
Abstract
Cancer is the disease with highest public health impact in developed countries. Particularly, breast cancer has the highest incidence in women worldwide and the fifth highest mortality in the globe, imposing a significant social and economic burden to society. The disease has a complex heterogeneous etiology, being associated with several risk factors that range from lifestyle to age and family history. Breast cancer is usually classified according to the site of tumor occurrence and gene expression profiling. Although mutations in a few key genes, such as BRCA1 and BRCA2, are associated with high breast cancer risk, the large majority of breast cancer cases are related to mutated genes of low penetrance, which are frequently altered in the whole population. Therefore, understanding the molecular basis of breast cancer, including the several deregulated genes and related pathways linked to this pathology, is essential to ensure advances in early tumor detection and prevention. In this review, we outline key cellular pathways whose deregulation has been associated with breast cancer, leading to alterations in cell proliferation, apoptosis, and the delicate hormonal balance of breast tissue cells. Therefore, here we describe some potential breast cancer-related nodes and signaling concepts linked to the disease, which can be positively translated into novel therapeutic approaches and predictive biomarkers.
Introduction
Cancer is one of the main causes of morbidity and mortality, being the second most prominent cause of deaths worldwide, with about 14 million new cases and 8.2 million deaths in 2012,Citation1,Citation2 representing one of the major issues in public health. According to the World Health Organization, breast cancer is the malignancy with the highest incidence among women, accounting for 25% of all female cancers diagnosed. Despite recent improvements in diagnosis and treatment, breast cancer remains the leading cause of cancer mortality among woman, representing 7% of all cancer deaths. The incidence and prevalence of breast cancer is increasing, especially in industrialized countries in North America, Western and Northern Europe, and Oceania.Citation3 From 2006 to 2010, breast cancer accounted for 19.8% of diagnosed cancers in the US.Citation4 For 2017, a total of 252,710 new cases and 40,610 deaths due to female breast cancer are estimated in the US alone.Citation1
The development of the mammary gland encompasses different stages, such as embryonic, prepubertal, pubertal, pregnancy, and lactation. In each of these stages, such processes as epithelial proliferation during pregnancy and secretory alveolar differentiation during lactation take place.Citation5 It has been proposed that breast cancer can develop through different mechanisms, but starts most frequently with imbalanced activity of pathways involved in patterning and morphogenesis of mammary gland developmental stages.
Several breast cancer classifications have been proposed, according to their invasive characteristics, occurrence, histology, or molecular profiling. Based on their site of occurrence, tumors can be classified as lobular (located at breast lobules) or ductal (at breast ducts). Carcinomas may also arise from invasive epithelial cells (medullary carcinoma), mucus-producing cells (mucinous carcinoma, also called colloid carcinoma), or a subtype of invasive ductal carcinoma (tubular carcinoma).Citation6,Citation7 Traditionally, breast cancer has been classified by immunohistochemical detection of hallmark receptors associated with distinct cell functions. Cancers derived from luminal cells are the most common types of breast cancer, expressing hormone receptors for ER, PR, or the amplified HER2/ErbB2 receptor for EGF-like growth factors. HER2 is a transmembrane glycoprotein belonging to a family of receptors that regulate cell growth, proliferation, survival, differentiation, and angiogenesis. Activation of the receptor and its subsequent phosphorylation lead to several signaling pathways, such as those of PI3K, MAPK, and PKC, and also to their pathophysiological functions.Citation7,Citation8 HER2-overexpressing tumors are considered to be more malignant, overexpressing other genes, such as GRB7 and PGAP3, and may display representative TP53 mutation. Each category is usually targeted by a specific drug therapy.
Tumors lacking expression of all three of these receptors (ER, PR, HER2) are referred to as triple-negative breast cancers, which most often derive from cells of basal origin.Citation9 Molecular gene expression profiling has also redefined breast cancer subtypes as luminal A, luminal B, HER2-rich, and basal-like, which roughly parallel the immunohistochemical categories.Citation10–Citation12 In addition, a more recently discovered claudin-low class of breast cancers has been defined.Citation13
Claudins are a family of tight junction proteins that are expressed exclusively in epithelial cells.Citation6,Citation7,Citation9 Claudin-low tumors, which lack or display low levels of E-cadherin and claudin 3, 4, and 7, are more heterogeneous than basal and luminal A subtypes, in addition to displaying more extensive lymphocytic infiltrates, larger tumors, and a high expression of mesenchymal markers as their main features. This tumor subtype is also associated with young age of onset and lower survival rates when compared with the luminal A subtypes.Citation14,Citation15 A summary of histological and molecular classification of the different breast cancer subtypes is presented in .
Table 1 Breast cancer subtype classifications, based on site of occurrence and/or biomarker status
Understanding the development of breast cancer, applying appropriate (and often patient-specific) treatment, and making an adequate prognosis is frequently mixed by the diverse histological patterns and biological/morphological features of the disease, as well as its clinical behavior and molecular characteristics. Additionally, the onset and progression of the disease are often related to lifestyle, race,Citation10 geographical variation, age at menarche or menopause, and family history,Citation16 attributable to high penetrance susceptibility genes such as BRCA1 and BRCA2.Citation17,Citation18 As a result, a combination of these factors leads to deregulation of signaling pathways that modulate the normal function and development of the mammary gland. Therefore, unraveling the molecular network that is behind this disease should provide guidance toward early (and more accurate) diagnosis, as well as better targets for possible therapy intervention. Here, we describe the roles of major signaling pathways in the normal development of the mammary gland and in breast cancer progression. A summary of the most important molecular alterations associated with breast cancer along these pathways is presented in .
Table 2 Main molecular alterations in pathways associated with breast cancer and respective targeted therapies
Estrogen receptor
The ER pathway
Estrogen comprises a set of steroid hormones involved in the development and maintenance of reproductive, cardiovascular, bone morphogenetic, and immune and central nervous systems.Citation19 ERs belong to the nuclear hormone receptor superfamily (or nuclear receptor family), which are highly conserved throughout evolution. These receptors are mostly present in the nucleoplasm, but may also be bound to the plasma membrane. Their early appearance in evolution contributed to the wide range of effects triggered by this pathway, such as ER regulating lipid and glucose metabolism in the liver,Citation20 whereas in cardiac tissue estrogen prevents apoptosis and necrosis of cardiac and endothelial cells.Citation21
The main mechanism of ER activation is through estrogen (E2) binding to its receptor (ER). E2–ER complexes translocate to the nucleus and bind to EREs present in the DNA, regulating transcription of a subset of ERE-modulated genes. Typically, an ERE encompasses a 15 bp palindromic RE containing two PuGGTCA motifs, separated by 3 bp.Citation22 Binding of the E2–ER complex also results in recruitment of coactivator complexes, with histones being further acetylated, leading to chromatin remodeling and finally to recruitment of the basal transcription machinery.Citation23 More than 230 estrogen-stimulated human genes with conserved and unconserved elements have been identified, and more than 70,000 potential ERE motifs have been found through genome wide screenings.Citation24 In humans, the main estrogen hormone is 17β-estradiol (E2), which binds to the ligand binding domain of the ER, a globular domain that harbors a hormone binding site and a dimerization interface and also contains coactivator–corepressor interaction capabilities. However, ERs display a ligand cavity slightly wider than their ligands, allowing them to be activated by a set of steroid hormones, environmental contaminants, xenoestrogens, phytoestrogens, and synthetic compounds with diverse structures.Citation25,Citation26
An alternative mechanism involving activation of ER in the absence of estrogen binding has been described,Citation27–Citation29 in which ER activation occurs through phosphorylation by cellular kinases cross-coupling with ER signaling. Phosphorylation of ERs on both serineCitation30 and tyrosineCitation31 residues has been observed, with serine phosphorylation of the N-terminal transcriptional activation function (AF)-1 domain (which influences the receptor’s transactivation activity) and phosphorylation of ER’s serine 118 (necessary for EGF response involving the MAPK pathway).Citation32 Further signaling studies revealed a new membrane-coupled receptor, namely GPR30, which can mediate a rapid response. GPR30 (also named GPER1) is a G-protein-coupled receptor located in the cell membrane and endoplasmic reticulum that stimulates intracellular calcium mobilization, activation of phosphoinositide kinase signaling, and inhibition of cell migration.Citation33,Citation34
ER variants
Two ERs are expressed in human cells: ERα (ERα66), encoded by the ESR1 gene, and ERβ (ERbβ1), encoded by ESR2. ERα displays six distinct domains, termed “A”–“F”, whereas ERβ has the same distribution but lacks the first “A” domain. In most cells, ERα and ERβ localize to the plasma membrane and to cytoplasmic organelles, such as mitochondria and the endoplasmic reticulum, exhibiting overlapping tissue distribution and functions under normal conditions.Citation35 However, ERα and ERβ integrate the estrogen pathway with opposing functions for some genes involved in cell proliferation.Citation36 For instance, in HeLa cells, estrogen activates ERα, whereas ERβ is activated by antiestrogens.Citation37 Additionally, ERα and ERβ have opposite actions at the cyclin D1 promoter,Citation36 where ERβ is able to inhibit cyclin D1 expression even when ERα is activated by saturating concentrations of 17β-estradiol. These opposing functions are purportedly carried out by the activation of functional domains (AF1) on both receptors. ERβ does not contain a strong AF1 domain, but contains a repressor domain that may inhibit the overall transcriptional activity of the receptor.Citation38 These studies support a potential modulatory role for ERβ in proliferation of mammary tissue induced by estrogen.Citation36
Although ERα and ERβ receptors have similar structures, their splice variants are much more diverse. In ERα, for instance, exon skipping generates isoforms with highly variable lengths and motif composition.Citation39 The variants generated by alternative splicing could play an important role in cancer development.Citation40 Many of these variants are translated as ERs with diverse responses to hormone stimulation. For example, ERβ increases ERα degradation, while the isoform ERβ2 is strongly correlated with high grade prostatic cancer and poor prognosis. Currently, a great deal of information linking splicing variants to susceptibility to disease is available, in addition to their roles in maintaining normal physiology. Different types of variants can exert diverse effects: some variants can fail to bind ligands, others display altered subnuclear localization and capacity for transcriptional activation by dimerization, while others bind to cofactors more weakly and fail to bind ERE to start transcription.Citation41
Genomic and nongenomic pathways
There are two main types of ER signaling pathways. The first, named genomic or classical mechanism, is initiated by activation of the ER in response to a given stimulus. ERs can be activated through binding of their specific ligand (E2) or by phosphorylation, independently of the ligand, prompting transcription of a group of genes, either by direct binding to promoter sequences or by activating complexes of transcription factors that bind to non-ERE sequences. The E2–ER complex recruits coregulatory proteins to promoter sequences, activating or inhibiting transcription, the result being determined by the type of ligand, the type of complex formed, and even by the presence of some splicing variants. Activation of this pathway from stimulus to response may take several hours.Citation42
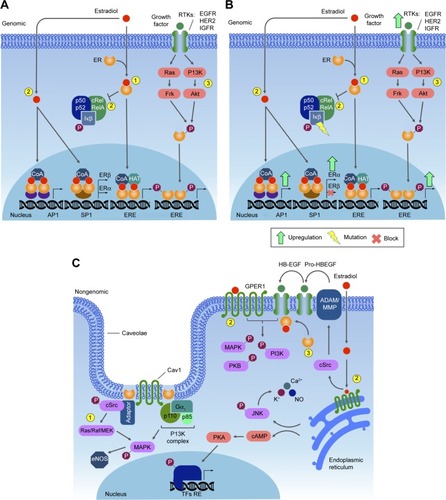
The second type of ER signaling pathway, nongenomic mechanism, triggers much faster responses, ie, within seconds to minutes. In this mechanism, ER activation on the plasma membrane or adjacent to receptors is mediated by various protein kinase cascades, generating such responses as nitric oxide (NO) release, increases in ion flux across membranes, and activation of RTK and the protein lipid kinase pathway.Citation34 Also, E2 stimulates Gβγ subunit protein-dependent transactivation of the EGFR–ERK signaling axis, through release of pro-HBEGF and suppression of the EGFR–ERK pathway.Citation43 The proposed mechanism involves membrane-localized GPR30, ERα, and ERβ associated with G-protein complexes, generating the earliest rapid signals.Citation44–Citation46 Specific G-proteins or proteins physically associated with ER can activate growth factor receptors, such as EGFR, which triggers secondary signals, like ERK and PI3K activation, calcium, PKC, or Src kinase.Citation47–Citation49 Most of this membrane protein complex is associated with scaffold anchor proteins, such as caveolin 1, striatin, and SHC, generally in caveolae.Citation50–Citation52 The outcome of these signaling pathways is the modulation of cell adhesion, migration, survival, and proliferation, thus playing an import role in the onset of cancerCitation53 ().
Figure 1 ER genomic pathway.
ER in mammary tissue development
The E2–ER complex has essential roles in morphogenesis in many female tissues, such as uterus,Citation54,Citation55 ovary,Citation56 and mammary glands,Citation57 as well as male tissues like gonadsCitation58 and the prostate.Citation59,Citation60 In normal mammary gland tissue, cell division starts during puberty, proceeding with cyclic cell proliferation, controlled by endocrine regulation through ovarian secretion of E2 and also by GH and IGF1 to complete branching morphogenesis.Citation61,Citation62 In adult tissue, progesterone plays an important role in mammary SC expansion.Citation63 E2, GH, and IGF1 receptors mediate pathways regulating ductal growth and morphogenesis.Citation64
The human breast tissue epithelium is arranged hierarchically, involving the participation of SCs, which do not express ERα or PR, differentiated hormone-sensing ERα+ cells, and myoepithelial cells. Morphologically, two main types of epithelial cell types may be distinguished: luminal cells lining the central lumen, where the majority of ER+ cells are found, and the myoepithelial cells, which lay adjacently to the basement membrane.Citation65,Citation66
A possible mechanism for the estrogen sensitivity of the mammary tissue is that ERα+ cells initiate proliferation by signaling in a paracrine manner. Therefore, ERα+ cells would be required to initiate proliferation,Citation67,Citation68 such that after estrogen stimulation, EGFs, such as Areg, are released by ERα+ cells and trigger cell proliferation.Citation69,Citation70 Areg is strongly induced by estrogen in mammary epithelial cells at puberty, binding to EGFR, which is located on stromal cells. This protein requires induction of the ADAM17 metalloproteinase to be released from mammary epithelial cells and reach the stromal cells, a mechanism that allows activation of stromal EGFR.Citation71
In the suggested model, GH induces the production of IGF1 in both the liver and locally in mammary stroma and epithelium. As such, E2 and IGF1 generate the first signal to trigger proliferation, required for ductal morphogenesis. The effect of E2 on ERα+ sensing cells then induces the release of Areg, which acts through stromal cells, recruiting additional growth factors that contribute to the rapid growth of mammary glands, and stimulating terminal end bud formation and ductal branching, which occurs at puberty.Citation72 Therefore, a paracrine circuitry model consisting of estrogen response via cross talk with the stroma and IGF1 as the primary effector downstream of E2 and GHCitation73,Citation74 is responsible for the sensitivity of breast tissue to estrogen. In adult mammary gland tissue, progesterone has a proliferative effect, which is activated via a paracrine mechanism. However, estrogen also upregulates PRs, playing a key role in maintaining differentiated ERα+ cells and the presence of populations of SCs in the adult mammary gland.Citation63
ER and breast cancer
In most cases of breast cancer, it is well known that the E2–ER complex is involved in malignant transformation and progression. Estrogen is important in the maintenance of breast tissue; however, the mechanism by which this hormone renders quiescent epithelial ERα+ cells into highly proliferative cells during tumorigenesis is still poorly understood. The answer may lie in the cross talk between epithelial ERα+ and stromal EGFR signaling, since some studies show that both ERα and Areg are upregulated in early hyperplastic precursors of breast cancer, which together may initiate tumorigenesis.Citation75 On the other hand, estrogen withdrawal ensues during menopause. The suppressive activity of ERα on some growth factor receptors in the normal mammary epithelium is affected by the decrease in estrogen level during menopause, allowing for expression of growth factor receptors on ER-positive cells, thus rendering these cells potentially proliferative and cancerous. Once this occurs, ER may be activated by growth factor-stimulated TKs, and normal regulation of cell proliferation is then lost.Citation76,Citation77
Breast cancer may arise by different mechanisms. Over-expression of HER2 in 12%–20% of breast cancers is often due to gene amplification.Citation78 In some types of breast cancers, there are specific translocations, such as those between chromosomes 12 and 15 (t[12;15]), commonly detected in secretory carcinomas of mammary tissue,Citation79 as well as that between chromosomes 11 and 19 (t[11;19][q21;p13]), which is characteristic of breast mucoepidermoid carcinoma.Citation80 In some subtypes of ER+ breast cancer, somatic mutations have been identified by whole genome sequencing. These mutations are present in a set of genes that impact important signaling pathways, which possibly affect ER function.Citation81 For instance, the luminal A breast cancer subtype displays mutations in PIK3CA (49%), MAP3K1 (14%), GATA3 (14%), TP53 (12%), and MAP2K4 (12%) and loss of PTEN (13%), among others. The luminal B breast cancer subtype has mutations in TP53 (32%), PIK3CA (32%), MAP3K1 (5%) and other genes.Citation82 These data reinforce the concept that breast cancer is etiologically diverse, with the current classification based on histology and morphology reflecting the heterogeneity of this disease.
