and/or metastatic), well-differentiated, or moderately differentiated (low or intermediate grade) NETs with documented disease progression in the prior 12 months, independent of primary tumor location and functional status, were enrolled in the study. A key exclu-sion criterion included patients with baseline glycosylated hemoglobin (HbA1c) >7.0%.
MTD/dose-limiting toxicity (DLT) analysis
MTD was defined as the highest dose with expected DLT in ≤33% of the treated patients in the first two treatment cycles. A treatment cycle was typically 28 days long, starting with each injection of pasireotide LAR and ending right before the next injection. Patients who received two injections of pasireotide LAR and underwent sufficient safety evaluation or who experienced DLT within the first two treatment cycles were considered evaluable for DLT (dose-determining set). Dose-escalation decisions were taken based on the observed number of DLTs (via BLRM)Citation22 and observed safety profile of the patients in the dose-determining set. Other safety and efficacy analyses included all patients who received one or more doses of study medication; patients were grouped according to the first dose received. AEs and laboratory abnormalities were assessed according to the Medical Dictionary for Regulatory Activities (MedDRA) version 18.0 and Common Terminology Criteria for Adverse Events (CTCAE) version 3.0, respectively.
PKs and biomarker analysis
Descriptive statistics and graphical depiction for pasireotide plasma concentrations were performed for PK analysis. PK/pharmacodynamic analysis evaluated the relationship between pasireotide plasma concentrations and levels of efficacy and safety markers. Biomarkers for efficacy included insulin-like growth factor 1 (IGF-1), glucagon and NET-specific tumor biomarker chromogranin A (CgA), and neuron-specific enolase (NSE). Among safety markers, heart rate (HR) was assessed using 24 hours Holter monitoring assessment on Cycle 1 (C1) Day 1 and Cycle 2 (C2) Day 1 in all the patients. Analyses of bradycardia were performed by combining HR data from assessment of vital signs, 12-lead electrocardiograms (ECGs), and 24-hour Holter ECGs with bradycardia defined as minimum HR <40 beats per minute (bpm).
Tumor evaluation
Tumor evaluation was based on Response Evaluation Criteria In Solid Tumors (RECIST) 1.0 by site investigator; 95% CIs were based on the Clopper–Pearson method. It was not required for tumor assessment to be blinded in this study.
The study was approved by the respective institutional review boards of participating sites (Cedars-Sinai Cancer Center, Los Angeles; Dana–Farber Cancer Institute, Boston; H. Lee Moffitt Cancer Center, Tampa; and MD Anderson Cancer Center, Houston), and all patients provided written informed consent to participate.
Results
Patient characteristics
As of July 2015, a total of 29 patients, 15 patients in the dose-escalation phase and 14 patients in the dose-expansion phase, were treated with pasireotide LAR in five successive cohorts. In the dose-escalation phase, the initial dose was 80 mg in Cohort 1 (n=6), and the dose was then escalated to 120 mg in Cohort 2 (n=4). The dose of 120 mg was maintained in Cohort 3 (n=5). The dose-expansion phase started with 120 mg in cohort 4 (n=7). However, after safety evaluation, the dose was reduced to 80 mg in Cohort 5 (n=7). Overall, 13 patients received the 80 mg dose and 16 patients were treated with the 120 mg dose. In the 80 mg dose group, dose de-escalation (from 80 to 60 mg) was performed in one patient due to AEs. Primary tumor sites included small intestine (45%; 13/29), pancreas (24%; 7/29), lung (17%; 5/29), colon (3%; 1/29), rectum (3%; 1/29), and unknown (7%; 2/29). Most common sites of metastases included liver in 83% (24/29) and lung in 31% (9/29) of patients. Median time since initial diagnosis of primary site was 50 months (range, 1–218 months). Most patients had undergone prior treatments, including prior surgery, 97% (28/29), and prior radiotherapy, 21% (6/29). A total of 38% (11/29) of patients received systemic treatment beyond SSAs. Most patients (90%; 26/29) received prior SSA therapy. Detailed patient characteristics are listed in .
