
Abstract
Inhibitors of poly(ADP-ribose) polymerases (PARPs), which play a key role in DNA damage/repair pathways, have been developed as antitumor agents based on the concept of synthetic lethality. Synthetic lethality is the idea that cell death would be efficiently induced by simultaneous loss of function of plural key molecules, for example, by exposing tumor cells with inactivating gene mutation of BRCA-mediated DNA repair to chemically induced inhibition of PARPs. Indeed, three PARP inhibitors, olaparib, rucaparib and niraparib have already been approved in the US or Europe, mainly for the treatment of BRCA-mutant ovarian cancer. Clinical trials of various combinations of PARP inhibitors with cytotoxic or molecular-targeted agents are also underway. In particular, expanded applications of PARP inhibitors are anticipated following recent reports that defects in homologous recombination repair (HRR) are associated with mutations in repair genes other than BRCA1/BRCA2, such as ATM, ATR, PALB2, RAD51, CHEK1 and CHEK2, as well as with epigenetic loss of BRCA1 function through promoter methylation or overexpression of the BRCA2-interacting transcriptional repressor EMSY. Current topics of interest include selection of the best agent in each clinical context, identification of new treatment targets for HRR-proficient cases, and development of PARP inhibitor-based regimens that are less toxic and that prolong overall survival as well as progression-free survival. In addition, potential long-term side effects and suitable biomarkers for predicting efficacy and mechanisms of clinical resistance are in discussion. This review summarizes representative preclinical and clinical data for PARP inhibitors and discusses their potential for future applications to treat various malignancies.
Introduction
Poly(ADP-ribose) polymerases (PARPs) are important components of DNA damage/repair pathways, playing key roles in genomic stability and tumor cell survival. PARP inhibitors have attracted great interest as potential antitumor agents since the concept of synthetic lethality was introduced by Bryant et alCitation1 and Farmer et alCitation2 in 2005. This concept is based on the idea that simultaneous loss of function of two or more key gene products can cause cell death, even if a deficiency in only one of them is not lethal. Thus, for example, tumor cells with inactivating mutation of the BRCA DNA-repair genes, which is not lethal, might be killed if they are also exposed to chemical inhibition of PARPs. Indeed, preclinical and clinical studies have verified antitumor effects of PARP inhibition in BRCA-mutant tumors.Citation1–Citation8 So far, three agents (olaparib, rucaparib and niraparib) have been approved by western regulatory agencies, and >70 clinical trials evaluating PARP inhibitors for various kinds of malignancies are underway.Citation9 Hereditary breast and ovarian cancer (HBOC) is a syndrome associated with germline mutations of BRCA1/BRCA2 and is considered as the most suitable target for PARP inhibitors. A pooled analysis of 26 observational studies with a total of 3,879 ovarian cancer patients showed that germline BRCA mutations were associated with better survival, supporting the position of HBOC as a distinct clinical subtype.Citation10 Triple-negative breast cancer (TNBC), characterized by lack of expression of estrogen receptor, progesterone receptor and HER2 gene amplification, is a pathologically and clinically aggressive entity and is considered as the most problematic for breast cancer patients and clinicians, in which hormone treatments or anti-HER2 agents are ineffective. Defects of DNA-repair pathways including BRCA1 are reported in many TNBC cases, providing a rationale for PARP inhibitor treatment.Citation11 Defects in homologous recombination repair (HRR) genes other than BRCA1/BRCA2 and acquired loss of BRCA1 function through promoter methylation have also opened up the possibility of new indications for PARP inhibitor treatment.Citation12,Citation13 However, only limited information is yet available about mechanisms of clinical resistance to these inhibitors or biomarkers to identify suitable cases for treatment. In this review, we summarize the current status of basic research and clinical trials on PARP inhibitors and discuss prospects for extending their applicability and improving clinical outcomes.
Mechanisms of action
PARP is a nuclear protein first reported in 1963 as a DNA-dependent polyadenylic acid-synthesizing nuclear enzyme, and to date, 17 members of the PARP protein family have been identified.Citation14,Citation15 PARP members are involved in various cellular processes, such as DNA repair, cellular differentiation, gene transcription, inflammation, mitosis, cell death and metabolism.Citation16 Among them, PARP-1 accounts for >90% of the total PARP activity and is considered to be a key player in DNA base excision repair and repair of DNA single-strand breaks (SSBs).Citation16–Citation18 PARP-1 binds to SSBs through a series of N-terminal zinc finger DNA-binding domains and catalyzes the polymerization of ADP-ribose (PARylation) with NAD+ as a substrate, resulting in the production of variable-sized polymers of ADP-ribose (PAR).Citation15,Citation19,Citation20 The amount of PAR production is regulated by the balance between PARP-1 and poly(ADP-ribose) glucohydrolase, which hydrolyzes PAR.Citation21 PAR recruits SSBs repair scaffolding proteins, such as X-ray repair cross-complementing protein 1 (XRCC1), DNA ligase III and DNA polymerase beta, and concurrently modifies chromatin structure around lesions.Citation15,Citation19 PARP-2, which accounts for 5%–10% of the total PARP activity, is also essential for DNA base excision repair, in collaboration with PARP-1.Citation22 Accumulation of SSBs due to inhibition of PARylation or base excision repair produces DNA double-strand breaks (DSBs) through replication fork collapse.Citation23 HRR and nonhomologous end joining (NHEJ) are the main types of repair mechanisms for DSBs.Citation3,Citation15,Citation21,Citation24 HRR is an accurate process conducted using a homologous DNA sequence and is composed of gene conversion and single-strand annealing pathways. On the other hand, NHEJ is an inaccurate process in which broken DNA ends are directly connected without using a homologous sequence. The former process occurs exclusively during the S and G2 phases of the cell cycle, while the latter is cell cycle independent.Citation15 While HRR normally takes a leading role in the repair of DSBs, deficiency of this process often leads to alternative NHEJ activation, and some of the genetic alterations introduced by NHEJ promote carcinogenesis by cancer-driver genes.Citation12,Citation24,Citation25 PARP-1 affects repair of DSBs and SSBs through activating ATM in the HRR pathway and inactivating DNA-dependent protein kinase in the NHEJ pathway, and thus PARP occupies a central position in the whole picture of DNA damage control and repair.Citation26,Citation27 Interactions between PARP, HRR and NHEJ are concisely illustrated in a review by O’Sullivan et al.Citation26 PARP-1 expression and activity are increased in actively proliferating cells, and high PARP-1 expression is a feature of various tumors.Citation28 A preclinical study using 318 cell lines from 30 cancer types showed that PARP-1 mRNA expression was extremely high in small cell lung cancer (SCLC), in addition to hematologic malignancies. These results might provide clues to new treatment approaches for intractable tumors such as SCLC or neuroendocrine carcinomas derived from other organs, which are unresponsive to conventional molecular-targeting agents.Citation29