An emerging group of nuclear receptors involved in key processes of mammary tissue development are orphan nuclear receptors.Citation83 These receptors share functional domains with the ER, wherein their AF1 sites are ligand-independentCitation84 and thus constitutively active. Several studies have demonstrated a correlation between the clinical outcomes of different types of breast cancer with the expression of these receptors, such as COUP-TFI,Citation85,Citation86 NGFIB,Citation87 rRORα,Citation88,Citation89 ERRα, ERRβ, and ERRγ. Due to their high degree of homology with ERs, more emphasis has been given to the study of ERRs and particularly to ERRα, a master regulator of cellular energy metabolism in both normal and cancer cells.Citation90–Citation92 Interestingly, high ERRα expression positively correlates with HER2 status and poor outcome in breast tumors, suggesting a tentative possibility for a new prognostic biomarker.Citation93,Citation94 Recent studies found that ERRα is part of the AMPK–PGC1α–ERRα axis, a key regulator in reprogramming of cellular metabolism and cellular adaptation to metabolic stress.Citation95,Citation96 PGC1α–ERRα is a known repressor of folate metabolismCitation97 and one of the main pathways upregulated in cancer cells; however, several studies have associated the PGC1α–ERRα transcriptional axis with increased tumor growth in breast cancer.Citation97,Citation98 One possible explanation for this apparent contradiction is the number of metabolic pathways regulated by the PGC1α–ERRα axis,Citation99–Citation101 acting in conjunction and cross talking to achieve energy balance in response to stress and several signals in the tumor microenvironment. These results highlight the use of ERRα as a putative biomarker in breast cancer, and reinforce the idea that directing specific drugs to the folate pathway in tumors overexpressing PGC1α/ERRα is a powerful tool to improve patient prognosis.Citation83
Clinical targeting of the ER pathway
Overexpression and activation of ERα increase cell proliferation and malignant transformation of luminal-type breast cancers,Citation75 prompting the development of antiestrogen treatments. The most successful therapies for ERα+ breast cancer have relied on synthetic molecules designed to block mainly ERα, such as selective ER modulators like tamoxifen, raloxifene, and toremifene, aromatase inhibitors, and selective ER degraders, such as fulvestrant. Though highly effective, these drugs have unwanted side effects in nontarget tissues, with approximately 50% of patients acquiring resistance and developing further metastases.Citation102,Citation103 Combined drug therapies, such as tamoxifen and aromatase inhibitors, have been shown to improve disease-free survival substantially.Citation104 Expression of ER, PRs, and HER2 determines the tumor hormone receptor status. In fact, these molecular markers are valuable to determine prognosis and predict response to anti-ERα therapy.Citation105
In normal breast tissue, the predominant ER is ERβ. However, ERβ levels are reduced in breast tumors, compromising the potential efficacy of targeted therapies.Citation106 Most compounds that selectively target ERβ elicit anti-inflammatory effects, but show no therapeutic effect in cancer. Alternative targets are also currently under development, such as G1 (agonist) and G15 (antagonist), specifically targeting GPR30. Although drugs targeting ER are widely used, caution should be applied, since the mechanisms by which these drugs operate, their effects on specific tissues and cell types, and their specificity toward each receptor type, including GPR30, remain to be determined.Citation107
Phosphatidylinositide-3 kinase
The PI3K pathway
PI3K signaling is central for a number of cellular processes related to cell growth, proliferation, motility, survival, and apoptosis. This pathway mediates metabolism by modulating both glycolytic flux and fatty acid synthesis. Activation of PI3K signaling consistently leads to cell mass accumulation and cell proliferation, as well as differentiation events by promoting protein synthesis, cell cycle progression, and actin rearrangement.Citation108 PI3K signaling also modulates autophagy, a nonapoptotic programmed cell death mechanism, as well as protein and organelle recycling into metabolic intermediates through lysosomes.Citation108–Citation110
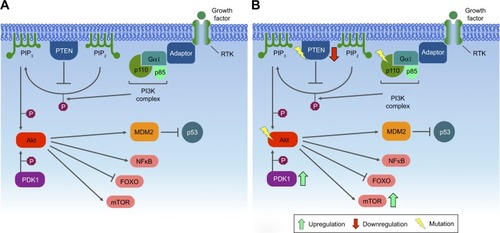
PI3Ks are lipid-based protein kinases, usually divided into three distinct classes (I, II, and III). Class I is subdivided into IA and IB, according to their structural characteristics and substrate specificity. Class IA consists of heterodimers with regulatory and catalytic subunits, the former mediating activation of the latter by RTKs, G-protein-coupled receptors, and adaptor proteins, such as the Ras oncoprotein.Citation111–Citation113 Upon activation, PI3K phosphorylates PIP2 in the plasma membrane, converting it into PIP3. Dephosphorylation of PIP3 is an important modulatory step in this pathway, due to the activity of the PTEN tumor suppressor, which acts as a negative regulator of this pathway.Citation114 PIP3 recruits and activates Akt, also known as PKB. This activation stimulates the catalytic activity of Akt, resulting in phosphorylation of other downstream proteins that regulate cell growth, survival, and entry into the cell cycle. The Akt protein has three isoforms: Akt1, Akt2, and Akt3. During breast cancer progression, Akt1 promotes cell proliferation by S6 and cyclin D1 upregulation, while Akt2 promotes cell migration and invasion through F-actin and vimentin induction.Citation108,Citation113 Akt3 is the less studied isoform, but is known to be mainly involved in triple-negative breast cancers, in which it may play a role in migration, invasion, and tumor growth.Citation115
One of the major downstream effectors of the PI3K signaling pathway is a serine/threonine protein kinase complex called mTOR. mTOR is subdivided into two complexes – mTORC1 and mTORC2 – which are structurally similar but functionally distinct.Citation116,Citation117 Autophagy is regulated by mTORC1, which can either phosphorylate or dissociate the ULK1 complex to block or initiate autophagy, respectively. When nutrients are limited, mTORC1 dissociates from the ULK1 complex, initiating autophagy, in which PIP3 plays an important role, establishing the autophagosomes and translocating the cytoplasmic material to lysosomes for degradation.Citation109 mTORC2 organizes the cellular actin cytoskeleton and regulates Akt phosphorylationCitation116 ().
Figure 2 The PI3K signaling pathway.
PI3K in mammary tissue development
In vivo studies suggest that the p110 catalytic isoform p110α, the catalytic subunit of class IA PI3K, is involved in duct outgrowth and branching during puberty, postpartum lactation, EGF signaling in mammary epithelial cells, and mammary gland development. The p110β isoform may play a regulatory role in refinement of PI3K downstream signaling, responding to insulin and EGF, while its absence induces a moderate hypermorphic mammary gland phenotype.Citation118
mTORC1 requires RAPTOR as a main cofactor, while mTORC2 requires RICTOR.Citation119 With mammary epithelial cells, it has been observed that Akt inhibition decreases branching and colony size, suggesting that Akt is involved in signaling growth control and branching morphogenesis dependent on RICTOR. Moreover, inhibition of mTORC2 reduces ductal branching and lengthening. Finally, it has been suggested that the mTORC2–RICTOR complex is necessary for PKCα signaling through the small GTPase Rac1, in order to trigger mammary morphogenesis cell survival, cell junctions, and motility.Citation120
PI3K and breast cancer
Mutations in PIK3CA, the gene encoding the p110α catalytic subunit of class IA PI3K, are among the most common alterations observed in human malignancies, found in approximately 25% of breast cancers. This mutation confers a gain of function to p110α and may render HER2-based therapy ineffective.Citation117,Citation121 Most of the mutations in PIK3CA occur in three sites: two in the helical domain (E542 and E545) and one in the kinase domain (H1047).Citation122 Other mutations along the PI3K pathway are frequently found in human cancers, especially breast cancer. PTEN mutations are frequent in basal-like (67%) and HER2+ (22%) breast cancers, while AKT1 mutations are often found in luminal/HR+ (2.6%) tumors.Citation116
Notch activation may render breast cancer cells resistant to PI3K-mTORC1 inhibition, and loss of PTEN may lead to resistance to Notch inhibition.Citation123 The Notch signaling pathway is evolutionarily conserved, and mainly regulates cell fate determination during development and maintains adult tissue homeostasis.Citation124 In mammals, four Notch proteins are present (Notch1, Notch2, Notch3, And Notch4), which are defined by a large extracellular domain and a cytoplasmic domain. In particular, Notch1 is involved in breast cancer tumorigenesis and metastasis, apparently triggering EMT in epithelial cancer cells.Citation124
With regard to the function of Akt during breast tumorigenesis, Akt1 and Akt2 regulate cell migration and invasion in different manners. While Akt2 promotes cell migration and invasion through F-actin and vimentin induction, Akt1 inhibits cell migration and invasion through downregulation of β1 integrin and FAK.Citation125 Akt may be directly phosphorylated and activated by PIP3 or by the putative PDPK1/PDK1. Activation of PIP3 provides a docking site (containing pleckstrin homology domains) for both Akt and PDK1.Citation113 In breast cancer, PDK1 is overexpressed and amplified in approximately 20% of tumors. PDK1 phosphorylates Akt1 at ThrCitation308, suggesting that another protein kinase, namely PDK2, is responsible for Ser473 phosphorylation. However, it is still unclear whether PDK2 is an alternative kinase or a modification of PDK1 by the PDK1-interacting fragment.Citation126
Clinical targeting of the PI3K pathway
Several preclinical and initial clinical trials have shown great potential in targeting the PI3K–Akt pathway in cancer-related conditions.Citation127 Inhibition of the PI3K pathway has been associated with improved antitumor T-cell response and tumor angiogenesis, since: 1) the activity of p110β is involved in tumors displaying PTEN loss; 2) p110α drives angiogenesis; 3) p110γ, p110δ, and p110β have important functions in inflammatory cells in the tumor microenvironment; and 4) p110δ and mTOR control important aspects of adaptive immunity, which include lymphocyte activation and differentiation.Citation128,Citation129 For breast cancer treatment, PI3K inhibitors can be very effective, especially when combined with HER2 inhibitors, since PIK3CA mutations seem to be associated with resistance to these drugs. Therefore, a combinatorial treatment by inhibiting PI3K, in addition to HER2 blockade, may constitute a powerful therapy.Citation121 Targeting mutated kinases and RTKs may be interesting for drug development, considering that similar therapies have demonstrated promising results in other cancer types, such as non-small cell lung cancer.Citation122,Citation130 LY294002 and wortmannin comprise the first generation of PI3K inhibitors and, thus lack selectivity for particular PI3K isoforms and present toxic effects. Some other drugs, such as BEZ235, BKM120, and BGT226 (from Novartis), XL765 and XL147 (from Exelixis), or SF1126 (from Semaphore), which inhibit nonselective PI3K/mTOR, have been employed in preclinical or in clinical trials. Other drugs, such as PX316 (from Pro1X Pharmaceuticals), GSK690693 (from Glaxo-SmithKline), Akti1/2 and MK2206 (both from Merck), and XL418 (from Exelixis), inhibit Akt activity. Another viable target in the PI3K pathway is the mTOR node, blocked by the anticancer drugs rapamycin (also known as sirolimus [Rapamune, from Pfizer]), CCI779 (Torisel, from Pfizer), RAD001 (Afinitor, from Novartis), and AP23573 (from Merck/Ariad).Citation113
Mitogen-activated protein kinase
The MAPK pathway
MAPKs are phosphoproteins stimulated by mitogens.Citation131 Most of the signals directly involving MAPKs are evolutionarily conserved and biologically versatile, and found in animals, plants, fungi, and protists.Citation132–Citation135 MAPK activity is regulated by a cascade composed of a MAPK, MAPKK/MKK/MEK, and MAPKKK or MEKK. The MAPK pathway may be activated by STE20 kinase or small GTP-binding proteins,Citation136 and is responsible for controlling cell survival and adaptation in response to chemical and physical stress.Citation135,Citation136
MAPKs are key components in the control of embryogenesis, cell differentiation, growth, proliferation, migration, and apoptosis.Citation135,Citation137 Six groups of MAPKs have been characterized in mammals: 1) ERK1/2, 2) ERK3/4, 3) ERK5, 4) ERK7/8, 5) JNK1/2/3, and 6) the isoforms of p38 a/b/g/d.Citation135,Citation138 MAPKs are composed of 80 kDa homodimers containing a threonine/x/tyrosine domain, where the X represents glutamate (E), proline (P), or glycine (G), in ERK, JNK, or p38, respectively,Citation139–Citation141 with N- and C-terminal regions of different lengths.Citation138
Generally, the initial stimulus in this cascade comes from an extracellular signal, such as a peptide growth factor, binding to its receptor protein embedded in the plasma membrane. This binding induces the receptor, such as RTKs, to initiate an autophosphorylation process that elicits the adaptor protein SHC, which in turn binds RTK and becomes self-phosphorylated. The receptor SHC complex then recruits the GRB2 adapter protein and the SOS guanine nucleotide exchange factor, catalyzing the conversion of GDP-Ras to GTP-Ras, which leads to MAPKKK phosphorylation. MAPKKK phosphorylates MAPKK in two serine residues, Ser218 and Ser222, thereby rendering it active and capable of phosphorylating MAPK in a threonine and tyrosine residue.Citation132–Citation135,Citation142–Citation146 Depending on the stimulus, phosphorylation of MAPKKK may be carried out by other small GTPases, such as Rac, CDC42, and Rho, which in turn modulate MAPK activation status downstreamCitation138,Citation147 ().