Table 1 Patient baseline characteristics
Patient disposition
At the time of data cutoff (July 2015), one of 13 patients was still undergoing treatment with the 80 mg dose and four patients of 16 were undergoing treatment with the 120 mg dose. The median duration of follow-up was 6 months in the 80 mg dose group and 9 months in the 120 mg dose group from enrollment to data cutoff date. The overall median duration of exposure to study treatment was 8.7 months with a median of 6.7 months in the 80 mg group and 10.1 months in the 120 mg dose group. The primary reason for treatment discontinuation was disease progression in the majority of the patients (). In the pasireotide 80 mg group, four patients discontinued the study treatment due to AEs (leukocytosis, diarrhea, fatigue, lipase increase, and weight decrease). Of these AEs, leukocytosis and lipase increase were Grade 3/4 AEs; diarrhea and lipase increase were suspected to be drug-related AEs. In the 120 mg group, four patients discontinued the study treatment due to AEs (atrioventricular block complete, performance status decrease, blood creatinine increase, gamma-glutamyl transferase increase, and hyperglycemia). Of these AEs, atrioventricular block complete, performance status decrease, and gamma-glutamyl transferase increase were Grade 3/4 AEs; gamma-glutamyl transferase increase and hyperglycemia were suspected to be drug related.
Table 2 Primary reasons for treatment discontinuation
MTD/DLTs
A total of 15 patients were recruited and treated in three dose cohorts in the dose-escalation phase. There was no protocol-defined DLTs reported in the study. In a post hoc analysis of the safety data from dose-escalation cohort and one cohort (the 120 mg cohort) in the dose-expansion phase, greater incidence of bradycardia (HR <40 bpm) was identified in the 120 mg group compared to that in the 80 mg group. At the time of cutoff, considering both the dose-escalation and dose-expansion cohorts, the incidence of bradycardia with HR <40 bpm was 31% (5/16) in the 120 mg group and 0% (0/13) in the 80 mg group based on Holter assessment. Subsequently, 120 mg was considered to be the MTD for pasireotide LAR in patients with advanced NETs.
Safety
The safety set included all patients (N=29) from the study. AEs of any grade were seen in 92% of patients who received the 80 mg dose and 100% of patients who received the 120 mg dose. AEs leading to discontinuation with suspected drug relationship were seen in two (15%) and two (13%) patients in the 80 mg and 120 mg dose groups, respectively. Grade 3/4 AEs suspected to be drug related were seen in two (15%) and five (31%) patients who received 80 mg and 120 mg doses, respectively.
Most common AEs of any grade regardless of drug relationship were hyperglycemia, diarrhea, and fatigue in both the 80 mg and 120 mg dose groups (). There were two deaths reported in the study, including one on-treatment death (within 56 days of last treatment). The on-treatment death occurred in a patient treated with the 120 mg dose who died 25 days after the end of treatment due to hepatorenal failure; this was considered by the investigator to be unrelated to the study drug. One patient in the 80 mg group died 345 days after the end of treatment due to disease progression in the posttreatment follow-up phase.
Table 3 AEs regardless of study drug relationship (≥15% in the 120 mg dose)
Efficacy
PFS by local radiological review
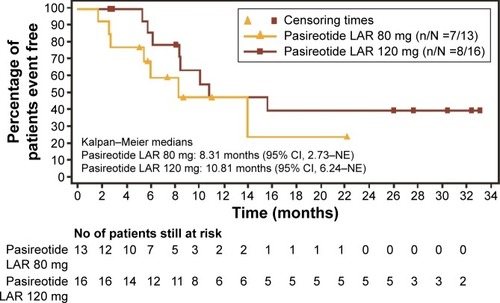
PFS events were reported in 54% (7/13) of the patients in the 80 mg arm and 50% (8/16) of patients in the 120 mg arm. The median PFS (95% CI) was 8.3 months (2.7, not estimable [NE]) and 10.8 months (6.2, NE) in the 80 mg dose group and 120 mg dose group, respectively ().
Figure 2 Kaplan–Meier estimates of PFS by local radiological review.
Best overall response as per local radiological review
Two partial responses (PRs) were observed as best overall response in the 16 patients receiving the 120 mg dose. Both these patients had pancreas as their primary site of tumor. One of the patients (58 years of age; female) had a well-differentiated tumor and mediastinum diaphragmatic lymph node metastases; the second patient (44 years of age; female) had a moderately differentiated tumor and liver metastases. In these patients, PRs were reported from Day 168 and Day 198, respectively, and in both patients, PRs were confirmed on subsequent visits. No complete response (CR) was seen in either dose group. Most patients (>75%) had stable disease (SD) in both dose groups. Disease control rate (DCR; CR, PR, or SD) was 77% (95% CI, 46%, 95%) in the 80 mg group and 94% (95% CI, 70%, 100%) in the 120 mg group ().