In contrast to the limited efficacy of PARP inhibition alone, the combination of PARP inhibition with DNA-repair defects involving other signaling pathways is often lethal to tumor cells.Citation30 This synthetic lethality is powerful, because BRCA-deficient tumor cells have shown >1,000-fold greater sensitivity to PARP inhibitors in preclinical models.Citation1,Citation2 Inactivation of BRCA1 and BRCA2, which are essential for HRR, increases the importance of PARP1 for maintaining genomic stability, and addition of PARP inhibition thus leads to cell cycle arrest and apoptosis through stimulating inaccurate NHEJ.Citation30,Citation31 In addition to germline BRCA-mutant tumors, BRCA deficiency is observed in somatic BRCA-mutant tumors or tumors with epigenetic loss of function through BRCA1 promoter methylation or overexpression of the BRCA2 transcription suppressor EMSY.Citation12,Citation32 In addition, large-scale genomic data showed that some BRCA-proficient tumors have defects in HRR genes such as ATM, ATR, CHK1, CHK2, PALB2, RAD51 and PTEN.Citation13 These gene products are linked in the HRR signaling pathway as follows: ATM recruited in response to DSBs produces γH2AX and concurrently phosphorylates and activates CHK2, while ATR activated by replication protein A phosphorylates and activates CHK1, and PALB2 facilitates invasion of the BRCA2–RAD51 complex into damaged DNA by localizing BRCA2 to DSBs.Citation12,Citation16 As mentioned earlier, tumors with acquired loss of BRCA1 function or defects in HRR genes share clinical and biological features with germline BRCA-mutant tumors and are known as BRCAness phenotype tumors.Citation12,Citation13,Citation24,Citation33 Recent genomic data indicate that the frequency of BRCAness phenotype tumors is not negligible, and thus, the concept of synthetic lethality is adaptable to more cases than might have been expected. Pennington et alCitation34 analyzed germline or somatic mutations of 30 genes, including BRCA1/BRCA2 and other HRR genes, in 390 ovarian cancer patients and showed that 31% of patients possess some deleterious alterations in HRR genes. Mateo et alCitation35 also conducted targeted sequencing of DNA-repair genes, including BRCA1/BRCA2, ATM, and CHEK2 and Fanconi anemia genes in 49 patients with castration-resistant prostate cancer and identified homozygous deletions or deleterious alterations in 33% of the cases. Large-scale integrated genomic analyses of ovarian cancer by The Cancer Genomic Atlas (TCGA) project found gene alterations in HRR genes in 51% of 316 cases.Citation36 These data highlight BRCAness phenotype tumors as a clinically promising target of PARP inhibitors. In addition to inhibiting PARylation, the inhibitors trap PARP1/PARP2 enzymes at sites of DNA damage and induce cytotoxic PARP–DNA complexes.Citation30 The effects of these agents depend mainly on the degree of PARP trapping rather than the ability to inhibit PARylation.Citation22,Citation24,Citation37 However, it remains unclear whether an agent with stronger PARP-trapping activity would have greater clinical benefits, and prospective clinical trials are warranted to address this question.
It should be noted that DNA-repair function is intrinsically decreased in tumor cells compared with normal cells, and this disparity accounts for the high selectivity of PARP inhibitors.Citation3,Citation38 Nevertheless, it remains important to develop inhibitors with high potency and low toxicity. The clinical efficacy of PARP inhibitors as single agents is actually only moderate, except for BRCA-mutant or BRCAness tumors, and therefore, combination therapy is a reasonable approach to improve the utility of these inhibitors, which should sensitize cells to DNA damage caused by various cytotoxic agents, exhibiting synthetic lethality in the clinical context.Citation3 Some combined regimens have been tested, but it appears that the combination effects are dependent on the partner cytotoxic agent used.Citation24,Citation39 In in vitro preclinical studies, olaparib showed better synergy than veliparib with temozolomide (TMZ), whereas rucaparib showed the strongest combination effect with topotecan but the weakest effect with paclitaxel (PTX) or gemcitabine (GEM).Citation39–Citation41 In terms of mechanisms, accumulated DNA damage and increased PARP dependency for repair are suggested for the combinations with platinum agents or topoisomerase-1 inhibitors, elevated PARP1 trapping for the combinations with TMZ, and induced BRCAness phenotype for the combinations with taxanes.Citation42 The next step in evaluating such combinations would be in vivo studies or clinical trials.
Some preclinical studies have recently demonstrated a unique and promising combination effect between PARP inhibitors and alkylating agent TMZ in isocitrate dehydrogenase 1 (IDH1)-mutant tumors.Citation43,Citation44 The synthesis of NAD+ as a substrate PARylation is reduced in IDH1-mutant cases, and this depletion of NAD+ is speculated to result in enhancement of PARP inhibition. Here, TMZ promotes PARP recruitment through inducing DNA damage, reasonably explaining interaction of above two agents, which indicates a broader application of PARP inhibitors in various malignancies with NAD metabolic defects, including glioma.
Clinical applications of PARP inhibitors
Clinical applications of PARP inhibitors are the most advanced in ovarian cancer, and so far, olaparib, rucaparib and niraparib are commercially available in the US or Europe. However, many clinical trials are underway, including malignancies such as breast cancer and prostate cancer, as well as ovarian cancer, and tumors without BRCA mutations, such as BRCAness phenotype tumors. In this section, we review clinical evidence of the efficacy of five PARP inhibitors (olaparib, talazoparib, rucaparib, veliparib and niraparib; ).