Figure 3 MAPK signaling pathway.
Abbreviation: TPA, tetradecanoyl phorbol acetate.
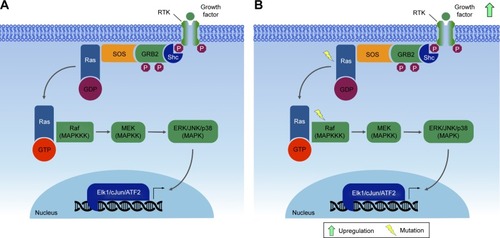
A single MAPK cascade may lead to different cellular responses. Every MAPK is activated by specific MAPKKs (MEK1/2 for ERK1/2, MKK3/6 for p38, MKK4/7 [JNKK1/2] for the JNKs, and MEK5 for ERK5); however, each MAPKK may be activated by more than one MAPKKK.Citation136 For instance, the cascade comprising Raf1 or Mos (MAPKKK), MEK1/2 (MAPKKs), and ERK1/2 (MAPKs)Citation133,Citation142,Citation148 may induce mitogenesis, transdifferentiation (PC12 cells) and even activation of the CDC2 cyclin B complex (oocytes), depending on the cell type and the stimuli.Citation133,Citation149
The Raf–MEK–ERK-MAPK cascade, present in various animal cell types, can be activated by the GTPase Ras.Citation133,Citation142 Activated Ras induces the protein kinase activity of Raf kinases. The Raf family of protein kinases is composed of A-Raf, B-Raf, and Raf1,Citation150 and each of the isoforms contains three conserved regions: CR1, CR2, and CR3. The CR3 region corresponds to the kinase domain, whereas the CR1 and CR2 regions are located in the amino-terminal end, being involved in regulation of the catalytic domain. Deletion of CR1 and CR2 generates constitutively active Raf1 mutants, which may be activated independently of Ras.Citation150,Citation151
MAPK and breast cancer
Mutations in RAS have been observed in many tumors.Citation152 These mutations are mainly associated with the constitutive activation of ERK1/2, thus promoting tumor cell proliferation. Abnormalities in MAPK signaling affect most of the cellular processes commonly associated with the development of cancers:Citation135 independence from proliferation checkpoints, evasion of apoptotic signals, unlimited replicative potential, invading and metastatic capacities, and the ability to attract and sustain angiogenesis. In particular, recent studies have reported higher frequency of modified cells with activated MAPK in breast cancer.Citation153
Mutations along the MAPK pathway, which may lead to the development of cancer, are often found in Ras and B-Raf, whereas JNK and p38 appear to be poorly related to malignant transformation.Citation135 The ERK pathway has been the best studied to date, with deregulations being described in approximately a third of human cancer cases. In cancer cells, the constitutive activation of ERK signaling occurs in the early steps of the pathway, and may be due to a series of factors, such as overexpression of RTK, activating mutations in TK receptors, autocrine or sustained paracrine production of activating ligands, and mutations of RAS and BRAF. However, this amplification and/or deregulation may also occur in targeted nuclear transcription factors, such as c-Myc and AP1.Citation135
Due to the high frequency of activating mutations, the Ras–Raf axis is suggested as a regulatory node of the pathway. Mathematical modeling predicts that activation of this node leads to general activation in the cell.Citation135 Ras GTPases control the activity of various signaling pathways, and mutations in KRAS and NRAS have already been described for several types of cancers, since they lead to inefficient GTP hydrolysis, causing Ras to be in an active state and leading to continuous activation of the MAPK route. This deregulated Ras activation can also recruit scaffold proteins, such as KSR and SUR8/SHOC2, which modulate Raf activation by Ras.Citation135
Human breast cancer may be subdivided into estrogen-dependent and -independent types. Estrogen typically activates growth factors which increase the levels of MAPK activity. In estrogen-independent tumors, MAPK stimulates growth upon activation by other peptide hormones.Citation153 The functions of MAPK in breast cancer appear to be complex, owing to several cellular responses that they modulate and also their interaction with different pathways (ie, ER and HER2). Generally, MAPKs are associated with good prognosis across most classifications of breast cancer, notably in those with positive ER.Citation154
Studies demonstrate that ERK1/2 and phospho-ERK1/2 are associated with good clinicopathological status and that good prognosis may be related to their roles in inducing apoptosis. Phosphorylated JNK1/2 has also been shown to be stimulated by stress or growth factors, activating apoptosis and even augmenting cell death signaling in MCF7 breast cancer-derived cells under the influence of high estradiol levels. Finally, growing evidence suggests a role for phospho-p38 in inducing apoptosis in breast cancer. Also, MAPKs have been associated with longer survival of ER+ patients.Citation154
Activated MAPK can also phosphorylate ER, either directly or indirectly, and increase its transcriptional efficiency (an important feature of hormone-dependent breast cancer). A recent report also suggested that RSK (downstream target of MAPK) can phosphorylate ER, an effect that increases its own transcriptional efficiency.Citation155,Citation156 Additionally, MAPK can phosphorylate PR at sites that act as ubiquitination signals, leading to degradation by the 26S proteasome.Citation157
In the absence of estrogen and progesterone, breast cancer cells could be stimulated by growth factors (ie, EGF, EGF1, insulin, prolactin, TGFα, or TGFβ), increasing MAPK activation. Cells overexpressing ErbB2 (a type of RTK) exhibit increased activated MAPK, which is mediated through ErbB2 interaction with endogenous ligands, such as EGF and neuregulin.Citation153 The tumor suppressor PTEN, with dual-specificity phosphatase activity, blocks phosphorylation of the insulin-stimulated MAPK in MCF7 cells by inhibiting IRS1 phosphorylation and inhibiting the formation of IRS1–GRB2–SOS complexes. Consequently, cell growth is suppressed and cyclin D1 downregulated, halting cell cycle progression.Citation158
MAPKs have an important role in cell cycle arrest and sequestration of ERK in the cytoplasm. Feedback to upstream mediators is common in inhibitory signals mediated by mutated RAS and RAF. Further stimulation through this pathway is omitted by HDM2 and FOXO3. The MAPK cellular function is context-specific and cell type-specific, in order to mediate signals that can lead to diverse cellular functions. Furthermore, the function of MAPKs is affected by their cross talk and interaction with other pathways, which can influence their behavior.Citation154
Clinical targeting of the MAPK pathway
Since MAPK signaling regulates both physiological and pathological processes, upstream inhibition is not advised as a good method for treatment. Each MAPK could be involved in several responses; therefore, blocking an MAPK for treatment of a specific disease could be deleterious for another physiological process, while inhibiting an upstream protein could cause negative downstream effects. Sorafenib, a drug that can inhibit the Raf kinase, is approved for treatment of several cancer types.Citation159,Citation160 However, no targeted therapy is available for other MAPKs. Many p38 blocking drugs have been tested, but so far none has reached approval for use, due to their low efficiency and/or hepatoxicity.Citation161
Janus kinase/signal transducer and activator of transcription
The JAK–STAT pathway
Several extracellular signals may cause rapid changes in the expression of specific genes. The JAK–STAT pathway is one of example of how cells are able to recognize signals to generate rapid and accurate responses. Studies of interferon-induced intracellular signaling have led to the discovery of this pathway, which relates to the major signaling mechanism used by cytokines and growth factors.Citation162 Among the main cytokines that activate this pathway are interferons, interleukins, and colony-stimulating factors. Among growth factors, such hormones as prolactin, leptin, erythropoietin, and thrombopoietin are the main activators.Citation163 Activation of this pathway stimulates important cellular events, such as proliferation, differentiation, migration, and apoptosis, as well as critical processes, such as hematopoiesis, immunodevelopment, mammary gland development, and lactation.Citation164,Citation165
This pathway is relatively simple, comprising a few major components: cell surface receptors, JAKs (TKs that are constitutively associated with the receptor), and STATs (latent cytoplasmic transcription factors).Citation162 JAKs are cytoplasmic TKs that participate in downstream signaling of various cell surface receptors that have no intrinsic TK activity. In mammals, four members of the JAK family are known: JAK1, JAK2, JAK3, and Tyk2.Citation166,Citation167 JAKs share seven domains (JH1–JH7), including: the four-point-one protein–ezrin–radixin–moesin (FERM) domain, which is important for association of JAKs with receptors; the SH2 motif, with scaffold function; the pseudokinase motif, which regulates JAK kinase activity; and the TK motif, which is catalytically active, being important for phosphorylation of receptors and STATs.Citation168
STATs are latent cytoplasmic transcription factors, which are activated upon recruitment to an activated receptor complex. To date, seven members of the STAT family have been identified in mammals: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6.Citation163 Six well-defined domains compose STAT structure, including: a conserved N-terminal domain involved in regulation of STAT activity, such as the formation of STAT tetramers and tyrosine dephosphorylation;Citation169 a coiled-coil domain, involved in receptor binding and association with regulatory proteins;Citation170 a DNA binding domain; the SH2 domain, which is important for recruitment of STATs to the activated receptor complex; and a variable C-terminal transactivation domain, which is important in transcriptional modulation of target genes. Interaction among the different types of JAK and STAT proteins will depend on the cytokine receptor that has been activated.Citation171
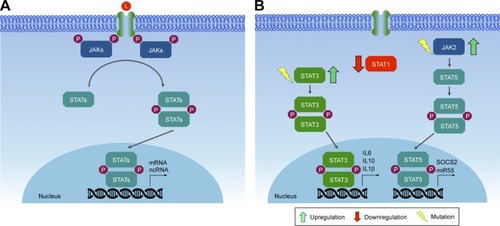
Binding of cytokines or growth factors to their specific receptors on the cell surface leads to receptor dimerization and subsequent activation of JAKs by transphosphorylation (ie, JAK molecules phosphorylate each other). Activated JAKs have the ability to phosphorylate specific tyrosine residues present in the receptor, which will then serve as anchoring sites for STATs. Anchored STATs are phosphorylated by JAKs, leading to their homodimerization or heterodimerization. Phosphorylated and dimerized STATs leave the receptor and translocate to the nucleus by an importin α5-dependent mechanism and the Ran nuclear import pathway, where they bind DNA at specific regulatory regions in promoters, activating transcription of target genes ().Citation172,Citation173
Figure 4 JAK–STAT signaling pathway.
JAK/STAT in mammary tissue development
The STAT family of transcription factors participates significantly in the development of the mammary gland, since its members play important roles in the regulation of cell proliferation and apoptosis. Deletion of STAT3 results in fetal lethality, preventing further investigation into the development of the gland, although some authors believe that STAT3 is important in the self-renewal of mammary SCs.Citation174 Other members of the STAT family (1, 3, 5, and 6) have important functions in the adult mammary gland. For instance, STAT1 is phosphorylated in the virgin gland (a gland that did not undergo physiological changes) and late involution during remodeling of the gland, but not during pregnancy and lactation. STAT3 is a mediator of cell death and inflammatory signaling in gland involution, becoming phosphorylated on the day of birth and in the first 10 days of involution. STAT5 is essential for lobuloalveolar development and expression of milk protein genes, being predominantly activated during pregnancy. Finally, STAT6 induces the expression of cytokines and important transcription factors in the maintenance of luminal alveolar cells.Citation175
During lactation, mammary gland cells maintain upregulated growth to produce milk through activation of STAT5. However, upon weaning, the mammary glands undergo an involution process. During involution, STAT5 is inactivated and STAT3 triggers the apoptosis process to reduce cell numbers.Citation175 One of the transcriptional targets of STAT3 is the PI3K regulatory subunits p55α and p50α, which in turn inhibit or downregulate PI3K/Akt survival signaling to promote apoptosis. In addition, STAT3 downregulation of Akt, which blocks activation of STAT5, might represent an additional mechanism of mammary gland involution.Citation176–Citation178
JAK/STAT and breast cancer
Due to the high number of cytokines and growth factors that activate this pathway, gain-of-function, loss-of-function mutations, and polymorphisms in JAK and/or STAT genes are associated with several human diseases. Constitutive activation of this pathway may arise by several mechanisms, including production of autocrine/paracrine cytokines, activating mutations (point mutations that generate amino-acid substitution) of receptors, JAKs and/or other upstream oncogenes, which then activate STATs.Citation179 Due to the important roles in regulating cell proliferation and survival, as well as in inflammation, it is not surprising that most JAKs and STATs are also implicated in tumorigenesis of breast tissues, acting either as oncogenes or as tumor suppressors.
Somatic mutations in members of the JAK family are rare in cancer.Citation180 Despite this, somatic mutations in JAK1, JAK2, and JAK3 have already been described in breast cancer samples.Citation181,Citation182 Recent research shows the possible involvement of JAK2 and TYK2 in the development of breast cancer. Studies on epithelial mammary cells show that constitutive activation of JAK2 caused by the V617F point mutation generates hyperactivation of STAT5, leading to increased proliferation and resistance to cell death. Transgenic mice expressing JAK2V617F presented an accelerated tumorigenic process.Citation182 Tyk2−/− mice inoculated with breast cancer cells show increased tumor growth and metastasis compared to wild-type mice, suggesting a possible tumor suppressor role, mediated in part by myeloid-derived suppressor cells.Citation183
A great deal of evidence indicates the involvement of STAT1, STAT3, STAT5, and STAT6 in the formation, progression, prognosis and prediction of breast cancer.Citation177 Depending on the physiological context, STAT1 may act as an oncogene or as a tumor suppressor. Approximately 45% of ER+ and 22% of ER− breast cancer cases exhibit low levels of STAT1 in neoplastic cells, while high levels of STAT1 are detected in benign breast tissue adjacent to the tumor. This may indicate that STAT1 is repressed during breast tumor progression.Citation184 Conversely, high STAT1 expression is associated with metastasis and drug resistance.Citation185 It appears that in postmenopausal ER+ breast tumors, STAT1 acts as a tumor suppressor, while in ER− tumors or ER+ premenopausal malignancies, STAT1 promotes tumor progression.Citation177
STAT3 is constitutively activated in a wide range of solid tumors, including breast cancer.Citation186 Its aberrant activation may promote invasion and metastasis, in addition to regulating the inflammatory response in breast tumorigenesis. Human studies have shown that activation of STAT3 is frequently observed in primary breast cancers, being associated with poor prognosis and tissue invasion.Citation187,Citation188 Constitutive activation of STAT3 appears to affect the tumor microenvironment through secretion of various cytokines, such as IL-10, IL-6, IL-1β, by tumor cells, which stimulate nontumor cells, T-helper (TH)-17 cells, and tumor associated macrophages to secrete even more cytokines, thereby creating a positive feedback loop. This promotes growth and differentiation of tumor cells. STAT3-directed secretion of IL-10 by tumor cells also results in inhibition of antitumor immunity, eg, by inhibiting maturation of dendritic cells.Citation189
Like STAT3, STAT5 is also constitutively activated in breast cancer, but is considered weakly oncogenic in breast cancer mouse models. According to the data currently available, activated STAT5 may promote tumorigenesis, but it is not a genuine proto-oncogene, at least in breast cancer. Activated STAT5 promotes tumorigenesis by expanding the population of mammary alveolar cells, which have been suggested to be especially susceptible to tumorigenesis.Citation190 Finally, STAT6, which is important in the balance between TH1 and TH2 cells, also affects tumor progression, facilitating evasion of the immune system by tumor cells.Citation189 T lymphocytes can differentiate into either TH1 cells or TH2 cells, depending on the cytokines secreted in their environment. TH1 cells identify tumor antigens and trigger an immunoresponse, whereas TH2 cells display oncogenic potential, promoting invasion and tumor metastasis. STAT6 is essential for the differentiation of T lymphocytes in TH2 cells mediated by IL4.Citation177
Clinical targeting of the JAK–STAT pathway
Mounting data showing the relevance of the JAK–STAT pathway in diseases related to the immune system and various types of cancers render the members of this pathway attractive as therapeutic targets. The effectiveness of targeting JAK–STAT signaling has been demonstrated in clinical trials in patients with solid tumors.