Table 4 Best overall response and DCR
Tumor analysis
The mean percentage change in tumor size (standard deviation) was 1.6% (±9.38%) in the 80 mg group and −10.8% (±30%) in the 120 mg group. The median percentage change in tumor size was −2.7% and 3.5% in the 80 mg and 120 mg groups, respectively (two-sided P-value =0.609, as per exact Wilcoxon two-sample test).
PKs/pharmacodynamics
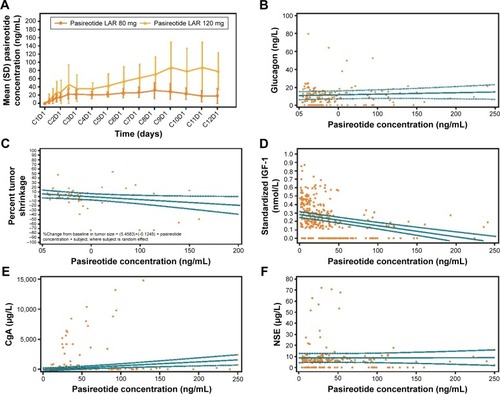
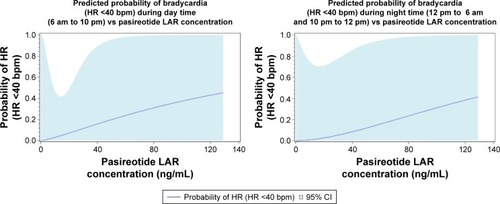
The PK profiles of pasireotide LAR 80 mg and 120 mg doses are shown in . Day 1 pre-dose concentrations on C2 through Cycle 12 (C12) represent Cmin concentrations; day 22 concentration from the C1 represents the Cmax concentration (mean ± SD: 80 mg, 13.7±11.5 ng/mL; 120 mg, 23.2±31.4 ng/mL). For PK/safety analysis, the logistic regression analysis suggested that the probability of bradycardia (HR <40 bpm) tends to increase with increasing pasireotide concentrations, although statistical significance was not reached (odds ratio [95% CI] for a 1.5-fold increase in pasireotide concentration during day time [6 am through 10 pm]: 1.67 [0.38–7.34]; during night time [10 pm through 6 am]: 2.02 [0.49–8.29]). The thyroid-stimulating hormone (TSH) levels among five patients experiencing bradycardia (HR <40 bpm) ranged from 0.8 mU/L at base-line to 7.91 mU/L at the last visit assessment; in four out of five patients, the levels were <5 mU/L at the last visit. No significant association was observed between pasireotide concentrations and glucagon levels (P=0.42; ).
Figure 3 Results of the PK/pharmacodynamic analysis.
Abbreviations: PK, pharmacokinetic; LLOQ, lower limit of quantitation; C2, Cycle 2; C12, Cycle 12; C1, Cycle 1; CgA, chromogranin A; NSE, neuron-specific enolase; LAR, long-acting release.
Regarding concomitant medications among patients with bradycardia (HR <40 bpm), two patients were prescribed medication for hyperglycemia and hypertension, and one patient had intermittent heart blockage during the study for which a pacemaker was inserted.
In the PK/efficacy analysis, higher pasireotide concentrations appeared to have a weak correlation with higher percentage tumor shrinkage, and this trend did not achieve statistical significance (slope =−0.012; P=0.08; ). IGF-1 levels showed an expected decreasing trend with increasing pasireotide concentrations, which confirmed the biochemical suppression of IGF-1 in patients with advanced NETs (). There was no exposure–response relationship between NET-specific tumor biomarkers CgA and NSE levels and pasireotide concentrations ().
Discussion
This phase I study evaluated the MTD of pasireotide LAR in patients with advanced NETs with primary tumor sites, including the gastrointestinal (GI) tract, pancreas, and lung. No protocol-defined DLTs were reported in the study, and the MTD was defined at 120 mg for pasireotide LAR in patients with advanced NETs.