Table 1 Summary of Phase II/III trials of PARP inhibitors
Olaparib
Fong et alCitation45 conducted the first Phase I trial of olaparib (AZD-2281; AstraZeneca plc, Cambridge, UK) in 60 patients including 22 BRCA-mutant cases, and the maximum tolerated dose (MTD) was established as 400 mg twice daily. An additional cohort study of 50 patients with BRCA-mutant ovarian cancer showed that the overall clinical benefit rate (CBR) as a composite radiological and tumor marker factor and the overall response rates (ORRs) were 46% and 28%, respectively, and there was a significant association between CBR and platinum-free interval, defined as the period from last platinum administration to disease progression (platinum sensitive, 69%; platinum resistant, 45%; platinum refractory, 23%).Citation46 In a Phase II trial in 57 patients with recurrent BRCA-mutant ovarian cancer, ORR was 33% in the 400 mg twice daily cohort and 13% in the 100 mg twice daily cohort; ie, higher dose administration of olaparib gave a better clinical outcome.Citation47 Kaufman et al also conducted a Phase II trial in 298 patients with recurrent BRCA-mutant cancers, including 193 ovarian cancers, 62 breast cancers, 23 pancreatic cancers and 8 prostate cancers. They reported that ORR was 26% in the entire cohort, while the values were 31%, 13%, 22% and 50% in patients with ovarian, breast, pancreatic and prostate cancers, respectively.Citation4 Another Phase II trial in 64 patients with recurrent high-grade serous ovarian cancer (HGSOC) or TNBC showed a higher ORR in patients with BRCA-mutant ovarian cancer than in ovarian cancer patients without mutation (41% vs 24%).Citation5 These clinical data led to accelerated approval of olaparib by the US Food and Drug Administration (FDA) in 2014 for fourth-line or later treatment of BRCA-mutant ovarian cancer. Some impressive data also support the value of olaparib maintenance treatment. Ledermann et alCitation6 conducted a randomized placebo-controlled Phase II trial in 265 patients with recurrent platinum-sensitive HGSOC and reported that progression-free survival (PFS) as a primary end point was significantly longer in olaparib-receiving patients (median PFS: 8.4 months vs 4.8 months; hazard ratio [HR], 0.35; 95% confidence interval [CI], 0.25–0.49; P<0.001). Moreover, the preplanned retrospective analysis for this cohort revealed that PFS benefit was greater in BRCA-mutant ovarian cancer (median PFS: 11.2 months vs 4.3 months; HR, 0.18; 95% CI, 0.10–0.31; P<0.0001).Citation48 Based on the above results, the European Medicines Agency (EMA) approved olaparib as a maintenance therapy in 2014 for platinum-sensitive relapsed BRCA-mutant HGSOC. Currently, three Phase III trials, termed SOLO1 (NCT01844986), SOLO2 (NCT01874353) and SOLO3 (NCT02282020), are in progress for BRCA-mutant ovarian cancer using the primary end point of PFS. Briefly, SOLO1 is a placebo-controlled Phase III trial in which newly diagnosed patients are randomized to olaparib maintenance (300 mg twice per day) or placebo after a platinum-based regimen. In SOLO2, the usefulness of olaparib maintenance for recurrent platinum sensitive-patients is being evaluated. SOLO3 also targets recurrent platinum-sensitive cases, comparing olaparib maintenance (300 mg twice per day) with physician’s choice single-agent chemotherapy (PTX, topotecan, pegylated liposomal doxorubicin or GEM). These Phase III trials are expected to yield definite conclusions as to the clinical efficacy of olaparib.
Clinical investigations of olaparib are in progress for patients with breast and prostate cancers, as well as ovarian cancer. Tutt et alCitation49 conducted a Phase II trial in 54 patients with recurrent BRCA-mutant breast cancer and reported that the ORR was 41% in the 400 mg twice daily cohort and 22% in the 100 mg twice daily cohort. While ORR in BRCA-mutant cases was generally similar for patients with ovarian cancer and those with breast cancer, the Phase II trial by Tutt et alCitation49 found no responders among BRCA-wild breast cancer patients, in contrast to an ORR of 24% in BRCA-wild ovarian cancer patients.Citation5 The poor sensitivity of BRCA wild-type breast cancer patients might be explained by the biological heterogeneity of breast cancer. Mateo et alCitation35 performed a Phase II trial in 50 cases with metastatic castration-resistant prostate cancer, resulting in an ORR of 33% in the entire cohort. Among them, 16 cases with homozygous deletions, deleterious alterations or both in DNA-repair genes had an ORR of 88%, including 100% in seven patients with BRCA2 loss and 80% in five cases with ATM aberrations. Based on these clinical data, olaparib received FDA breakthrough designation in 2016 for patients with BRCA or ATM-mutant castration-resistant prostate cancer, who have progressed on prior taxane-based chemotherapy and abiraterone or enzalutamide as a new-generation hormonal agent.
Talazoparib
Talazoparib (BMN-673; Medivation, San Francisco, CA, USA) has stronger PARP-trapping activity than other PARP inhibitors.Citation50 In the Phase I trial for patients with advanced solid tumor, MTD was established at 1.0 mg/day and gave ORR values of 50%, 42%, 9%, 20% and 0% in patients with BRCA-mutant breast cancer, BRCA-mutant ovarian cancer, SCLC, pancreatic cancer and Ewing sarcoma, respectively.Citation51 The Phase III EMBRACA trial comparing talazoparib with physician’s choice is underway for patients with locally advanced and/or metastatic germline BRCA-mutant breast cancer (NCT01945775).
Rucaparib
In a Phase I/II study in patients with advanced solid tumors performed by Kristeleit et al,Citation52 the MTD was established as 600 mg twice per day and the disease control rate (complete response [CR] + partial response [PR] + stable disease [SD] ≥24 weeks) was 70% in germline BRCA-mutant breast cancer patients receiving rucaparib (AG014699; Clovis Oncology, Boulder, CO, USA) ≥300 mg once per day. A Phase II trial in 35 patients with recurrent BRCA-mutant ovarian cancer gave an ORR of 65% in the entire cohort.Citation53 Swisher et alCitation7 conducted the Phase II ARIEL2 trial in 206 patients with recurrent, platinum-sensitive, high-grade ovarian cancer, where patients were classified into BRCA mutant, BRCA wild type plus genomic loss of heterogenity (LOH) high and BRCA wild-type plus LOH low groups using next-generation sequencing. In this trial, the cutoff value of LOH was defined as 14%. PFS as a primary end point was significantly longer in the BRCA-mutant group and the LOH high/BRCA wild group compared with the LOH low/BRCA wild group (HR, 0.27; 95% CI, 0.16–0.44; P<0.0001, 0.62; 95% CI, 0.42–0.90; P=0.011, respectively). Their study is clinically useful in demonstrating that genomic LOH, as well as BRCA-mutant status, is helpful for identifying rucaparib responders. Based on these trial data, rucaparib obtained FDA breakthrough designation in 2015 for BRCA-mutant ovarian cancer in third-line or later treatment. The Phase III ARIELE3 trial (NCT01968213) is currently in progress for patients with platinum-sensitive ovarian cancer, in which rucaparib maintenance is being compared with placebo with PFS as the primary end point.
Veliparib
Veliparib (ABT-888; AbbVie, North Chicago, IL, USA) is an oral inhibitor possessing relatively weak PARP-trapping activity.Citation50 In a Phase I trial in patients with BRCA-mutant cancer, BRCA-wild type platinum-refractory ovarian cancer or basal-like breast cancer, MTD was established at 400 mg twice per day and the ORR values were 23% in BRCA-mutant and 4% in BRCA wild-type patients.Citation54 In a Phase II trial by Coleman et al,Citation55 50 patients with persistent or recurrent BRCA-mutant epithelial ovarian, fallopian tube or primary peritoneal cancer were enrolled to receive veliparib 400 mg twice per day; ORR was 26% in the entire cohort, 20% in platinum-resistant patients and 35% in platinum-sensitive cases.