JAK inhibitors
The first studies with JAK inhibitors (ie, tyrphostin AG490) led to inhibition of recurrent leukemia B-cell growth in vitro and in vivo.Citation191 In the early 2000s, another JAK inhibitor (pyridone 6) was introduced by Merck, with activity against all members of the JAK family in vitro; however, this compound did not present significant results in vivo.Citation192 Analysis of its crystallographic structure showed that pyridone 6 binds to adenosine triphosphate pocket in the JH1-kinase domain of the JAK2 active conformation. This information served as the basis for the development of various JAK inhibitors described thus far.Citation193
Several selective JAK inhibitors are currently in clinical trials for the treatment of solid tumors. These inhibitors focus mostly on two members of the JAK family: JAK1 and JAK2. Ruxolitinb (Novartis), which targets both JAK1 and JAK2, is one of the most promising compounds, used in various clinical trials (Phase I, II, and III) for a wide variety of solid tumors (breast, lung, gastric, colon, colorectal, and pancreatic cancer). Another interesting inhibitor targeting JAK1 and JAK2 (momelotinib; Gilead Sciences) has also been tested for lung, colon, and pancreatic cancers. The INCB047986 and INCB39110 inhibitors (Incyte Corp.), which specifically target JAK1 by blocking its phosphorylation, are also in advanced phases of clinical trials.Citation194
STAT inhibitors
Constitutive activation of STATs is well described in several types of cancer.Citation195–Citation197 STATs are important transcription factors for pathology progression, by controlling genes with key roles in cell proliferation and modulation of the tumor microenvironment.Citation198 Therefore, the STAT family is an increasingly attractive therapeutic target in cancer. However, great challenges persist in developing STAT inhibitors, since their blockade is much more complex than blocking kinases, such as JAKs.Citation179 One of the challenges lies in target specificity, which may generate off-target effects, resulting in undesired side effects.Citation199 Another challenge is redundancy in the action of different STATs. For example, selective STAT3 blockade, which is critical for IL-6 downstream signaling, may not be fully effective since IL-6 can also act through STAT1 signaling.Citation200
Despite these limitations, a wide range of molecules that mainly inhibit STAT3 and STAT5 have been developed and tested both in vivo and in vitro. These include peptidomimetics, small-molecule inhibitors, and oligonucleotides.Citation201 As an example of peptidomimetics, CJ1383, a STAT3 inhibitor, has shown promising results in two breast cancer cell lines with high levels of phosphorylated STAT3.Citation202 Several small inhibitory molecules targeting the SH2 domain in STAT3 and STAT5 should also be mentioned. LLL12, S31201 (both from Biovision, Inc.) and SF-1066, which inhibit STAT3 activity in different ways, have shown important results using the MDA-MB-231 breast cancer line as a model.Citation203,Citation204 FLL32 and IS3295, which inhibit STAT5, have also shown promising in vitro results in several breast cancer cell lines, such as MDA-MB-231, SUM159, and SK-BR-3.Citation205 Finally, as an example of nucleotide-based inhibitors, siRNAs and G-quartets (guanine-rich oligonucleotides that form inter- or intramolecular four-stranded structures) against STAT3 have shown positive results, based on studies using the MCF7 and MDA-MB-231 breast cancer cell lines.Citation206–Citation208
Wingless-type MMTV-integration-site family
The Wnt pathway
Human and most mammalian genomes harbor 19 WNT genes, falling into 12 evolutionarily conserved WNT subfamilies.Citation209 Wnt proteins are secreted ~40 kDa cysteine-rich glycoprotein ligands, activating a complex mechanism of signal transduction via multiple pathways: the canonical β-catenin-dependent pathway and several noncanonical β-catenin-independent pathways.Citation210 These Wnt cascades play an important role in embryonic development processes, as well as in carcinogenesis.Citation211 The noncanonical Wnt signaling pathway, which operates independently of β-catenin-mediated transcription (), is separated into the Wnt planar cell polarity and Wnt/Ca2+ signaling branches.Citation212 Moreover, this pathway appears to function independently of transcription ().
Figure 5 Wnt pathways.
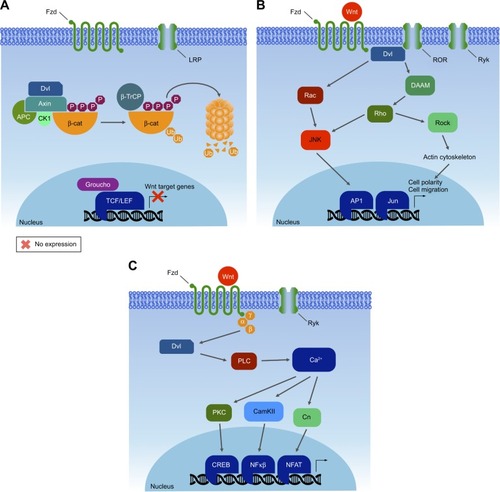
The canonical pathway was first identified and delineated from genetic screens in Drosophila melanogaster, and intensive studies in worms, frogs, fish, and mice have led to the identification of a basic molecular signaling framework.Citation213 Briefly, the canonical Wnt pathway is defined by the translocation of β-catenin into the nucleus, where it acts as a coactivator of transcription. Wnt ligands bind to an Fzd and a member of coreceptors LRP5/6. Intracellular signaling proceeds via the Axin and Dvl proteins (). These in turn lead to inactivation of the β-catenin destruction complex, which causes β-catenin to be stabilized and relocated into the nucleus. Within the nucleus, β-catenin forms a complex with the TCF/LEF transcription factors, triggering the expression of specific target genes.Citation214,Citation215 This transcriptional activity determines cell fate decisions, survival, and proliferation. The precise targets may vary among cell types, but in some cases can include the oncogenes c-Myc and cyclin D1.Citation216 In the new model, Axin stabilizes the destruction complex in both the presence and absence of WNT, and β-catenin is degraded through phosphorylation-mediated recognition by β-TrCP in the intact complex.Citation212 This allows newly synthesized β-catenin to accumulate in the cytosol before nuclear translocation (). Despite the fact that major pathway components have been characterized, the function of Wnt signaling within the context of cancer biology is intriguingly complex and remains only partially understood.
Figure 6 Canonical Wnt signaling pathway.
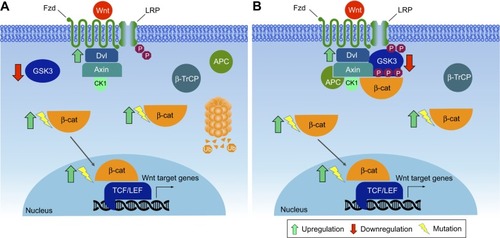
Wnt in mammary tissue development
Several components of Wnt signaling have been documented during various stages of morphogenesis of the mammary gland, including a wide range of Wnt ligands, receptors, downstream effectors, and DNA-binding proteins.Citation212 Functionally, it is clear that canonical Wnt signaling is necessary for the initiation of mammary development. Dkk1 expression prevents localized expression of all mammary placode markers.Citation217 Wnt6 is initially expressed on the surface ectoderm as a broader band surrounding the Wnt10-expressing mammary lines.Citation209,Citation218 In addition, Wnt4 mediates progesterone-induced ductal side branching in early pregnancy.Citation219 Contrarily, the expression of Axin, which acts to destabilize β-catenin, causes defective alveolar formation during pregnancy.Citation220 β-Catenin also plays important roles in mammary development, such as cell adhesion, signal transduction, and regulation of gene expression, which are essential for mammary SC biology during mammary development.Citation221
Noncanonical Wnt signaling has also been shown to take part in the negative regulation of mammary epithelial outgrowths, adding another level of complexity.Citation209 For example, loss of Wnt5a during other polarized morphogenic events increases cytoplasmic and nuclear β-catenin and accelerates ductal outgrowth.Citation222 Importantly, Wnt5a is an essential mediator of TGFβ, suggesting that low thresholds of β-catenin signaling are maintained during pubertal ductal morphogenesis through TGFβ and Wnt5a antagonism.
Wnt and breast cancer
Given the importance of Wnt signaling for adult SC biology, it is not surprising that Wnt pathway mutations are observed frequently in cancer, most notably in tissues that normally depend on Wnt for self-renewal or repair.Citation223 Mutations including β-catenin, Axin, or other Wnt pathway components, which result in β-catenin accumulation, are found in breast tumorigenesis.Citation224–Citation226
Wnt signaling is activated in over 50% of breast cancer and linked to reduced overall survival.Citation227 Deregulated Wnt signaling leads to increased SC renewal, proliferation, migration, and survival.Citation228 Studies have shown that molecular subtypes of breast cancer exhibit different levels of Wnt pathway activity.Citation229 Solid human tumors, including those of the breasts, prostate, liver, and ovaries, have increased levels of cytoplasmic and nuclear β-catenin, indicating over-expression of Wnt signaling.Citation230,Citation231 In mice, overexpression of Wnt1, Wnt3, or Wnt10 leads to mammary carcinomas.Citation209,Citation232 Increased expression of Dvl1, a cytosolic positive regulator of Wnt signaling, is prominent in primary breast cancers.Citation233,Citation234
Investigations have shown that noncanonical Wnt members, such as Wnt5a receptors, are connected to breast cancer metastases,Citation235 as well as to shorter overall survival of breast cancer patients.Citation183,Citation236,Citation237 Immunohistochemistry expression profiling in 90 triple-negative breast cancer specimens has shown that low levels of Wnt5a expression are associated with positive lymph node metastasis, enhanced cell motility, Ki67 proliferation, and significantly worse recurrence-free survival.Citation238,Citation239 Modulation of receptor activation could be another factor for hyperactive Wnt signaling in breast cancer.Citation209 An alternative splicing of LRP5, which removes the coding region of LRP5 that interacts with the secreted Wnt signaling repressor Dkk1, has been reported in parathyroid and breast cancer.Citation240
Metastasis is a trademark of late-stage cancer and a main challenge to therapy. A main adaptive change of tumors during therapy is EMT.Citation241 EMT is a known example of epithelial plasticity that is important in cancer metastasis.Citation242 During tumor progression, EMT allows benign tumor cells to infiltrate the surrounding tissue and metastasize to distant sites.Citation243 Activation of canonical Wnt signaling stabilizes Snai2 by inhibiting GSK3βactivity and initiates EMT transcriptional programs in breast cancer cells.Citation244 Another candidate gene that regulates EMT is ASPP2, which encodes a protein that binds and inhibits the N-terminal phosphorylation of β-catenin, leading to its stabilization. Decreased expression of ASPP2 leads to EMT and is correlated with low survival in hepatocellular and breast cancer.Citation242
Identification of novel biomarkers would be a promising approach for developing new diagnostic and therapeutic strategies. Markers that can predict the site of metastasis are scarce, and there are few reports on the utility of evaluating Wnt signaling members in the diagnosis of cancer.Citation245 Research has suggested Dkk1 as a diagnostic biomarker for a wide variety of cancers, including breast cancer, implicating the therapeutic potential of anti-Dkk1 antibody to neutralize the activity of Dkk1 function for cancer cell invasion and growth.Citation246 The majority of cancer patients presented elevated Dkk1 levels compared to healthy controls, and thus confirmed previous data supporting the usefulness of Dkk1 as a serological biomarker of cancer.
Wnt1-expressing mouse mammary epithelial cells show transcriptional upregulation of COX2; however, it is not clear whether this is due to direct regulation of the COX2 promoter by β-catenin.Citation247 β-catenin is expressed in the nucleus, cytoplasm, and/or mesenchyme of 36.1% of breast cancer patients.Citation248 Therefore, β-catenin status might serve as a predictive biomarker in breast cancer.
Clinical targeting of the Wnt pathway
Despite significant effort, there are no drugs currently approved for clinical use in breast cancer, which mainly target members of the Wnt pathway. However, a few small-molecule groups that target modulators of Wnt-related genes has emerged. Among these, the leading group are the so-called Porcupine inhibitors (LGK974 [Novartis]).Citation249 Porcupine is a membrane-bound O-acetyltransferase that is required for palmitoylation of Wnt ligands, a necessary step in the processing and secretion of Wnt ligands.Citation249 LGK974 is now in clinical Phase I trials for melanoma, breast cancer (triple-negative), pancreatic adenocarcinoma, colorectal cancer, and head and neck cancers.
Transforming growth factor beta
The TGFβ pathway
TGFβ was first isolated in 1982 in a search for secreted autocrine growth factors capable of transforming normal fibroblasts into malignant cells, which can proliferate in the absence of normal growth controls.Citation250,Citation251 The purified factor promoted wound healing by stimulating production of extracellular matrix factors and vascularization. At the same time, growth inhibitory effects of purified TGFβ were clearly demonstrated,Citation252 leading to the paradox of TGFβ playing a role in both promoting and inhibiting cell growth.Citation253 TGFβ is expressed in nearly all tissue, and the cellular response to TGFβ is highly dependent on the different contexts. The role of TGFβ has been most often studied in models for immunosuppression and inflammation, extracellular remodeling, cell proliferation, differentiation, survival, and invasion, all of which are paramount in cancer onset and progression.Citation254
TGFβ signal transduction is carried by canonical and noncanonical pathways. In the canonical pathway, the TGFβ extracellular ligand binds the TGFβ2-membrane receptor complex, which contains a cytoplasmic serine/threonine kinase domain that in turn phosphorylates receptor-regulated SMADs (homologue of Drosophila MAD), notably SMAD2 and SMAD3, to transduce the activating signal. SMAD2/3 dimerizes with the common mediator SMAD4, and this activated complex translocates into the nucleus to participate as a coactivator or corepressor of targeted gene transcription,Citation255 leading to the propagation of a diverse variety of TGFβ-induced responses in their specific contexts. TGFβ also triggers the Snail family of transcriptional repressors, which includes Snai1, Snai2/Slug, Twist1, ZEB1, and ZEB2.Citation256
TGFβ in mammary tissue development
Because the ubiquitously expressed TGFβ provokes many different phenotypes in different tissues, attributing specific outcomes to TGFβ is a complex exercise that largely depends on cell context and timing. Much of the insight into the role of TGFβ in mammary gland development has been achieved through experiments using ectopic TGFβ treatment of explants and transgenic mouse models. Early experiments showed that implanted slow-release pellets of TGFβ inhibited mammary ductal branching and proliferation at the mammary end buds.Citation257,Citation258 Consistent with this observation, transgenic mice expressing TGFβ driven by an MMTV enhancer/promoter showed suppressed mammary ductal tree development, though lactation was unaffected and no spontaneous tumors emerged in this model.Citation259 The converse inhibition of TGFβ using dominant-negative transgenic mice resulted in increased ductal tree side branching and proliferation.Citation260 In heterozygous TGFβ mice, which express <10% of normal TGFβ expression levels, mammary ductal and alveolar development is accelerated,Citation261 consistent with a fundamentally inhibitory role for TGFβ during mammary development. When TGFβ expression was directed specifically to alveolar cells by WAP gene regulatory elements, lobuloalveolar development was impaired and milk production inhibited.Citation262 Transgenic transplantation experiments revealed that the mammary epithelium itself was defective, ruling out possible trans-signaling effects.Citation263 Mammary explants treated with TGFβ also show suppression of milk production.Citation264 After lactation, the involution process of the mammary gland correlates with TGFβ expression and other apoptotic regulatory genes.Citation265,Citation266 An inhibitory role for TGFβ in ductal morphogenesis and alveolar development and function and a promotional role for TGFβ in mammary gland involution and remodeling have been clearly established; however, mechanisms and downstream intracellular factors remain unclear.