In a post hoc safety analysis, a higher incidence of bradycardia (HR <40 bpm; five patients; 31%) was observed in the 120 mg dose group. Potential causes for this observation were investigated with respect to pasireotide concentrations and imbalances in hormone levels. In the PK analysis, higher pasireotide concentrations did suggest a moderate increase in risk of bradycardia (HR <40 bpm); however, this observation was not statistically significant (). Modulation of glucagon levels by pasireotide (a known effector of the cardiovascular axis and often used in treatment of bradycardia caused by beta-blockersCitation23,Citation24) doses was further examined as a potential cause of bradycardia (HR <40 bpm). However, no clinically significant changes in the levels of glucagon were observed with increasing pasireotide concentrations. Bradycardia (HR <40 bpm) thus appears to be a dose-limiting effect; however, the mechanism and clinical significance remain uncertain and need further study. Incidence of bradycardia (HR <40 bpm) at 120 mg dose during the study was the reason not to test higher doses of pasireotide. Future studies could explore whether different dosing schedules could achieve higher exposure while minimizing bradycardia (HR <40 bpm).
Figure 4 Predicted probability of bradycardia vs pasireotide LAR concentration.
Preliminary efficacy observations showed encouraging PFS and DCRs with pasireotide LAR in patients with advanced progressive NETs. In the preliminary assessment of tumor responses, pasireotide LAR showed evidence of antitumor activity as observed by the high rate of disease stabilization with both the 120 mg and the 80 mg doses in this patient population. Although objective radiographic responses are rarely observed with SSAs, two PRs were observed among 16 patients in the 120 mg cohort, providing further evidence of the promising antitumor efficacy of pasireotide LAR in patients with advanced NETs.
In the biomarker analysis, IGF-1, an established biochemical activity marker in pituitary tumors,Citation25 and NET-specific tumor biomarkers CgA and NSECitation26,Citation27 were further investigated to gain insights into the PKs of pasireotide LAR. As expected, pasireotide LAR showed a dose-dependent reduction in IGF-1 levels, confirming the biochemical suppression of IGF-1 in patients with advanced NETs. The levels of tumor burden markers CgA and NSE did not show any significant change, correlating with the SD seen in the majority of the patients upon pasireotide LAR treatment.
Furthermore, the phase II COOPERATE-2 study was conducted to assess the efficacy and safety of pasireotide (LAR; 60 mg/28 days, IM) in combination with everolimus (10 mg/day, oral) in patients with advanced, well-differentiated, progressive pNETs. Although a benefit with regard to PFS was not observed in this study, an antisecretory effect associated with pasireotide was confirmed by the suppression of tumor growth factors IGF-1 and IGF-2 and corresponding regulation of the IGF-binding proteins IGFBP2 and IGFBP3.Citation21
Conclusion
Pasireotide at a dose of 120 mg every 4 weeks appears to show encouraging antitumor activity. The drug is generally well tolerated at this dose; however, the bradycardia (HR <40 bpm) rate of 31% represents a potential concern. The clinical significance of pasireotide-associated bradycardia (HR <40 bpm) is unclear. Phase II studies evaluating pasireotide at a dose of 120 mg are warranted in patients progressing on conventional SSAs.
Acknowledgments
We thank Anamika Gulati from Novartis Healthcare Pvt. Ltd. for providing medical editorial assistance and Christelle Darstein for her strong biostatistics analysis support. This study was funded by Novartis Pharmaceuticals Corporation. This paper was presented at the American Society of Clinical Oncology (ASCO) Annual Meeting 2016 as a poster presentation with interim findings as “Phase I, multi-center, open-label, dose-escalation study of pasireotide LAR (PAS) in patients with advanced neuroendocrine tumors (NET),” http://meetinglibrary.asco.org/content/169565-176.
Disclosure
JCY has received consulting or advisory fees from Ipsen, Lexicon, and Novartis and research funding from Novartis. MGK, KHR, KH, and SR are employees of Novartis. JRS has received honoraria from Novartis; consulting or advisory fees from Ipsen, Lexicon, and Novartis; and research funding from Novartis and Pfizer and is on the speaker’s bureau for Bayer and Genentech. EMW has received consulting or advisory fees from Celgene, Ipsen, and Novartis. ACM is a member of the Speaker Bureau for Genentech. JAC has received consulting or advisory fees from Ipsen, Lexicon, Novartis, and Oxigene and has stock in Merck. The authors report no other conflicts of interest in this work.