Niraparib
In the Phase I trial in patients with advanced solid tumor, 300 mg once per day was established as the MTD.Citation56 Mirza et al conducted the randomized Phase III NOVA trial for a maintenance situation in 553 recurrent platinum-sensitive ovarian cancer patients, who were divided into two groups based on germline BRCA mutation and HRR deficiency (HRD) status using BRACAnalysis and myChoice HRD test (Myriad Genetics, Salt Lake City, UT, USA), respectively. Niraparib (MK4827; Tesaro, Waltham, MA, USA) maintenance (300 mg once per day) was compared with placebo with PFS as the primary end point.Citation8 Remarkably, patients receiving niraparib had a longer PFS, irrespective of germline BRCA mutation and HRD status (BRCA-mutant cohort: median PFS 21.0 months vs 5.5 months; HR, 0.27; 95% CI, 0.17–0.41; P<0.001; non-BRCA-mutant and HRD-positive cohort: 12.9 months vs 3.8 months; HR, 0.38; 95% CI, 0.24–0.59; P<0.001 and overall non-BRCA-mutant cohort: 9.3 months vs 3.9 months; HR, 0.45; 95% CI, 0.34–0.61; P<0.001). Based on the promising results of the NOVA trial, the FDA approved niraparib for recurrent platinum-sensitive ovarian cancer regardless of BRCA mutation and HRD status in March 2017.
In summary, olaparib is currently the best-established agent for BRCA-mutant cases, and this agent is available for prostate cancer and ovarian cancer cases. Rucaparib is promising due to its higher ORR for BRCA-mutant cases, and niraparib is the first agent demonstrating PFS prolongation for BRCA-wild cases in a Phase III trial. Veliparib has modest activity as a single agent, and so combination therapies are expected to become the mainstream of its clinical development. Talazoparib showed strong cytotoxicity in preclinical models, and further evidence is needed in this respect. In addition to these representative agents, clinical evidence has been gradually accumulated for other PARP inhibitors, including iniparib.Citation57
Combination therapy with PARP inhibitor and cytotoxic or molecular-targeted agents
Combination trials of PARP inhibitors are actively underway, usually with cytotoxic agents. Veliparib is considered the most suitable for combination trials because of its modest hematopoietic toxicity.Citation58 Currently, the Phase III GOG PARTNERS 3005 trial (NCT02470585) is in progress for newly diagnosed ovarian cancer cases. Patients are randomized to the combination of carboplatin (CBDCA)/PTX and veliparib (150 mg twice per day) or CBDCA/PTX alone, followed by maintenance monotherapy with veliparib (400 mg twice per day) or placebo, with PFS as the primary end point.
There are also some impressive data for olaparib combinations. Oza et alCitation59 conducted a Phase II trial for patients with recurrent platinum-sensitive ovarian cancer, in which patients were randomized to the combination of CBDCA/PTX and olaparib (200 mg twice per day), followed by maintenance monotherapy with olaparib (400 mg twice per day) or CBDCA/PTX alone, with PFS as the primary end point. PFS was significantly longer in the combination group (median PFS 12.2 months vs 9.6 months; HR, 0.51; 95% CI, 0.34–0.77; P=0.0012), and it was prominent in BRCA-mutant cases (HR, 0.21; 95% CI, 0.08–0.55; P=0.0015). In the randomized Phase II trial by Bang et al,Citation60 123 patients with recurrent or metastatic gastric cancer were randomized to the combination of olaparib (200 mg twice per day) and PTX or PTX alone, followed by maintenance monotherapy with olaparib (200 mg twice per day) or placebo. While PFS prolongation (primary end point) was not clearly detected, overall survival (OS) was significantly better in the combination group (median OS: 13.1 months vs 8.3 months; HR, 0.56; 80% CI, 0.41–0.75; P=0.005), and the benefit was especially marked in cases with low ATM protein level (median OS: not reached vs 8.2 months; HR, 0.35; 80% CI, 0.22–0.56; P=0.002). Although further consideration of the discrepancy between PFS and OS is needed, this trial is noteworthy for demonstrating the impact of BRCAness phenotype in the clinical situation.
In spite of the low PARP-inhibitory activity of iniparib (BSI201, Sanofi S.A., Paris, France) as a single agent, the open-label Phase II trial by O’Shaughnessy et alCitation61 found that combination therapy with iniparib and GEM/CBDCA improved not only CBR as a primary end point but also OS compared with GEM/CBDCA alone (56% vs 34%; P<0.01, median OS: 12.3 months vs 7.7 months; HR, 0.57; 95% CI, 0.36–0.90; P=0.01).Citation62,Citation63 However, the subsequent Phase III trial for patients with metastatic TNBC failed to demonstrate prolongation of OS (median OS: 11.8 months vs 11.1 months; HR, 0.88; 95% CI, 0.69–1.12; P=0.28), and further evaluation of the survival benefits of this agent is needed.Citation63,Citation64
Combinations with molecular-targeted agents and cytotoxic agents are under investigation and in some cases have moved to the clinical trial phase. Angiogenesis inhibitors seem to be attractive, in that this class of agents downregulates HRR genes through constructing a hypoxic tumor environment.Citation65 Liu et alCitation66 reported a randomized Phase II trial of cediranib, which is an oral tyrosine kinase inhibitor of vascular endothelial growth factor (VEGF) receptor (VEGFR)1, VEGFR2 and VEGFR3. In their trial, 90 patients with recurrent platinum-sensitive ovarian cancer were randomized to the combination group of olaparib (200 mg twice per day) and cediranib (30 mg per day) or the olaparib (400 mg twice per day) alone group. PFS was significantly longer in the combination group (median PFS: 17.7 months vs 9.0 months; HR, 0.42; 95% CI, 0.23–0.76; P=0.005), and in response to this promising result, Phase III trials of a combination regimen of olaparib and cediranib are underway (NCT2502266, NCT02446600, ICON9). Clinical trials of combination regimens of PARP inhibitors with cytotoxic agents, molecular-targeted agents or immunotherapies that are currently in progress are summarized in .