TGFβ and breast cancer
In cultured cells, TGFβ is a potent inducer of EMT, a prerequisite for metastasis and cancer progression. EMT precedes invasion of the vasculature, transport in circulation, penetration of the basement membrane of a distal tissue, and finally metastatic tumor formation. During the complex, stepwise process of EMT, changes in tissue architecture correspond to changes in cellular functions. Epithelial cells lose cell–cell adherens junctions and apical–basal polarity. The resulting mesenchymal cells acquire a spindle-like morphology, with increased migration and invasive potential. Expression of specific structural and differentiation genes changes as well, such as from cytokeratins and E-cadherin in epithelial cells to vimentin and N-cadherin in mesenchymal cells.
Exposure of mammary carcinoma cells to exogenous TGFβ in the absence of inhibitory antibodies increases cell invasion potential in migration assays and lads to lung metastasis in rodents.Citation267 Immunohistopathological analysis of human breast carcinomas reveals higher levels of extracellular TGFβ protein, especially at the advancing edges of infiltrating mammary duct carcinomas and in lymph node metastases.Citation268,Citation269
TGFβ has been shown directly to promote EMT in breast cancer in both cell cultures assays and mouse models. Exogenous TGFβ induces Ras-transformed mammary epithelial cells into EMT and secretion of TGFβ in these cells in an autocrine loop, and maintains the mesenchymal state.Citation270 TGFβ1 and TGFβ3 ligands have been identified in a screen for genes that cooperate with ErbB2/HER2 in activating EMT in a mammary epithelial cell migration assay. Addition of ectopic TGFβ recapitulated this migration in a dose-dependent manner.Citation271 Transgenic mouse models expressing both activated ErbB2/HER2 and TGFβ have also revealed modulation of the invasiveness and metastasis of mammary tumors in vivo that was dependent on the expression or repression of TGFβ.Citation272–Citation274
TGFβ affects EMT via downstream canonical SMAD-dependent pathways, as well as through noncanonical signaling and cross talk with other pathways.Citation255 In mammary epithelial cells, the TWIST1 and SNAI1 genes are induced by activated SMADs, and subsequently repress transcription of key genes involved in maintaining epithelial identity and integrity, notably E-cadherin, provoking an EMT transition.Citation275,Citation276 Snail protein expression is found in duct-infiltrating breast carcinomas, presenting lymph node metastases and is inversely correlated with the grade of tumor differentiation.Citation277 Knocking down TWIST inhibits metastases of mammary carcinomas to the lung, and high levels of Twist expression are found in highly invasive infiltrating lobular carcinomas: breast cancers that have lost E-cadherin expression.Citation278
A clear role for noncoding RNA in regulating TGFβ-induced EMT in breast cancer has been established.Citation279,Citation280 Several members of the miR200 family (miR-200f) play a tumor suppressor role by inhibiting the Snail family transcriptional repressors ZEB1 and ZEB2.Citation281–Citation283 High expression of miR200f sequences targets the TGFβ pathway genes ZEB1/ZEB2, SMAD2, SMAD5, SNAI1, and others, increases E-cadherin expression, reduces cell motility, and restores an epithelial phenotype to mammary carcinoma cells in vitro.Citation282,Citation284 Conversely, ZEB1 and ZEB2 are capable of suppressing miR200f clusters, creating a negative feedback loop with TGFβ promoting EMT and miR-200 antagonizing.Citation281 A similar feedback loop has been established between miR34 and Snail in a breast carcinoma cell line.Citation285
Breast carcinomas exhibit a spectrum of both epithelial and mesenchymal phenotypes, and this nonbinary mix of features in tumors has given rise to the notion of partial EMT, describing an intermediate state where conversion to either epithelial or mesenchymal states is possible and reversible.Citation286 Such plasticity between EMT states complicates our understanding of in vivo cellular processes during metastatic invasion, as the context of each specific tumor microenvironment often determines the outcome. The advent of molecular gene expression profiling has led to a narrowing identification of the cells of origin of breast cancer and suggested new ways to classify them. This research has intersected with studies on EMT, mammary SCs, and TGFβ.
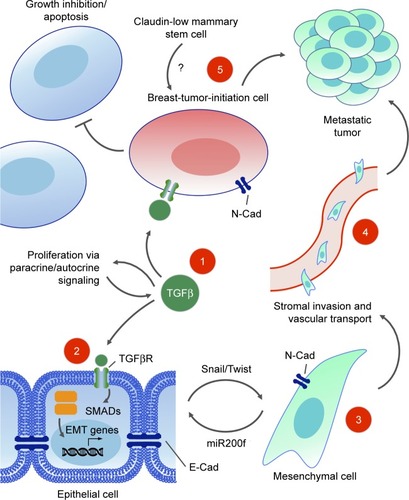
A subpopulation of breast cancer cells with increased tumorigenic potential has been isolated by cell sorting using cell surface markers. These CD44+/CD24− cells were called tumor-initiating cells, due to their higher potency in initiating tumors when xenografted into immunocompromised mice.Citation287 A gene expression profile revealed that the TGFβ pathway genes were specifically expressed and activated in this CD44+/CD24− cell population. Furthermore, the gene expression signature of CD44+/CD24− cells resembled that of normal mammary SCs and putative breast cancer SCs (CSCs) more than they did other subpopulations of sorted breast cancer cells.Citation288
Gene expression signatures from the CD44+/CD24− tumor-initiating cell population and a mammary SC population were compared in cells forced to undergo EMT, and found to share specific expression of SC markers, as well as EMT mesenchymal markers. SC-like properties, such as ability to undergo multipotent tissue differentiation, were observed in both populations.Citation289 An autocrine–paracrine feedback loop maintains the mesenchymal, stemlike state in these tumor-initiating cells.Citation290 From this point forward, the concept of a breast CSC or breast tumor-initiating cell (BTIC) was proposed, describing a distinct subpopulation of cells with SC-like self-renewing properties, expressing mesenchymal markers, including those involved in TGFβ signaling, which are capable of effecting tumor progression through invasion, migration, intravasation, and metastasis.
Sorting of cell populations by flow cytometry and grouping by functional gene signature profiles has allowed researchers to begin reconciling apparent contradictions in the growth-promoting vs -inhibiting effects of TGFβ signaling. Instead of a temporal switch from a cytostatic role of TGFβ toward a tumorigenic role in EMT and metastasis, both activities can take place in different cells of the same tumor cell population. CD44+/CD24−/claudin-low subpopulations of tumor and normal mammary epithelial cells respond selectively to exogenous TGFβ treatment by expansion of their BTIC or basal/SC population, whereas the BTICs of other sorted populations remain static or are reduced.Citation291 In ER+ breast cancers, differing TGFβ/SMAD3-driven gene expression signatures are generated that are able to uncouple the tumor suppressive effects of TGFβ in patient cohorts with good clinical outcomes from the tumor-promoting activities of TGFβ with poorer outcomes.Citation292 In the near future, greater knowledge about specific gene signatures from individual tumors will be informative for the choice of therapy, such as employing TGFβ antagonists or not. The epigenetic landscape will play an increasingly important role in determining TGFβ-induced phenotypes within specific cell contexts and cancer subtypes. The epigenomes of BTIC-promoting vs BTIC-suppressing breast cancer cells differentially determine the subset of target genes that can be bound and activated by TGFβ/SMAD3, adding another layer of complexity to context-dependent signaling by TGFβ ().Citation293
Figure 7 TGFβ signaling in breast cancer progression
Clinical targeting of the TGFβ pathway
TGFβ has become a popular target for drug development in cancer therapy. In addition to EMT and growth inhibition, TGFβ signaling is also involved in normal tissue homeostasis, extracellular matrix regulation, and immunoresponse modulation. With such highly pleiotropic activities of TGFβ, greatly influenced by specific contexts of cells, tissues, and architecture in complex processes, developing specific therapies to target TGFβ for a single disease presents a formidable challenge.Citation294 The attraction of targeting TGFβ would be to antagonize its EMT-promoting activities and BTIC-promoting properties, as well (perhaps) as to reduce interstitial fluid pressure to improve the efficiency of drug delivery.Citation295 Reducing TGFβ ligands or TGFβ signaling is seen as the goal, rather than direct ablation of TGFβ-inducing or -responding cells, so the efficacy of a therapy can only be assessed in a systemic in vivo setting, rather than via cytotoxicity screens on cells in vitro.
Two therapies against metastatic breast cancer have entered clinical trials. Fresolimumab, a humanized inhibitory antibody directed against the TGFβ ligand, is currently in a Phase II trial (NCT01401062) and administered in conjunction with radiotherapy. Eli Lilly has developed the drug LY2157299 (galunisertib), which targets the TGFβ1 receptor.Citation296 This small-molecule inhibitor is also administered in conjunction with radiotherapy, and is currently recruiting participants (NCT02538471). LY2157299 has been able to inhibit TGFβ signaling and consequently tumor formation in a human-derived mouse model of glioblastoma.Citation297 In another promising study, the TGFβ inhibitor LY2157299 was used in parallel with inhibitory antibodies against TGFβ2 receptors and SMAD4 RNAi. Following chemotherapy with the taxane paclitaxel, these TGFβ inhibitors blocked tumor reinitiation in a mouse model of triple-negative breast cancer by suppressing the expansion of BTICs.Citation298 Such therapies combining TGFβ inhibitors with chemotherapeutic drugs show the most promise, as TGFβ activation in BTICs may promote drug resistance and cytotoxic chemotherapeutic drugs may enrich drug-resistant TGFβ-dependent BTIC populations, which contribute to tumor reinitiation.Citation299–Citation301
Nuclear factor-κ light-chain enhancer of activated β-cells
The NFκB pathway
NFκB was originally discovered as a nuclear factor that specifically binds to a 10-base-pair DNA sequence (5′-GGGA CTTTCC-3′) within the enhancer of the Igκ light chain of activated β-cells.Citation302 Currently, NFκB is largely known to be involved in cell cycle regulation, immunoresponse, inflammation, proliferation, and cell death. In mammals, the NFκB transcription factor family is composed of five members, divided in two classes based on the sequences of the C-terminal domain. Members of one class – the NFκB proteins NFκB1/p105 and NFκB2/p100 – have long C-terminal domains that contain multiple copies of ankyrin repeats, which inhibit these molecules until they are processed by either limited proteolysis or arrested translation into p50 and p52, respectively.Citation303 Although p52 and p50 lack the transcription activation domains, they can positively regulate transcription through heterodimerization with members of the second class: the Rel subfamily, composed of cRel, RelB, and RelA (p65), which contain transcription activation domains that confer the ability to initiate transcription.Citation304 They all share an Rel homology region, which mediates specific DNA binding to the NFκB consensus sequence and dimerization and interaction with IκB inhibitory molecules.Citation305–Citation308
Prior to stimuli, NFκB dimers are bound to inhibitory molecules of the IκB protein family (eg, IκBα, IκBβ, IκBγ, IκBε), which keep NFκB complexes inactive in the cytoplasm and block their binding capacity toward DNA.Citation309 The classical (or canonical) NFκB pathway is induced by inflammatory cytokine signaling, DNA damage, or infectious agents. This stimulation leads to activation of the IκB kinase (IKK) complex (composed of IKKα/IKK1 and IKKβ/IKK2 and the scaffolding protein IKKγ/NEMO),Citation310,Citation311 which phosphorylates Iκα and then allows the E3 ubiquitin ligase βTrCP to promote polyubiquitination and further proteasome-dependent degradation of IκB.Citation312–Citation314 As a result, liberated NFκB rapidly enters the nucleus, where it can activate the expression of specific genes that contain DNA binding sites for NFκB (κB enhancer sites).Citation315 An alternative (or noncanonical) pathway of NFκB activation is activated by signals transmitted by a subset of immunorelated receptors, including BAFFR, LTβR, CD40, RANKL, TNFR2, and Tweakr.Citation316 This alternative induction mechanism leads to activation of NFκB inducing kinase (NIK), which pre-dominantly phosphorylates and activates IKKα. Activated IKK phosphorylates p100, resulting in its ubiquitination and partial processing to p52.Citation317 Following processing, alternate NFκB dimers (mainly composed of cRel and p52) are released to translocate further to the nucleus and act as transcription factors, thereby promoting expression of NFκB-related target genes. Nevertheless, binding of NFκB to the respective κB sites does not ensure transcriptional initiation, which in fact requires the interaction of NFκB with a few mediators, such as transcriptional adaptors and coactivator proteins like CBP and its paralogue p300, for assembly of the transcription machineryCitation318 ().
Figure 8 NFκB pathway.
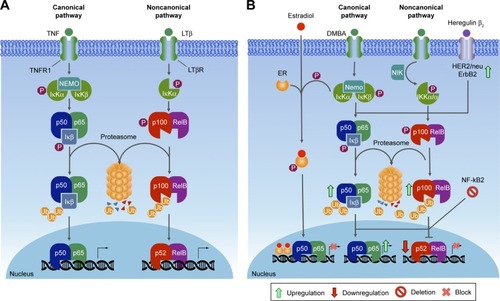
NFκB in mammary tissue development
Expression of the NFκB subunits, such as p50, p52, p65 (RelA), and RelB, as well as the prototypical inhibitor IκBα, follows a consistent pattern during mouse mammary tissue development, being typically high during pregnancy, diminished during lactation, and elevated again in breast involution.Citation319–Citation323 NFκB acts as a negative regulator of β-casein gene expression during pregnancy in mice by interfering with tyrosine phosphorylation of STAT5, blocking a premature differentiation of the gland.Citation320 Ablation of IκBα in the mammary epithelium results in hyperplasia and increased lateral ductal branching in virgin mice, suggesting that NFκB modulates proliferation, branching, and normal structural development of the mammary epithelium during early postnatal morphogenesis.Citation324 IKKαAA/AA mice bearing a serine–alanine mutation that prevents its activation, fail to develop lobuloalveolar tissue properly, which is caused by impaired RANKL-mediated induction of cyclin D1, which is encoded by an NFκB target gene.Citation325 Furthermore, a deletion of the gene for IKKβ in the mammary gland reduces apoptosis and delays involution, suggesting a proapoptotic role for IKKβ,Citation326 whereas its constitutive expression leads to a reduction β-casein levels, accompanied by detection of cleaved caspase 3 during lactation and involution.Citation327 In immortalized cultures of KIM2 cells, lactogenic hormone deprivation leads to increased NFκB activity, suggesting a survival role for NFκB in mammary involution.Citation321 The p65–p300 complex is associated with modulation of inflammatory responses 48 hours after weaning.Citation328 High levels of nitric oxide (NO) are detected during the first hours of involution, when NFκB binds to the NOS2 promoter to modulate its transcription.Citation329 Collectively, these data demonstrate the importance of NFκB signaling in the development of normal mammary glands.