References
- Gardner-RoehneltNMUpdate on the management of neuroendocrine tumors: focus on somatostatin antitumor effectsClin J Oncol Nurs2012161566422297008
- di BartolomeoMBajettaEBuzzoniRClinical efficacy of octreotide in the treatment of metastatic neuroendocrine tumors. A study by the Italian Trials in Medical Oncology GroupCancer19967724024088625251
- ObergKManagement of neuroendocrine tumoursAnn Oncol200415suppl 4iv293iv29815477324
- KvolsLKMoertelCGO’ConnellMJSchuttAJRubinJHahnRGTreatment of the malignant carcinoid syndrome. Evaluation of a long-acting somatostatin analogueN Engl J Med1986315116636662427948
- RubinJAjaniJSchirmerWOctreotide acetate long-acting formulation versus open-label subcutaneous octreotide acetate in malignant carcinoid syndromeJ Clin Oncol199917260060610080605
- RinkeAMullerHHSchade-BrittingerCPlacebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study GroupJ Clin Oncol200927284656466319704057
- CaplinMEPavelMCwiklaJBLanreotide in metastatic enteropancreatic neuroendocrine tumorsN Engl J Med2014371322423325014687
- BerardiRRinaldiSTorniaiMGastrointestinal neuroendocrine tumors: searching the optimal treatment strategy – a literature reviewCrit Rev Oncol Hematol20169826427426643525
- SchmidHASchoeffterPFunctional activity of the multiligand analog SOM230 at human recombinant somatostatin receptor subtypes supports its usefulness in neuroendocrine tumorsNeuroendocrinology200480suppl 1475015477717
- de HerderWWHoflandLJvan der LelyAJLambertsSWSomatostatin receptors in gastroentero-pancreatic neuroendocrine tumoursEndocr Relat Cancer200310445145814713257
- VillaumeKBlancMGouysseGVEGF secretion by neuroendocrine tumor cells is inhibited by octreotide and by inhibitors of the PI3K/AKT/mTOR pathwayNeuroendocrinology201091326827820389030
- TulipanoGStummRPfeifferMKreienkampHJHölltVSchulzSDifferential beta-arrestin trafficking and endosomal sorting of somatostatin receptor subtypesJ Biol Chem200427920213742138215001578
- CescatoRSchulzSWaserBInternalization of sst2, sst3, and sst5 receptors: effects of somatostatin agonists and antagonistsJ Nucl Med200647350251116513620
- LiMLiWKimHJYaoQChenCFisherWECharacterization of somatostatin receptor expression in human pancreatic cancer using real-time RT-PCRJ Surg Res2004119213013715145694
- RongaGSalernoGProcacciniE111In-octreotide scintigraphy in metastatic medullary thyroid carcinoma before and after octreotide therapy: in vivo evidence of the possible down-regulation of somatostatin receptorsQ J Nucl Med1995394 suppl 1134136
- PlockingerUWiedenmannBTreatment of gastroenteropancreatic neuroendocrine tumorsVirchows Arch2007451suppl 1S71S8017684765
- SchmidHAPasireotide (SOM230): development, mechanism of action and potential applicationsMol Cell Endocrinol20082861–2697417977644
- ChenXShenGJiangJPharmacokinetics and safety of subcutaneous pasireotide and intramuscular pasireotide long-acting release in Chinese male healthy volunteers: a phase I, single-center, open-label, randomized studyClin Ther20143681196121025012727
- WolinEMJarzabBErikssonBPhase III study of pasireotide long-acting release in patients with metastatic neuroendocrine tumors and carcinoid symptoms refractory to available somatostatin analoguesDrug Des Devel Ther2015950755086
- CivesMKunzPLMorseBPhase II clinical trial of pasireotide long-acting repeatable in patients with metastatic neuroendocrine tumorsEndocr Relat Cancer20152211925376618
- KulkeMRuszniewskiPVan CutsemEA randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-differentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trialAnn Oncol Epub201736
- NeuenschwanderBBransonMGsponerTCritical aspects of the Bayesian approach to phase I cancer trialsStat Med200827132420243918344187
- LoveJNHowellJMGlucagon therapy in the treatment of symptomatic bradycardiaAnn Emerg Med19972911811838998104
- BindonMJBarlottaKGlucagon treatment for bradycardia and a junctional rhythm caused by excessive beta-blockadeResuscitation20098011132719744763
- SinghBSmithJAAxelrodDMInsulin-like growth factor-I inhibition with pasireotide decreases cell proliferation and increases apoptosis in pre-malignant lesions of the breast: a phase 1 proof of principle trialBreast Cancer Res201416646325385439
- GutPCzarnywojtekAFischbachJChromogranin A – unspecific neuroendocrine marker. Clinical utility and potential diagnostic pitfallsArch Med Sci20161211926925113
- BaudinEGigliottiADucreuxMNeuron-specific enolase and chromogranin A as markers of neuroendocrine tumoursBr J Cancer1998788110211079792158