Table 2 Ongoing trials of PARP inhibitor combinations
Toxicity profiles of PARP inhibitors
The grade 3–4 toxicity profiles in patients receiving five PARP inhibitors at their MTDs are shown in . According to previous reports on the aforementioned five inhibitors, nausea and fatigue were detected in >50% of cases in common, but the percentage of grade 3–4 events was <10%. The frequency of hematological toxicity varied depending on the agent and was conspicuous in clinical trials of talazoparib and niraparib.Citation8,Citation51 In a Phase III trial of niraparib, grade 3–4 thrombocytopenia, anemia and neutropenia were detected in >20% of the patients.Citation8 On the other hand, in a Phase II trial of veliparib, these events were detected in <2% of the cases. Thus, the moderate hematopoietic toxicity of veliparib could be advantageous for developing combinations with cytotoxic agents.Citation55 Moreover, rucaparib increased aspartate transaminase (AST) and alanine aminotransferase (ALT), but these changes were asymptomatic and normalized over time during continuous administration.Citation7 PARP inhibitors are newcomers in the field of oncology, and detailed toxicity profiles for long-term administration of PARP inhibitors remain to be established. Clinical trial data indicate that the onset of treatment-related myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are serious issues.Citation67 According to the US Package Insert for olaparib, MDS/AML was reported in 22 of 2,618 patients (0.8%), of which 17 cases were fatal, and the duration from olaparib initiation to the onset of MDS/AML ranged from <6 months to >2 years.Citation68 Because all patients had received previous chemotherapy with platinum agents and/or other DNA-damaging agents, it is not clear whether olaparib directly caused MDS/AML. It is possible that the combined effects of PARP inhibition and DNA damage due to cytotoxic agents or the innate defect of BRCA might promote the onset of MDS/AML. DNA-repair inhibitors such as PARP inhibitors might increase intratumor genomic mutation rates and induce evolution of metastatic or resistant clones.Citation69 As for other agents, MDS/AML was reported in two of 377 patients (0.5%) treated with rucaparib and seven of 751 patients (0.9%) treated with niraparib.Citation70,Citation71 Therefore, clinicians should monitor each patient’s complete blood count carefully and stop the inhibitor administration immediately if MDS/AML is clinically suspected. Moreover, preclinical models suggested an association of PARP1 with cardiovascular diseases and long-term memory formation, and long-term observation is required to examine the effects on the cardiovascular system and on mental health.Citation31
Table 3 Grade 3–4 toxicity profiles of PARP inhibitors
Identification of biomarkers predicting suitable cases
Identification of responders to PARP inhibitors is important for providing maximum clinical benefit to suitable cases and for preventing development of drug resistance and is also desirable from a cost–benefit point of view. However, there are no established strategies for identifying relevant candidates, although combinations of various kinds of biomarkers with clinicopathological features have been investigated with a focus on detecting HRR-deficient cases.Citation33 According to the Phase I trial conducted by Fong et al,Citation46 platinum-free interval was significantly associated with CBR for olaparib administration, and Cochrane’s review of 599 patients with epithelial ovarian cancer noted that PARP inhibitors appeared to improve PFS in recurrent platinum-sensitive disease, whereas clinical significance for platinum-resistant disease was undetermined.Citation72 These data highlight platinum sensitivity as a clinical surrogate marker predicting PARP inhibitor’s responsiveness. Assays for the HRR pathway include targeted sequencing, whole-genome sequencing, copy number analyses, gene-expression profiling, protein expression assays and functional assays.Citation69
Genomic analysis is a direct and comprehensive approach for predicting suitable cases and has already been embedded in clinical trials, but it is relatively expensive for routine use in many institutions and requires frozen or formalin-fixed paraffin-embedded (FFPE) tumor specimens for analysis, as well as bioinformatic support from experts.Citation33 In addition, this approach is often affected by different analytical approaches and genomic heterogeneity among tumor specimens; also, genomic alterations do not necessarily cause functional changes in proteins.Citation73,Citation74 Genomic scars (accumulated patterns of DNA damage and repair) are unique in HRR-deficient cases, and these characteristic signatures are caused by large genomic deletions and LOH owing to the activation of inaccurate NHEJ. Telli et alCitation75 defined an HRD score composed of LOH, telomeric allelic imbalance and large-scale state transitions and reported that this index predicted response to neoadjuvant platinum-based regimen in cases with TNBC, regardless of BRCA-mutant status. In a prospective trial setting, two indexes (LOH and HRD score) were adopted for the ARIEL2 trial of rucaparib and the NOVA trial of niraparib, confirming the position of HRD score as an independent prognostic factor.Citation7,Citation8 Previous studies indicate that the total number of exome alterations, including non-synonymous ones, was also associated with clinical outcomes in BRCA-mutant ovarian cancers.Citation69
In terms of functional biomarkers, the detection of RAD51 foci with immunofluorescence is useful because of its rapidity. While administration of PARP inhibitors increases expression of RAD51 foci in HRR-proficient tumors, RAD51 foci remain absent in HRR-deficient tumors, indicating a role of RAD51 as a key player in the HRR signaling pathway.Citation12,Citation76 One weakness of this method is that we cannot predict the change in RAD51 foci prior to actual drug exposure.Citation12 PARP1 is an essential substrate for PARylation, and immunohistochemical analysis by von Minckwitz et alCitation77 showed that high cytoplasmic PARP expression predicted high sensitivity to chemotherapy in breast cancer samples. Moreover, elevated PAR levels detected by Western blot or immunohistochemistry or E26 transformation-specific sequence (ETS) gene fusions observed in Ewing’s sarcoma and prostate cancer are promising biomarkers.Citation78,Citation79 However, the usefulness of the aforementioned factors for the identification of PARP inhibitor responders has mainly been demonstrated in preclinical models, and their clinical relevance needs to be properly assessed in clinical trials.
Mechanisms of clinical resistance
Development of drug resistance is a major problem in patients receiving the same treatment regimen for a long period. Resistance to PARP inhibitors can appear at many steps of the DNA-repair pathways. A conceptual diagram of resistance mechanisms, classified into increased HRR capacity, decreased activity of PARP-1 and decreased intracellular availability of PARP inhibitor, is shown in .Citation80
Figure 1 Conceptual diagram of resistance mechanisms to PARP inhibitors.
Abbreviations: PARP, poly(ADP-ribose) polymerase; HRR, homologous recombination repair.
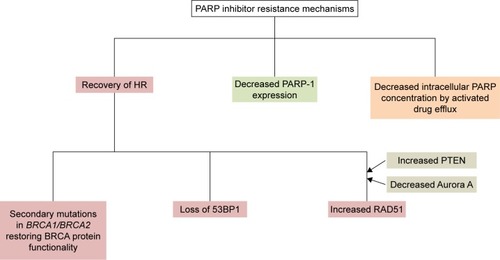
Recovery of HR is an important mechanism. Edwards et alCitation81 examined BRCA2 c.6174delT mutation of a BRCA2-deficient human pancreatic cancer cell line (CAPAN1) and reported that this frameshift mutation resulted in normalization of the functions of BRCA2 protein. Sakai et alCitation73 established seven subclones with restored BRCA2 protein and seven subclones without BRCA2 protein from CAPAN1 and found that the sensitivity to PARP inhibitors was lower in clones with secondary BRCA2 mutation than in those without BRCA2 mutation. Barber et alCitation82 compared genomic information for primary and metastatic samples from a male breast tumor patient developing resistance to olaparib and detected a secondary BRCA2 c.9106C > G mutation leading to the functional restoration of BRCA2 protein. In the context of cisplatin resistance, Swisher et alCitation83 compared recurrent platinum-resistant and platinum-sensitive tumors and reported that four of six in the former group developed secondary genetic changes in BRCA1 leading to functional BRCA1 protein restoration, whereas none of three in the latter group developed BRCA1 sequence alterations. Norquist et alCitation84 reviewed 46 recurrent ovarian cancer patients and reported that the percentage of secondary mutations restoring BRCA1/BRCA2 was significantly higher in platinum-resistant cases than in platinum-sensitive ones (46% vs 5%, P=0.003). Dhillon et alCitation85 proposed an interesting model of secondary BRCA restoration from a primary chemotherapy-sensitive tumor. First, BRCA-restored subclones coincidentally arise from initially BRCA-deficient or chemotherapy-sensitive tumor as a result of increased mutation rates associated with DNA-damaging agents. Next, these clones spread through selective pressure from repeated drug administrations. This theory might reasonably explain the induction of PARP inhibitor resistance during repeated cisplatin administrations.