NFκB and breast cancer
Abnormally high NFκB activity is a clinical hallmark of chronic inflammation, being detected in a vast number of cancers. However, this information has to be taken with caution, since NFκB may act either as a tumor suppressor or a tumor promoter, depending on the type of cancer.Citation330,Citation331 The role for NFκB as a tumor promoter arises from its aberrant activation and nuclear localization as a result of defects in regulation of the pathway, loss of negative feedback mechanisms, resistance to cell death, and influence of the tumor microenvironment.Citation331 However, other observations suggest that NFκB can also inhibit tumor growth. Tumor suppressor proteins, such as p53 and ARF, can induce the association of NFκB subunits with HDAC1 corepressor complexes.Citation332 This results in NFκB-dependent repression of target gene expression, providing a model through which NFκB subunits can facilitate apoptosis and cell cycle arrest and thus function as tumor suppressors themselves. This suggests a dual function for NFκB during tumor progression, inhibiting tumor growth, but as further mutations lead to a loss of tumor suppressor expression, the oncogenic functions of NFκB become unleashed, contributing to tumorigenesis.Citation330 The tumor inflammatory microenvironment maintains NFκB constitutively active in most tumor cells, and NFκB has been found to control multiple cellular processes in cancer, such as inflammation, transformation, proliferation, angiogenesis, invasion, metastasis, chemoresistance, and radioresistance. Therefore, NFκB suppression may have a positive impact by inhibiting the growth of tumor cells.
Though NFκB is required for normal mammary gland morphogenesis, its deregulated activation has been linked to tumor progression via stimulation of cell proliferation and survival and angiogenesis pathways and metastasis, ultimately driving breast carcinogenesis. In vivo and in vitro experiments in female rats, mice, and human mammary epithelial cell lines treated with a carcinogen, such as 7,12-dimethylbenz[a]anthracene, showed elevated activity of NFκB prior to malignant transformation and tumor formation.Citation333,Citation334 Elevated NFκB expression has been widely reported in human breast cancer cell lines and tissues, with abnormal constitutive expression of both canonical and noncanonical NFκB subunits, such as cRel, p65, p50, and p52.Citation335–Citation338 Several reports have observed a positive correlation between NFκB activation and the ErbB2 receptor, also known as HER2/Neu.Citation338–Citation340 In addition, NFκB is predominantly activated in ER− and ER−/HER2+ breast cancers, suggesting its importance in specific classes of breast cancers.Citation335,Citation340 Progression of ER-dependent to ER-independent breast cancer phenotype is correlated with an increase in NFκB activity. In an E2-independent breast ductal carcinoma cell line (MCF7/LCC1) model, constitutive activation of NFκB was observed prior to the loss of ER expression.Citation341 In ErbB2-expressing cell lines, administration of the ErbB2 activator heregulin β1 led to increased NFκB activation, whereas herceptin, an anti-ErbB2 monoclonal antibody, blocked this activation and led to apoptosis.Citation340 Phosphorylation of a tumor suppressor protein that regulates NFκB activation (CYLD) by the breast cancer oncogene kinase IKBKECitation342 promotes cell transformation.Citation343 In vitro assays have revealed that constitutive NFκB activity in breast cancer cells stimulates osteoclastogenesis by upregulating GM-CSF. Blocking NFκB suppressed osteolytic lesion development through osteoclastic bone resorption in vivo.Citation344
Together with oncogenic RAS, NFκB activity is required for protection of mammary epithelial cells from TGFβ-induced apoptosis. This protection is a prerequisite for these cells to undergo EMT toward an invasive, metastatic tumor phenotype.Citation345 In a triple transgenic model in which NFκB activity can be inhibited by doxycycline treatment in defined windows during PYMT-induced tumorigenesis, inhibition of NFκB signaling leads to increased tumor latency and decreased tumor burden.Citation327
BRCA1-deficient breast tumors exhibit aggressive behavior and are associated with poor survival.Citation346 Within BRCA1-deficient mammary glands, there is a subset of cells named luminal progenitor cells, which can form 3D colonies in a hormone-independent fashion.Citation347 In these cells, constitutive NFκB (cRel/p52) activation due to DNA damage caused by progesterone leads to hormone-uncoupled proliferation, increasing the risk of genomic instability and transformation.Citation348
Self-renewing breast CSCs are key players in perpetuating tumor maintenance and in treatment resistance and relapse. Interestingly, in HER2-dependent tumors, the proportion of CD44+ cells (marker for SC population) dramatically decreased upon NFκB suppression, indicating that this transcription factor is important in the maintenance of progenitor cell expansion.Citation349 Another study showed that repression of CD44, a cell surface glycoprotein, via NFκB inhibition consequently decreased proliferation and invasiveness of breast cancer cells.Citation350 Silencing of NFκB2/p100 in mammary cancer cell lines led to loss of p100 and p52 and to context-dependent effects on tumorigenicity of these cells.Citation351 In 4T1 mammary carcinoma cells, increased activity of NFκB (which induces EMT and CSCs) was a result of p100 loss, whereas in N202.1A mammary carcinoma cells, CSCs and tumorigenicity were positively regulated by p52, suggesting an opposite role for this protein in regulation of breast CSCs, and indicating that inhibiting p100 processing may be a potential therapeutic strategy to suppress CSC activity in a subset of breast tumors.Citation351
Clinical targeting of the NFκB pathway
Targeting the NFκB pathway seems to be a reasonable objective in view of the numerous implications of this pathway in diverse diseases, including breast cancer. Combinatorial therapeutic strategies, targeting members of the PI3K/Akt and MAPK pathways that lead to NFκB activation, have already been employed in several clinical trials.Citation352 Association between ErbB2 expression and NFκB activation and the inverse correlation found between the latter and ER status may be exploited in human breast cancer therapy.Citation353 Other agents, such as specific IKK inhibitors, are under investigation. Inhibition of TBK1, a noncanonical IKK, with TBK1-II, a drug that efficiently inhibits TBK1 and IKKε, suppresses growth of human HER2+ breast cancer cells and induced cellular senescence.Citation354 Inhibition of IKKs sensitizes cells to the cytotoxic effect of doxorubicin in different breast cancer cell lines.Citation355 In addition, inhibition of NFκB DNA binding is a good putative approach to inhibit NFκB activity, since this would prevent transactivation of prosurvival and antiapoptotic downstream targets, being highly selective. More than 780 compounds described to have NFκB inhibitory activityCitation356 could be used in combinatorial therapy for breast cancer. Compounds such as the proteasome inhibitor bortezomib have undergone clinical trials in breast cancer patients with some success.Citation357,Citation358 However, unselective pathway disruption potentially leads to adverse effects, such as immunosuppression, which could be particularly harmful in cancer patients, who are likely to be immunocompromised due to previous treatments.Citation359
Signaling integration (crosstalk) within breast cancer
Breast cancer is heterogeneous in nature, due to a slew of aberrations at the genomic and molecular levels affecting various signaling pathways. To understand the integrative role of these pathways in different physiological and pathological conditions, it is necessary to take into consideration their complex cross talk. The best example of cross talk in the ER signaling pathway is the signalosome, a large protein complex present at the membrane. Upon activation by HER2, ERK1/2 phosphorylates ER, thus increasing its sensitivity for its ligands or leading to ligand-independent activation. In addition, ER coregulators can be modified by kinases, thereby indirectly affecting ER activity.Citation107,Citation360,Citation361 Also GPR30, which colocalizes with ERα in the membrane, collaborates in transmission of signals to downstream kinase cascades.Citation45
The importance of signaling cross talk in cancer is also illustrated by the influence of estrogen signaling in EMT. Cross talk between ERα and several EMT regulators, such as Snail and Slug, leads to increased EMT.Citation362 Additionally, ER signaling can mediate delocalization of E-cadherin and cytoskeleton reorganization via the ER–Src–PELP1–PI3K–ILK1 pathway. Therefore, this cross talk can affect breast cancer progression, leading to metastasis via EMT and contributing to breast cancer cell motility.Citation102 Most of the nongenomic mechanisms in the ER pathway are implicated in breast cancer metastasis, with ERK and Akt phosphorylation being involved in breast cancer cell migration and Src and ILK1 kinases playing critical roles in cell invasion and metastatis.Citation361
PI3K signaling also exerts downstream control on other transcriptional factors, which include members of the FOX protein family (FOXO1, FOXO2, FOXO4, and FOXO6) and NFκB. In addition, PI3K signaling can inhibit p53 activity by a mechanism that involves the E3 ubiquitin protein ligase MDM2, regulating the cell cycle, inflammation, apoptosis, and DNA repair.Citation122 LKB1, a kinase located upstream of AMPK, negatively regulates mTORC1 signaling by tuberous sclerosis 1 or 2.Citation116,Citation117 PKC (activated in the PI3K pathway) has the potential to phosphorylate Raf1. PAK1 stimulated by PI3K and/or CDC42 pathways can also phosphorylate Raf1. Upon stimulation by PI3K, Akt can phosphorylate and inhibit both B-Raf and Raf1.Citation153,Citation363
NFκB-dependent transcription is not only tightly controlled by positive and negative regulatory mechanisms but also closely coordinated with other signaling pathways. Since activation depends on IκB degradation, the IKK complex is the gatekeeper of NFκB signaling, representing a critical node for interaction within parallel signaling pathways. Other upstream molecules, such as receptor interacting proteins and TNFR, are critical to IKK activation, but also signal other pathways. Thus, TNFR-associated factors represent a central point of divergence for activation of NFκB and AP1 transcription factor pathways.Citation364
MAPKs, JNK, ERK1/ERK2, and p38 promote activation of AP1, and activated AP1 affects cell survival, apoptosis, and stress responses. Another molecule, RIP1, has been described as being involved in activation of the PI3K–Akt pathway through not only NFκB-dependent negative regulation of expression of the mTOR kinase but also through NFκB-independent downregulation of the PI3K antagonist PTEN.Citation365 The IKKβ-induced degradation of p105 may potentially influence several additional pathways, since IKKβ can also phosphorylate Dok1, an Ras GAP-associated TK substrate and cell growth inhibitor, leading to inhibition of ERK1 and ERK2, demonstrating the context dependence of IKKβ-mediated MAPK regulation.Citation366 JAKs can phosphorylate MAPK, PIK3, and TGFβ associated receptors, thereby creating a site for anchoring proteins displaying SH2 domains in their structure. Upon phosphorylation, these proteins are able to stimulate a signaling cascade in their respective pathways.Citation367
It has been shown that ER activation can inhibit TGFβ-induced transcriptional activity and cell migration. Several lines of investigation have suggested that ERα is a major modifier of the TGFβ signaling pathway, with ER and TGFβ pathways being able to promote breast cancer metastasis.Citation368 Moreover, it is important to consider that the cross talk between Notch, Wnt, and SHH signaling pathways, as well as their role in regulating TICs, may also modulate breast cancer onset and progession.Citation369
Conclusion
Clearly, the risk for breast cancer is sexually dimorphic. In fact, breast cancer is about 100 times less common among men than in women. Still, the molecular role of sex hormones modulating breast cancer risk remains elusive. Elucidation of the function of ER in breast cancer cells should provide critical new insights into the function of these receptors. Expanding the vision of the entire network involving ER may lead to better design of specific therapies to treat breast cancer, with fewer side effects. It should also provide a better understanding of the mechanisms behind antiestrogen resistance. In vitro and in vivo models, clinical trials, and innovative treatments should contribute to better understanding of the function of each type of hormone receptor.
Besides the classic hormone signaling mechanisms, the pathways discussed in this review are shown to modulate central cellular functions that are critical in the onset and progression of breast cancer. The PI3K signaling pathway is important for mammary morphogenesis and mammary gland involution; however, alterations in the PI3K pathway are very common in several diseases, including Parkinson’s, obesity, type 2 diabetes, and different cancer types. Since several dysregulations in key nodes of this pathway are known to be associated with various disease states, the ability to pinpoint a specific alteration and to understand its functional relevance would allow for more accurate choice of treatment, with minimal side effects. Direct and indirect involvement of the MAPK pathway has been demonstrated for the development of breast cancer, participating in promotion of cell proliferation or rendering other pathways more active. JAK–STAT is one of the main signaling pathways that promotes communication of the extracellular medium with the cell nucleus. Wnt signaling regulates cell differentiation, proliferation, and SC pluripotency, and thus deregulation of Wnt signaling may lead to cancer development. TGFβ plays a dual role in breast cancer progression by either its tumor suppressing or tumor enhancing effects. NFκB represents a central factor in inflammation, stress response, and cell death, being recognized as an important element in several steps of cancer initiation and progression, activated by a large variety of stimuli, and displaying several downstream targets, thereby generating broad and complex feedback loops.
Several aspects of regulation of these pathways remain to be clarified, especially regarding their cross talk with other pathways, feedback, tumor microenvironment interactions, cell metabolism, risk factors, and response to drug therapy. However, it is clear that all of these pathways act as a large orchestrated network, and that a broad scope needs to be taken in planning future therapy strategies and designing prognostic tools. In this context, an increasingly detailed study of these pathways becomes even more important, since it may generate relevant and transformative knowledge regarding the nature of cellular communication, as well as the molecular basis for gene expression regulation in both normal and cancer cells. Therefore, identifying the range of mutations and/or molecular changes occurring in these major signaling pathways and how they interact across pathways is crucial for understanding breast cancer development, as well as for implementation of molecular-based approaches for continuous improvement in diagnostics, prognostics, and treatment of breast cancer patients.
Abbreviations
| AF-1 | = | N-terminal activation function 1 domain |
| AKT | = | Serine/threonine protein kinase 1 |
| AMPK | = | AMP-activated protein kinase |
| AR | = | Androgen receptor |
| Areg | = | Amphiregulin |
| ARF | = | ADP ribosylation factor |
| ASPP2 | = | Apoptosis-stimulating of p53 protein 2 |
| ATP | = | Adenosine triphosphate |
| BAFF-R | = | B-cell activation factor |
| BRCA1 | = | Breast cancer 1 supressor gene |
| BRCA2 | = | Breast cancer 2 supressor gene |
| BTIC | = | Breast tumor initiating cell |
| CBP | = | CREB-binding protein |
| CDC42 | = | Cell division cycle 42 |
| COUP-TFI | = | Chicken ovalbumin upstream promoter-transcription factor-1 |
| COX2 | = | Cyclooxygenase-2 |
| CSC | = | Cancer stem cell |
| CSF | = | Colony stimulating factor |
| CYLD | = | Cylindro-matosis human gene |
| DAAM | = | Dvl-associated activator of morphogenesis |
| DKK1 | = | Dickkopf-related protein 1 |
| DMBA | = | 7,12-Dimethylbenz[a]anthracene |
| DNA | = | Deoxy-ribonucleic acid |
| dsDNA | = | Double-stranded DNA |
| Dvl | = | Dishevelled |
| E2 | = | Estradiol:17β-estradiol |
| EGF | = | Epidermal growth factor |
| EGFR | = | Epidermal growth factor receptor |
| EMT | = | Epithelial-mesenchymal transition |
| ER | = | Estrogen receptor |
| ErbB2 | = | Human epidermal growth factor receptor 2 |
| ERE | = | Estrogen response element |
| ERK | = | Extracellular signal–regulated kinase |
| ERRα | = | Estrogen-related receptor alpha |
| ERRβ | = | Estrogen-related receptor beta |
| ERRγ | = | Estrogen-related receptor gamma |
| FAK | = | Focal adhesion kinase |
| FERM | = | Four-point-one motif, ezrin, radixin, moesin |
| FN14 | = | Fibroblast growth factor-inducible 14 |
| FOXO | = | Forkhead box O |
| FZD | = | Frizzled receptor |
| GATA3 | = | GATA biding protein 3 |
| GH | = | Growth hormone |
| GM-CSF | = | Granulocyte macrophage colony-stimulating factor |
| GOF | = | Gain-of-function |
| GPCR | = | G protein-coupled receptor |
| GPER1 | = | G protein-coupled ER 1 |
| GRB-2 | = | Growth factor receptor-bound protein 2 |
| GRB-7 | = | Growth factor receptor-bound protein 7 |
| GSK3β | = | Glycogen synthase kinase 3β, Gβγ, G beta-gamma complex |
| HDAC1 | = | Histone deacetylase 1 |
| HDM2 | = | Human double minute 2 protein |
| IFN | = | Interferon |
| IGF1 | = | Insulin-like growth factor 1 |
| IKK | = | IκB kinase complex |
| IKKα | = | Inhibitor of nuclear factor kappa-B kinase subunit alpha |
| IKKβ | = | Inhibitor of nuclear factor kappa-B kinase subunit beta |
| IL-10 | = | Interleukin 10 |
| IL-1β | = | Interleukin 1 beta |
| IL-6 | = | Interleukin 6 |
| IRS-1 | = | Insulin receptor substrate 1 |
| IκB | = | Inhibitor of kappa B |
| JAK | = | Janus kinase |
| JNK | = | Jun amino-terminal kinase |
| LBD | = | Ligand-binding domain |
| LOF | = | Loss-of-function |
| LRP | = | LDL-related receptor protein |
| LRP5/6 | = | Lipoprotein receptor-related protein |
| LTβR | = | Lymphotoxin β receptor |
| MAPK | = | Mitogen-activated protein kinase |
| MAPKKK | = | Mitogen-activated protein kinases kinases kinase |
| MCF7/LCC1 | = | Breast ductal carcinoma cell line |
| MEK | = | Mitogen-activated protein kinase kinase (or MAPKK, MKK) |
| MMTV | = | Mouse mammary tumor virus |
| mTOR | = | Mammalian target of rapamycin |
| NF-κB | = | Nuclear factor kappa-light-chain-enhancer of activated B cell |
| NGFI-B | = | Nerve growth factor induced-B |
| NIK | = | NF-κB inducing kinase |
| NO | = | Nitric oxide |
| NOS-2 | = | Nitric oxide synthase, inducible |
| NR | = | Nuclear receptor family |
| PAK1 | = | P21 (RAC1) activated kinase 1 |
| PCP | = | Planar cell polarity |
| PDK | = | Putative 3-phosphoinositide-dependent kinase (or PDPK1) |
| PGAP3 | = | Post-GPI attachment to proteins 3 |
| PI3K | = | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| PIK3CA | = | Phosphoinositide 3-kinase catalytic subunit α |
| PIP2 | = | Phosphatidylinositol-4,5-bisphosphate |
| PIP3 | = | Phosphatidylinositol-3,4,5-trisphosphate |
| PKB | = | Protein kinase B |
| PKC | = | Protein kinase C |
| PR | = | Progesterone receptor |
| proHB-EGF | = | Membrane-anchored heparin-binding EGF like growth factor |
| PTEN | = | Phosphatase and tensin homologue |
| PyMT | = | Polyomavirus middle T-antigen oncogene |
| RANK | = | Receptor activator for nuclear factor kappa B |
| RAPTOR | = | Regulatory-associated protein of mammalian target of rapamycin |
| RHR | = | Rel homology region |
| RIP1 | = | Receptor Interacting Serine/Threonine Kinase 1 |
| ROCK | = | Rho-associated kinase |
| RORα | = | Retinoic acid-related orphan receptor alpha |
| RTK | = | Receptor tyrosine kinase |
| SC | = | stem cell |
| SERD | = | Selective estrogen receptor degrader |
| SERM | = | Selective estrogen receptor modulator |
| SH2 | = | Src homology-2 |
| SHC | = | Src Homology 2 domain-containing-transforming protein C1 |
| siRNA | = | Small interfering RNA |
| SMAD | = | Contraction of SMA (small body size) and MAD (Mothers against decapentaplegic) |
| SNAI2 | = | SNAIL family transcriptional repressor 2 |
| SOS | = | Son of sevenless |
| SRC | = | V-Src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog |
| STAT | = | Signal transducer and activator of transcription |
| TAD | = | Transcription activation domain |
| TBK1 | = | TANK-binding kinase 1 |
| TCF/LEF | = | T-cell factor/lymphoid enhancer factor |
| TGFβ | = | Transforming growth factor β |
| Th17 | = | Helper T cells 17 |
| TNBC | = | Triple-negative breast cancer |
| TNFR2 | = | Tumor necrosis factor receptor 2 |
| TP53 | = | tumor protein 53 (or p53) |
| TRAF | = | TNFR-associated factor |
| TSC | = | Tuberous sclerosis |
| TWEAK-R | = | TWEAK receptor |
| TWEAK | = | tumor necrosis factor-like weak inducer of apoptosis |
| TYK2 | = | Tyrosine kinase 2 or tyrosine-protein kinase 2 |
| ULK1 | = | Unc-51-like kinase 1 |
| WNT | = | Wingless (wg)/integration 1 gene (int) |
| ZEB1 | = | Zinc finger E-box-binding homeobox 1 |
| ZEB2 | = | Zinc finger E-box-binding homeobox 2 |
| β-TRCP | = | Beta-transducin repeat containing E3 ubiquitin protein ligase |
Acknowledgments
We are grateful to the Department of Biochemistry from the Institute of Chemistry (IQ) of the University of São Paulo (USP, São Paulo, Brazil) for the academic support and opportunity to write this review. We thank the following Brazilian research funding agencies for the financial support: FAPESP (São Paulo State Foundation for Research), CNPq (National Research Council), and CAPES (Federal Agency for Superior Education and Training). MCS was additionally supported by grants from BNDES (Brazilian National Bank for Economic and Social Development), FINEP (Financiadora de Estudo e Projetos), MCTI (Science, Technology, and Innovation Ministry), MS-DECIT (Science and Technology Department of the Health Ministry), and Ouro Fino Saúde Animal Ltd. LFZ was supported by the International Centre for Genetic Engineering and Biotechnology (ICGEB). RGC was supported by a Special Visiting Researcher (PVE) grant from the “Science Without Borders” program (CAPES).
Disclosure
The authors report no conflicts of interest in this work.
References
- SiegelRLMillerKDJemalACancer statistics, 2017CA Cancer J Clin201767173028055103
- World Health OrganizationCancer2017 Available from: http://www.who.int/cancer/enAccessed July 14, 2017
- ParkinDMBrayFFerlayJPisaniPGlobal cancer statistics, 2002CA Cancer J Clin20055527410815761078
- DeSantisCMaJBryanLJemalABreast cancer statistics, 2013CA Cancer J Clin2014641526224114568
- MedinaDThe mammary gland: a unique organ for the study of development and tumorigenesisJ Mammary Gland Biol Neoplasia19961151910887477
- SharmaGNDaveRSanadyaJSharmaPSharmaKKVarious types and management of breast cancer: an overviewJ Adv Pharm Technol Res20101210912622247839
- MalhotraGKZhaoXBandHBandVHistological, molecular and functional subtypes of breast cancersCancer Biol Ther2010101095596021057215
- IqbalNIqbalNHuman epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implicationsMol Biol Int2014201485274825276427
- DaiXLiTBaiZBreast cancer intrinsic subtype classification, clinical use and future trendsAm J Cancer Res20155102929294326693050
- CareyLAPerouCMLivasyCARace, breast cancer subtypes, and survival in the Carolina Breast Cancer StudyJAMA2006295212492250216757721
- PerouCMSørlieTEisenMBMolecular portraits of human breast tumoursNature2000406679774775210963602
- SørlieTPerouCMTibshiraniRGene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implicationsProc Natl Acad Sci U S A20019819108691087411553815
- PratAParkerJSKarginovaOPhenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancerBreast Cancer Res2010125R6820813035
- SabatierRFinettiPGuilleAClaudin-low breast cancers: clinical, pathological, molecular and prognostic characterizationMol Cancer20141322825277734
- DiasKDvorkin-GhevaAHallettRMClaudin-low breast cancer: clinical and pathological characteristicsPLoS One20171210168669
- McPhersonKSteelCMDixonJMABC of breast diseases – breast cancer: epidemiology, risk factors, and geneticsBMJ2000321726162462810977847
- MikiYSwensenJShattuck-EidensDA strong candidate for the breast and ovarian cancer susceptibility gene BRCA1Science1994266518266717545954
- EastonDFordDPetoJInherited susceptibility to breast cancerCancer Surv199318951138013003
- GustafssonJAWhat pharmacologists can learn from recent advances in estrogen signallingTrends Pharmacol Sci200324947948512967773
- ShenMShiHSex hormones and their receptors regulate liver energy homeostasisInt J Endocrinol2015201529427826491440
- KnowltonAALeeAREstrogen and the cardiovascular systemPharmacol Ther20121351547022484805
- SanchezRNguyenDRochaWWhiteJHMaderSDiversity in the mechanisms of gene regulation by estrogen receptorsBioessays200224324425411891761
- McDonnellDPNorrisJDConnections and regulation of the human estrogen receptorScience200229655731642164412040178
- BourdeauVDeschênesJMétivierRGenome-wide identification of high-affinity estrogen response elements in human and mouseMol Endocrinol20041861411142715001666
- BolgerRWieseTEErvinKNestichSChecovichWRapid screening of environmental chemicals for estrogen receptor binding capacityEnviron Health Perspect199810695515579721254
- RuffMGangloffMWurtzJMMorasDEstrogen receptor transcription and transactivation: structure-function relationship in DNA- and ligand-binding domains of estrogen receptorsBreast Cancer Res20002535335911250728
- McBryanJYoungLLigand-independent signalling through estrogen receptor pathways in breast cancerLarionovAResistance to Aromatase Inhibitors in Breast CancerHeidelbergSpringer2015115144
- WeigelNLZhangYLigand-independent activation of steroid hormone receptorsJ Mol Med19987674694799660165
- CenniBPicardDLigand-independent activation of steroid receptors: new roles for old playersTrends Endocrinol Metab1999102414610322393
- AliSMetzgerDBornertJChambonPModulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B regionEMBO J1993123115311608458328
- MigliaccioADi DomenicoMCastoriaGTyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cellsEMBO J1996156129213008635462
- BunoneGBriandPAMiksicekRJPicardDActivation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylationEMBO J1996159217421838641283
- BologaCGRevankarCMYoungSMVirtual and biomolecular screening converge on a selective agonist for GPR30Nat Chem Biol20062420721216520733
- RevankarCMCiminoDFSklarLAArterburnJBProssnitzERA transmembrane intracellular estrogen receptor mediates rapid cell signalingScience200530757151625163015705806
- TengJWangZYJarrardDFBjorlingDERoles of estrogen receptor α and β in modulating urothelial cell proliferationEndocr Relat Cancer200815135136418310301
- LiuMMAlbaneseCAndersonCMOpposing action of estrogen receptors α and β on cyclin D1 gene expressionJ Biol Chem200227727243532436011986316
- PaechKDifferential ligand activation of estrogen receptors ER and ER at AP1 sitesScience19972775331150815109278514
- HallJMMcDonnellDPThe estrogen receptor β-isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogensEndocrinology1999140125566557810579320
- PoolaIKoduriSChatraSClarkeRIdentification of twenty alternatively spliced estrogen receptor alpha mRNAs in breast cancer cell lines and tumors using splice targeted primer approachJ Steroid Biochem Mol Biol200072524925810822014
- HerynkMHFuquaSAEstrogen receptor mutations in human diseaseEndocr Rev200425686989815583021
- TaylorSEMartin-HirschPLMartinFLOestrogen receptor splice variants in the pathogenesis of diseaseCancer Lett2010288213314819608332
- LiCBriggsMRAhlbornTEKraemerFBLiuJRequirement of Sp1 and estrogen receptor α interaction in 17β-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expressionEndocrinology200114241546155311250935
- FilardoEJQuinnJAFrackeltonARBlandKIEstrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axisMol Endocrinol2002161708411773440
- KumarPWuQChamblissKLDirect interactions with Gαi and Gβγ mediate nongenomic signaling by estrogen receptor αMol Endocrinol20072161370138017405905
- VivacquaABonofiglioDRecchiaAGThe G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cellsMol Endocrinol200620363164616239258
- ProssnitzERArterburnJBSklarLAGPR30: a G protein-coupled receptor for estrogenMol Cell Endocrinol2007265–266138142
- MarquezDCLeeJLinTPietrasRJEpidermal growth factor receptor and tyrosine phosphorylation of estrogen receptorEndocrine2001162738111887937
- RazandiMPedramAParkSTLevinERProximal events in signaling by plasma membrane estrogen receptorsJ Biol Chem200327842701271212421825
- SongRXZhangZSantenRJEstrogen rapid action via protein complex formation involving ERα and SrcTrends Endocrinol Metab200516834735316126407
- BaloghPSzabóAKatzSLikóIPatócsAKissALEstrogen receptor alpha is expressed in mesenteric mesothelial cells and is internalized in caveolae upon Freund’s adjuvant treatmentPLoS One2013811e7950824244516
- ChaudhriRASchwartzNElbaradieKSchwartzZBoyanBDRole of ERα36 in membrane-associated signaling by estrogenSteroids201481748024252378
- ShaulPWRegulation of endothelial nitric oxide synthase: location, location, locationAnnu Rev Physiol200264174977411826287
- DavisNMSokoloskyMStadelmanKDeregulation of the EGFR/PI3K/PTEN/Akt/mTORC1 pathway in breast cancer: possibilities for therapeutic interventionOncotarget20145134603465025051360
- MurphyLJGhaharyAUterine insulin-like growth factor-1: regulation of expression and its role in estrogen-induced uterine proliferationEndocr Rev19901134434532226350
- CookePSBuchananDLYoungPStromal estrogen receptors mediate mitogenic effects of estradiol on uterine epitheliumProc Natl Acad Sci U S A19979412653565409177253
- BaiWOliveros-SaundersBWangQAcevedo-DuncanMENicosiaSVEstrogen stimulation of ovarian surface epithelial cell proliferationIn Vitro Cell Dev Biol Anim2000361065766611229598
- MallepellSKrustAChambonPBriskenCParacrine signaling through the epithelial estrogen receptor α is required for proliferation and morphogenesis in the mammary glandProc Natl Acad Sci U S A200610372196220116452162