As for other HRR-related factors, P53-binding protein 1 (53BP1) plays a key role in DNA-repair responses and checkpoint control, and loss of 53BP1 promotes ATM-dependent resection of broken DNA ends in BRCA1-deficient cells and finally restores HRR.Citation86,Citation87 An association between increased RAD51 activity and resistance to PARP inhibitors is also indicated, and Aurora A and PTEN modulate RAD51 activity indirectly.Citation80,Citation88,Citation89 Aurora A inhibits the recruitment of RAD51 to DNA damage sites, while PTEN deficiency causes a reduction in RAD51 expression. RAD51 forms a complex with BRCA2, and its combination with damaged DSB is indispensable to HRR, so that increased RAD51 activity leads to resistance to PARP inhibitors.Citation90 Kondrashova et alCitation91 recently sequenced HRR pathway genes in PARP inhibitor pretreatment and post-progression tumor samples from patients enrolled in the ARIEL2 trial and detected secondary mutations in RAD51C and RAD51D associated with PARP inhibitor resistance.
P-Glycoprotein (P-gp; multi-drug resistance protein) acts as a drug efflux pump to decrease intracellular drug concentration and is associated with resistance to various kinds of agents. An in vivo model by Rottenberg et alCitation92 showed upregulation of the P-gp-encoding ABCB1A/B genes after long-term PARP administration. In addition, preclinical studies suggest other possibilities, including regulation by micro RNA, epigenetic re-expression of BRCA1, phosphorylation of PARP1 by c-Met and overexpression of HOX family members.Citation93–Citation97
Large-scale genomic data also revealed the genomic backgrounds of refractory or resistant cases. Patch et alCitation98 performed whole-genome sequencing of 92 ovarian cancer patients with primary refractory, resistant, sensitive and matched acquired resistant disease and identified several molecular events associated with acquired resistance, including reverse germline BRCA1/BRCA2 mutation, loss of BRCA1 promoter methylation and ABCB1 rearrangement. The practical relevance of these mechanisms should be considered in future clinical trials.
Conclusion and prospects for the future
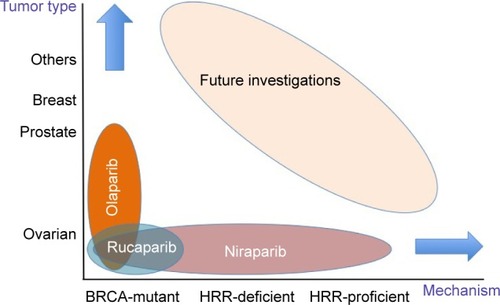
The concept of synthetic lethality led to great interest in PARP inhibitors as candidate antitumor agents, and clinical applications of this class of agents have so far focused mainly on BRCA-mutant ovarian cancer. Olaparib, rucaparib and niraparib are already available under US health insurance. Detailed assays for HRR repair pathway components, including genomic analysis, are broadening the applicability of PARP inhibitors in HRR-deficient cases, and future investigations are expected to focus on HRR-proficient cases and various kinds of malignancies other than ovarian, prostate and breast cancers (). Considering the variety of patients who may receive clinical benefit from PARP inhibitors, we consider that intergroup studies will be essential to improve and extend current clinical outcomes.
Figure 2 Current indications for olaparib, rucaparib, and niraparib.
Abbreviation: HRR, homologous recombination repair.
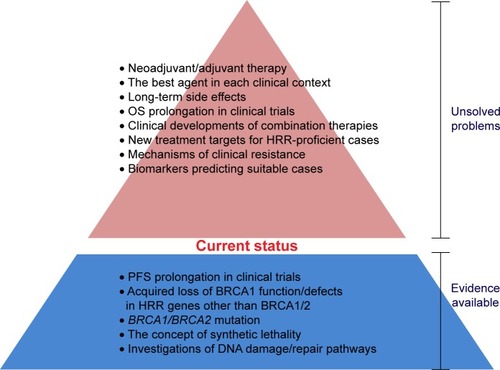
In addition to the need for well-established biomarkers to predict suitable cases and resistance mechanisms, there are several areas where further work is needed to realize the full potential of PARP inhibitors (). First, although clinical evidence for each agent has been accumulating gradually, direct comparative data of the available agents are lacking, and we cannot draw a clear conclusion as to which agent is the best option in each clinical context. In this sense, further accumulation of preclinical data and further randomized clinical trials are essential to establish the status of each agent. Second, the FDA approved the first PARP inhibitor, olaparib, in 2014, so that only about 3 years have passed since marketing. Therefore, potential long-term toxicities, including secondary malignancies, are not clear, and further follow-up data in clinical trials and postmarketing surveillance are indispensable to uncover the toxicity profile of this agent. Third, clinical effects on HRR-proficient tumors are not evident, except for niraparib. In addition, it would be desirable to discover new cascades other than the HRR pathway to provide additional indications for PARP inhibitors. Fourth, clinical trials so far performed have not demonstrated OS prolongation, in spite of PFS benefit. The current gold standard for assessment of efficacy of new agents is a statistically significant and clinically meaningful improvement in OS and quality of life (QOL). The surrogacy of PFS for OS is established only in advanced colorectal and ovarian cancers, and the clinical meaning of PFS in other types of tumors is not yet clear.Citation99 A longer follow-up period is required for assessment of OS, and at the same time, development of powerful combination regimens is expected to bring better clinical benefits. Interestingly, the olaparib trial data showed that high-dose olaparib gave a higher ORR, and the combination of high-dose olaparib with relatively low-dose cytotoxic agents seems to be an appropriate approach to concurrently achieve efficacy and tolerance.Citation42,Citation47,Citation49 However, clinical benefits need to be comprehensively evaluated from the viewpoints of QOL and cost-effectiveness, as well as OS. Finally, further accumulation of evidence about PARP inhibitors may lead to expanded adoption for neoadjuvant or adjuvant therapy in operable patients or preventive administration in BRCA mutation carriers in the future. Based on favorable trial data for pancreatic cancer, clinical development for intractable cancers, including this disease, might be promising.Citation4,Citation51 PARP inhibitors are considered advantageous for these adaptations in that the target population is small and well-defined and the risk is unlikely to outweigh the clinical benefit, since the complications resulting from the use of this class of agents are mild.Citation100
Figure 3 Current status of PARP inhibitors.
Abbreviations: PARP, poly(ADP-ribose) polymerase; HRR, homologous recombination repair; PFS, progression-free survival; OS, overall survival.
Disclosure
The authors report no conflicts of interest in this work.
References
- BryantHESchultzNThomasHDSpecific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymeraseNature2005434703591391715829966
- FarmerHMcCabeNLordCJTargeting the DNA repair defect in BRCA mutant cells as a therapeutic strategyNature2005434703591792115829967
- del RiveroJKohnECPARP Inhibitors: the cornerstone of DNA repair-targeted therapiesOncology (Williston Park)201731426527328412778
- KaufmanBShapira-FrommerRSchmutzlerRKOlaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutationJ Clin Oncol201533324425025366685
- GelmonKATischkowitzMMackayHOlaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised studyLancet Oncol201112985286121862407
- LedermannJHarterPGourleyCOlaparib maintenance therapy in platinum-sensitive relapsed ovarian cancerN Engl J Med2012366151382139222452356
- SwisherEMLinKKOzaAMRucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trialLancet Oncol2017181758727908594
- MirzaMRMonkBJHerrstedtJNiraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancerN Engl J Med2016375222154216427717299
- LinKYKrausWLPARP inhibitors for cancer therapyCell2017169218328388401
- BoltonKLChenevix-TrenchGGohCAssociation between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancerJAMA2012307438239022274685
- Gonzalez-AnguloAMTimmsKMLiuSIncidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancerClin Cancer Res20111751082108921233401
- LordCJAshworthABRCAness revisitedNat Rev Cancer201616211012026775620
- McCabeNTurnerNCLordCJDeficiency in the repair of DNA damage by homologous recombination and sensitivity to poly (ADP-ribose) polymerase inhibitionCancer Res200666168109811516912188
- ChambonPWeillJDMandelPNicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzymeBiochem Biophys Res Commun196311394314019961
- AshworthAA synthetic lethal therapeutic approach: poly (ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repairJ Clin Oncol200826223785379018591545
- BrownJSO’CarriganBJacksonSPYapTATargeting DNA repair in cancer: beyond PARP inhibitorsCancer Discov20177203728003236
- AméJCRolliVSchreiberVPARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymeraseJ Biol Chem199927425178601786810364231
- McLornanDPListAMuftiGJApplying synthetic lethality for the selective targeting of cancerN Engl J Med20143711725173525354106
- SchreiberVDantzerFAmeJCde MurciaGPoly (ADP-ribose): novel functions for an old moleculeNat Rev Mol Cell Biol20067751752816829982
- LordCJAshworthAThe DNA damage response and cancer therapyNature201248128729422258607
- PapaACarusoDStrudelMTomaoSTomaoFUpdate on Poly-ADP-ribose polymerase inhibition for ovarian cancer treatmentJ Transl Med20161426727634150
- MuraiJHuangSYDasBBTrapping of PARP1 and PARP2 by clinical PARP inhibitorsCancer Res201272215588559923118055
- PlummerERCalvertHTargeting poly (ADP-ribose) polymerase: a two-armed strategy for cancer therapyClin Cancer Res200713216252625617975135
- LordCJAshworthAPARP inhibitors: synthetic lethality in the clinicScience201735563301152115828302823
- AnnunziataCMO’ShaughnessyJPoly (ADP-ribose) polymerase as a novel therapeutic target in cancerClin Cancer Res201016184517452620823142
- O’SullivanCCMoonDHKohnECLeeJMBeyond breast and ovarian cancers: PARP inhibitors for BRCA mutation-associated and BRCA-like solid tumorsFront Oncol201444224616882
- ShrivastavMDe HaroLPNickoloffJARegulation of DNA double-strand break repair pathway choiceCell Res200818113414718157161
- BenafifSHallMAn update on PARP inhibitors for the treatment of cancerOnco Targets Ther2015851952825750544
- ByersLAWangJNilssonMBProteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1Cancer Discov20122979881122961666
- RatnamKLowJACurrent development of clinical inhibitors of poly (ADP-ribose) polymerase in oncologyClin Cancer Res20071351383138817332279
- RouleauMPatelAHendzelMJKaufmannSHPoirierGGPARP inhibition: PARP1 and beyondNat Rev Cancer201010429330120200537
- Hughes-DaviesLHuntsmanDRuasMEMSY links the BRCA2 pathway to sporadic breast and ovarian cancerCell2003115552353514651845
- LimDNgeowJEvaluation of the methods to identify patients who may benefit from PARP inhibitor useEndocr Relat Cancer2016236R267R28527226207
- PenningtonKPWalshTHarrellMIGermline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomasClin Cancer Res201420376477524240112
- MateoJCarreiraSSandhuSDNA-repair defects and olaparib in metastatic prostate cancerN Engl J Med20153731697170826510020
- The Cancer Genome Atlas Research NetworkIntegrated genomic analyses of ovarian carcinomaNature2011474735360961521720365
- PommierYO’ConnorMJde BonoJLaying a trap to kill cancer cells: PARP inhibitors and their mechanisms of actionSci Transl Med20168362s17
- Postel-VinaySVanheckeEOlaussenKALordCJAshworthASoriaJCThe potential of exploiting DNA-repair defects for optimizing lung cancer treatmentNat Rev Clin Oncol20129314415522330686
- MatulonisUAMonkBJPARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: does a development pathway forward exist?Ann Oncol20172844344728057663
- MuraiJZhangYMorrisJRationale for poly (ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibitionJ Pharmacol Exp Ther2014349340841624650937
- IhnenMzu EulenburgCKolarovaTTherapeutic potential of the poly (ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancerMol Cancer Ther20131261002101523729402
- DréanALordCJAshworthAPARP inhibitor combination therapyCrit Rev Oncol Hematol2016108738527931843
- LuYKwintkiewiczJLiuYChemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repairCancer Res20177771709171828202508
- TateishiKHiguchiFMillerJJThe alkylating chemotherapeutic temozolomide induces metabolic stress in IDH1-mutant cancers and potentiates NAD+ depletion-mediated cytotoxicityCancer Res201777154102411528625978
- FongPCBossDSYapTAInhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriersN Engl J Med2009361212313419553641
- FongPCYapTABossDSPoly (ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free intervalJ Clin Oncol201028152512251920406929
- AudehMWCarmichaelJPensonRTOral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trialLancet2010376973724525120609468
- LedermannJHarterPGourleyCOlaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trialLancet Oncol20141585286124882434
- TuttARobsonMGarberJEOral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trialLancet2010376973723524420609467
- HopkinsTAShiYRodriguezLEMechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitorsMol Cancer Res201513111465147726217019
- de BonoJRamanathanRKMinaLPhase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancersCancer Discov20177662062928242752
- KristeleitRSBurrisHALoRussoPPhase 1/2 study of oral rucaparib: final phase 1 resultsJ Clin Oncol201432suppl abstr2573
- Shapira-FrommerROzaAMDomchekSMA phase II open-label, multicenter study of single-agent rucaparib in the treatment of patients with relapsed ovarian cancer and a deleterious BRCA mutationJ Clin Oncol201533suppl abstr5513
- PuhallaSBeumerJHPahujaSFinal results of a phase 1 study of single-agent veliparib (V) in patients (pts) with either BRCA1/2-mutated cancer (BRCA+), platinum-refractory ovarian, or basal-like breast cancer (BRCA-wt)J Clin Oncol201432suppl abstr2570
- ColemanRLSillMWBell-McGuinnKA phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation – an NRG Oncology/Gynecologic Oncology Group studyGynecol Oncol2015137338639125818403
- SandhuSKSchelmanWRWildingGThe poly (ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trialLancet Oncol201314988289223810788
- Bell-McGuinnKMKonnerJATewWPA phase 2, single arm study of iniparib in patients with BRCA1 or BRCA2 associated advanced epithelial ovarian, fallopian tube, or primary peritoneal cancerInt J Gynecol Cancer201626225526026745694
- WagnerLMProfile of veliparib and its potential in the treatment of solid tumorsOnco Targets Ther201581931193926251615
- OzaAMCibulaDBenzaquenAOOlaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trialLancet Oncol201516879725481791
- BangYJImSALeeKWRandomized, double-blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancerJ Clin Oncol2015333858386526282658
- O’ShaughnessyJOsborneCPippenJEIniparib plus chemotherapy in metastatic triple-negative breast cancerN Engl J Med2011364320521421208101
- LichtSCaoHLiZMechanism of action of iniparib: stimulation of reactive oxygen species (ROS) production in an iniparib-sensitive breast cancer cell lineMol Cancer Ther201110supl 11 abstrA226
- GuhaMPARP inhibitors stumble in breast cancerNat Biotechnol20112937337421552220
- O’ShaughnessyJSchwartzbergLDansoMAPhase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancerJ Clin Oncol201432343840384725349301
- ChanNBristowRG“Contextual” synthetic lethality and/or loss of heterozygosity: tumor hypoxia and modification of DNA repairClin Cancer Res2010164553456020823145
- LiuJFBarryWTBirrerMCombination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 studyLancet Oncol201415111207121425218906
- O’Sullivan CoyneGChenAPMeehanRDoroshowJHPARP inhibitors in reproductive system cancers: current use and developmentsDrugs201777211313028078645
- LYNPARZA™ (olaparib) capsules, for oral use [prescribing information approved by the US FDA] Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206162lbl.pdfAccessed August 22, 2017
- StoverEHKonstantinopoulosPAMatulonisUASwisherEMBiomarkers of response and resistance to DNA repair targeted therapiesClin Cancer Res201622235651566027678458
- RUBRACA™ (rucaparib) tablets, for oral use [prescribing information approved by the US FDA] Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209115s000lbl.pdfAccessed August 22, 2017
- ZEJULATM (niraparib) capsules, for oral use [prescribing information approved by the US FDA] Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208447lbl.pdfAccessed August 22, 2017
- WiggansAJCassGKBryantAPoly(ADP-ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancerCochrane Database Syst Rev20155CD007929
- SakaiWSwisherEMKarlanBYSecondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancersNature200845171821116112018264087
- YachidaSJonesSBozicIDistant metastasis occurs late during the genetic evolution of pancreatic cancerNature201046773191114111720981102
- TelliMLTimmsKMReidJHomologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancerClin Cancer Res201622153764377326957554
- GraeserMMcCarthyALordCJA marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancerClin Cancer Res201016246159616820802015
- von MinckwitzGMüllerBMLoiblSCytoplasmic poly(adenosine diphosphate-ribose) polymerase expression is predictive and prognostic in patients with breast cancer treated with neoadjuvant chemotherapyJ Clin Oncol201129162150215721519019
- BrennerJCFengFYHanSPARP-1 inhibition as a targeted strategy to treat Ewing’s sarcomaCancer Res20127271608161322287547
- MichelsJVitaleIGalluzziLCisplatin resistance associated with PARP hyperactivationCancer Res20137372271228023554447
- MontoniARobuMPouliotEShahGMResistance to PARP-inhibitors in cancer therapyFront Pharmacol201341823450678
- EdwardsSLBroughRLordCJResistance to therapy caused by intragenic deletion in BRCA2Nature200845171821111111518264088
- BarberLJSandhuSChenLSecondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitorJ Pathol2013229342242923165508
- SwisherEMSakaiWKarlanBYWurzKUrbanNTaniguchiTSecondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistanceCancer Res20086882581258618413725
- NorquistBWurzKAPennilCCSecondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomasJ Clin Oncol201129223008301521709188
- DhillonKKSwisherEMTaniguchiTSecondary mutations of BRCA1/2 and drug resistanceCancer Sci201110266366921205087
- BuntingSFCallénEWongN53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaksCell2010141224325420362325
- BouwmanPAlyAEscandellJM53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancersNat Struct Mol Biol201017668869520453858
- SourisseauTManiotisDMcCarthyAAurora-A expressing tumour cells are deficient for homology-directed DNA double strand-break repair and sensitive to PARP inhibitionEMBO Mol Med20102413014220373286
- Mendes-PereiraAMMartinSABroughRSynthetic lethal targeting of PTEN mutant cells with PARP inhibitorsEMBO Mol Med200916–731532220049735
- LiuXHanEKAndersonMAcquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathwaysMol Cancer Res20097101686169219825992
- KondrashovaONguyenMShield-ArtinKSecondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinomaCancer Discov20177998499828588062
- RottenbergSJaspersJEKersbergenAHigh sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugsProc Natl Acad Sci U S A200810544170791708418971340
- KimYKimASharipAReverse the resistance to PARP inhibitorsInt J Biol Sci20171319820828255272
- Ter BruggePKristelPvan der BurgEMechanisms of therapy resistance in patient-derived xenograft models of BRCA1-deficient breast cancerJ Natl Cancer Inst201610811djw148
- ChoiYEMeghaniKBraultMEPlatinum and PARP inhibitor resistance due to overexpression of microRNA-622 in BRCA1-mutant ovarian cancerCell Rep201614342943926774475
- DuYYamaguchiHWeiYBlocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitorsNat Med20162219420126779812
- EspositoMTZhaoLFungTKSynthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitorsNat Med201521121481149026594843
- PatchAMChristieELEtemadmoghadamDWhole-genome characterization of chemoresistant ovarian cancerNature2015521755348949426017449
- BoothCMEisenhauerEAProgression-free survival: meaningful or simply measurable?J Clin Oncol201230101030103322370321
- SonnenblickAde AzambujaEAzimHAJrPiccartMAn update on PARP inhibitors – moving to the adjuvant settingNat Rev Clin Oncol2015121274125286972