- AschimELSætherTWigerRGrotmolTHaugenTBDifferential distribution of splice variants of estrogen receptor β in human testicular cells suggests specific functions in spermatogenesisJ Steroid Biochem Mol Biol2004921–29710615544935
- ChenMYehCRShyrCRLinHHDaJYehSReduced prostate branching morphogenesis in stromal fibroblast, but not in epithelial, estrogen receptor α knockout miceAsian J Androl201214454655522609821
- PrinsGSBirchLCouseJFEstrogen imprinting of the developing prostate gland is mediated through stromal estrogen receptor α: studies with αERKO and βERKO miceCancer Res200161166089609711507058
- MuellerSOMammary gland development in adult mice requires epithelial and stromal estrogen receptorEndocrinology200214362357236512021201
- RuanWKleinbergLDInsulin-like growth factor I is essential for terminal end bud formation and ductal morphogenesis during mammary developmentEndocrinology1999140115178518410537147
- JoshiPAJacksonHWBeristainAGProgesterone induces adult mammary stem cell expansionNature2010465729980380720445538
- RussoJAoXGrillCRussoIHPattern of distribution of cells positive for estrogen receptor a and progesterone receptor in relation to proliferating cells in the mammary glandBreast Cancer Res Treat199953321722710369068
- LimEVaillantFWuDAberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriersNat Med200915890791319648928
- AndersonEClarkeRBHowellAEstrogen responsiveness and control of normal human breast proliferationJ Mammary Gland Biol Neoplasia199831233510819502
- CunhaGRYoungPHomYKCookePSTaylorJALubahnDBElucidation of a role for stromal steroid hormone receptors in mammary gland growth and development using tissue recombinantsJ Mammary Gland Biol Neoplasia19972439340210935027
- ZepsNBentelJMPapadimitriouJMD’AntuonoMFDawkinsHJEstrogen receptor-negative epithelial cells in mouse mammary gland development and growthDifferentiation19986252212269566307
- LuettekeNCQiuTHFentonSETargeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland developmentDevelopment1999126122739275010331984
- WilsonCLSimsAHHowellAMillerCJClarkeRBEffects of oestrogen on gene expression in epithelium and stroma of normal human breast tissueEndocr Relat Cancer200613261762816728587
- SternlichtMDSunnarborgSWKouros-MehrHYuYLeeDCWerbZMammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulinDevelopment2005132173923393316079154
- KleinbergDLRuanWIGF-I, GH, and sex steroid effects in normal mammary gland developmentJ Mammary Gland Biol Neoplasia200813435336019034633
- HoveyRTrottJVonderhaarBEstablishing a framework for the functional mammary gland: from endocrinology to establishing a framework for the functional mammary gland: from endocrinology to morphologyJ Mammary Gland Biol Neoplasia200271173812160083
- RowsonARDanielsKMEllisSEHoveyRCGrowth and development of the mammary glands of livestock: a veritable barnyard of opportunitiesSemin Cell Dev Biol201223555756622504021
- LeeSMedinaDTsimelzonAAlterations of gene expression in the development of early hyperplastic precursors of breast cancerAm J Pathol2007171125226217591970
- El-AshryDMillerDKharbandaSLippmanMEKernFConstitutive Raf-1 kinase activity in breast cancer cells induces both estrogen-independent growth and apoptosisOncogene19971544234359242379
- PietrasRJArboledaJReeseDMHER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cellsOncogene19951012243524467784095
- RakhaEAPinderSEBartlettJMUpdated UK recommendations for HER2 assessment in breast cancerJ Clin Pathol2015682939925488926
- TognonCKnezevichSRHuntsmanDExpression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinomaCancer Cell20022536737612450792
- TononGModiSWuLt(11;19)(q21;p13) Translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathwayNat Genet200333220821312539049
- TroesterMAHerschkowitzJIOhDSGene expression patterns associated with p53 status in breast cancerBMC Cancer2006627617150101
- Cancer Genome Atlas NetworkComprehensive molecular portraits of human breast tumorsNature20124907418617023000897
- BenoitRCooneyAGiguereVIngrahamHLazarMMuscatGInternational Union of Pharmacology – LXVI: orphan nuclear receptorsPharmacol Rev200658479883617132856
- WärnmarkATreuterEWrightAPGustafssonJAActivation functions 1 and 2 of nuclear receptors: molecular strategies for transcriptional activationMol Endocrinol200317101901190912893880
- BoudotAKerdivelGLecomteSCOUP-TFI modifies CXCL12 and CXCR4 expression by activating EGF signaling and stimulates breast cancer cell migrationBMC Cancer20141440724906407
- XuMQinJTsaiSYTsaiMThe role of the orphan nuclear receptor COUP-TFII in tumorigenesisActa Pharmacol Sin2015361323625283503
- HedrickELeeSOKimGNuclear receptor 4A1 (NR4A1) as a drug target for renal cell adenocarcinomaPLoS One2015106e012830826035713
- DuJXuRRORα, a potential tumor suppressor and therapeutic target of breast cancerInt J Mol Sci20121312157551576623443091
- XiongGWangCEversBMZhouBPXuRRORα suppresses breast tumor invasion by inducing SEMA3F expressionCancer Res20127271728173922350413
- HongEJLevasseurMPDufourCRPerryMCGiguèreVLoss of estrogen-related receptor α promotes hepatocarcinogenesis development via metabolic and inflammatory disturbancesProc Natl Acad Sci U S A201311044179751798024127579
- GiguèreVTranscriptional control of energy homeostasis by the estrogen-related receptorsEndocr Rev200829667769618664618
- DebloisGGiguèreVOestrogen-related receptors in breast cancer: control of cellular metabolism and beyondNat Rev Cancer2012131273623192231
- AriaziEAClarkGMMertzJEEstrogen-related receptor α and estrogen-related receptor γ associate with unfavorable and favorable biomarkers, respectively, in human breast cancerCancer Res200262226510651812438245
- SuzukiSTakagiKMikiYNucleobindin 2 in human breast carcinoma as a potent prognostic factorCancer Sci2012103113614321988594
- HardieDGRossFAHawleySAAMPK: a nutrient and energy sensor that maintains energy homeostasisNat Rev Mol Cell Biol201213425126222436748
- Audet-WalshEPapadopoliDJGravelSPThe PGC-1α/ERRα axis represses one-carbon metabolism and promotes sensitivity to anti-folate therapy in breast cancerCell Rep201614492093126804918
- DebloisGSt-PierreJGiguèreVThe PGC-1/ERR signaling axis in cancerOncogene201332303483349023208510
- BiancoSSaillandJVanackerJMERRs and cancers: effects on metabolism and on proliferation and migration capacitiesJ Steroid Biochem Mol Biol20121303–518018521414406
- ZielloJEJovinISHuangYHypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemiaYale J Biol Med2007802516018160990
- KlimcakovaEChénardVMcGuirkSPGC-1α promotes the growth of ErbB2/neu-induced mammary tumors by regulating nutrient supplyCancer Res20127261538154622266114
- St-PierreJDroriSUldryMSuppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivatorsCell2006127239740817055439
- ChakravartyDNairSSSanthammaBExtranuclear functions of ER impact invasive migration and metastasis by breast cancer cellsCancer Res201070104092410120460518
- Garcia-BecerraRSantosNDiazLCamachoJMechanisms of resistance to endocrine therapy in breast cancer: focus on signaling pathways, miRNAs and genetically based resistanceInt J Mol Sci2013141108145
- LearyADowsettMCombination therapy with aromatase inhibitors: the next era of breast cancer treatment?Br J Cancer200695666166616926831
- HammondMEHayesDFDowsettMAmerican Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version)Arch Pathol Lab Med20101347e48e7220586616
- HayashiSIEguchiHTanimotoKThe expression and function of estrogen receptor α and β in human breast cancer and its clinical applicationEndocr Relat Cancer200310219320212790782
- ProssnitzERBartonMThe G-protein-coupled estrogen receptor GPER in health and diseaseNat Rev Endocrinol201171271572621844907
- VanhaesebroeckBGuillermet-GuibertJGrauperaMBilangesBThe emerging mechanisms of isoform-specific PI3K signallingNat Rev Mol Cell Biol201011532934120379207
- ShanwareNPBrayKAbrahamRTThe PI3K, metabolic, and autophagy networks: interactive partners in cellular health and diseaseAnnu Rev Pharmacol Toxicol20135318910623294306
- DeBerardinisRJChandelNSFundamentals of cancer metabolismSci Adv201625e160020027386546
- YuanTLCantleyLCPI3K pathway alterations in cancer: variations on a themeOncogene200827415497551018794884
- CantleyLCThe phosphoinositide 3-kinase pathwayScience200229655731655165712040186
- LiuPChengHRobertsTMZhaoJJTargeting the phosphoinositide 3-kinase pathway in cancerNat Rev Drug Discov20098862764419644473
- BaselgaJTargeting the phosphoinositide-3 (PI3) kinase pathway in breast cancerOncologist201116Suppl 11219
- ChinYRYoshidaTMarusykABeckAHPolyakKTokerATargeting Akt3 signaling in triple-negative breast cancerCancer Res201474396497324335962
- LeeJJLohKYapYPI3K/Akt/mTOR inhibitors in breast cancerCancer Biol Med201512434235426779371
- PaplomataEO’ReganRThe PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkersTher Adv Med Oncol20146415416625057302
- UtermarkTRaoTChengHThe p110α and p110β isoforms of PI3K play divergent roles in mammary gland development and tumorigenesisGenes Dev201226141573158622802530
- RosnerMHengstschlägerMCytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components Rictor and sin1Hum Mol Genet200817192934294818614546
- MorrisonMMYoungCDWangSmTOR directs breast morphogenesis through the PKC-α-Rac1 signaling axisPLoS Genet2015117e100529126132202
- RexerBNChanthaphaychithSDahlmanKBArteagaCLDirect inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cellsBreast Cancer Res2014161R924451154
- BaderAGKangSZhaoLVogtPKOncogenic PI3K deregulates transcription and translationNat Rev Cancer200551292192916341083
- MuellnerMKUrasIZGappBVA chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancerNat Chem Biol201171178779321946274
- BolósVMiraEMartínez-PovedaBNotch activation stimulates migration of breast cancer cells and promotes tumor growthBreast Cancer Res2013154R5423826634
- RiggioMPerroneMCPoloMLAKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteinsSci Rep201774424428287129
- VanhaesebroeckBAlessiDRThe PI3K-PDK1 connection: more than just a road to PKBBiochem J2000346356157610698680
- MassacesiCdi TomasoEUrbanPPI3K inhibitors as new cancer therapeutics: implications for clinical trial designOnco Targets Ther2016920321026793003
- OkkenhaugKGrauperaMVanhaesebroeckBTargeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapyCancer Discov20166101090110527655435
- FrumanDARommelCPI3K and cancer: lessons, challenges and opportunitiesNat Rev Drug Discov201413214015624481312
- PaezJGEGFR mutations in lung cancer: correlation with clinical response to gefitinib therapyScience200430456761497150015118125
- RossomandoAJPayneDMWeberMJSturgillTWEvidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine protein kinaseProc Natl Acad Sci U S A19898618694069432550926
- KosakoHGotohYMatsudaSIshikawaMNishidaEXenopus MAP kinase activator is a serine/threonine/tyrosine kinase activated by threonine phosphorylationEMBO J1992118290329081322292
- FerrellJEJrTripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputsTrends Biochem Sci199621124604669009826
- HuangCYFerrellJEJrUltrasensitivity in the mitogen-activated protein kinase cascadeProc Natl Acad Sci U S A1996931910078100838816754
- DhillonASHaganSRathOKolchWMAP kinase signalling pathways in cancerOncogene200726223279329017496922
- ChangLKarinMMammalian MAP kinase signalling cascadesNature20014106824374011242034
- PearsonGRobinsonFGibsonTBMitogen-activated protein (MAP) kinase pathways: regulation and physiological functionsEndocr Rev200122215318311294822
- CargnelloMRouxPPActivation and function of the MAPKs and their substrates, the MAPK-activated protein kinasesMicrobiol Mol Biol Rev20017515083
- PayneDMRossomandoAJMartinoPIdentification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase)EMBO J19911048858921849075
- AhnNGCampbellJSSegerRJensenALGravesLMKrebsEGMetabolic labeling of mitogen-activated protein kinase kinase in A431 cells demonstrates phosphorylation on serine and threonine residuesProc Natl Acad Sci U S A19939011514351478389470
- RobbinsDJZhenEOwakiHRegulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitroJ Biol Chem19932687509751068444886
- CrewsCMAlessandriniAEriksonRLThe primary structure of MEK, a protein kinase that phosphorylates the ERK gene productScience199225850814784801411546
- WuJHarrisonJKVincentLAMolecular structure of a protein-tyrosine/threonine kinase activating p42 mitogen-activated protein (MAP) kinase: MAP kinase kinaseProc Natl Acad Sci U S A19939011731778380494
- AlessiDRSaitoYCampbellDGIdentification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1EMBO J1994137161016198157000
- ZhengCFGuanKLActivation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residuesEMBO J1994135112311318131746
- FerrellJEJrBhattRRMechanistic studies of the dual phosphorylation of mitogen-activated protein kinaseJ Biol Chem19972723019008190169228083
- LiuYShepherdEGNelinLDMAPK phosphatases: regulating the immune responseNat Rev Immunol20077320221217318231
- KyriakisJMAppHZhangXFRaf-1 activates MAP kinasekinaseNature199235863854174211322500
- ZhengCFGuanKLCloning and characterization of two distinct human extracellular signal-regulated kinase activator kinases, MEK1 and MEK2J Biol Chem19932681511435114398388392
- MorrisonDKCutlerREThe complexity of Raf-1 regulationCurr Opin Cell Biol1997921741799069260
- WhitehurstCEOwakiHBruderJTRappURGeppertTDThe MEK kinase activity of the catalytic domain of RAF-1 is regulated independently of Ras binding in T cellsJ Biol Chem199527010559455997534298
- HobbsGADerCJRossmanKLRAS isoforms and mutations in cancer at a glanceJ Cell Sci201612971287129226985062
- SantenRJSongRXMcPhersonRThe role of mitogen-activated protein (MAP) kinase in breast cancerJ Steroid Biochem Mol Biol200280223925611897507
- AhmadDANegmOHAlabdullahMLClinicopathological and prognostic significance of mitogen-activated protein kinases (MAPK) in breast cancersBreast Cancer Res Treat2016159345746727592113
- KatoSEndohHMasuhiroYActivation of the estrogen receptor through phosphorylation by mitogen-activated protein kinaseScience19952705241149114947491495
- JoelPBSmithJSturgillTWFisherTLBlenisJLanniganDAPP90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167Mol Cell Biol1998184197819849528769
- LangeCAShenTHorwitzKBPhosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasomeProc Natl Acad Sci U S A20009731032103710655479
- WengLPSmithWMBrownJLEngCPTEN inhibits insulin-stimulated MEK/MAPK activation and cell growth by blocking IRS-1 phosphorylation and IRS-1/Grb-2/Sos complex formation in a breast cancer modelHum Mol Genet200110660561611230180
- KimDHSimTNovel small molecule Raf kinase inhibitors for targeted cancer therapeuticsArch Pharm Res201235460561522553052
- MatsudaYFukumotoMSorafenib: complexities of Raf-dependent and Raf-independent signaling are now unveiledMed Mol Morphol201144418318922179180
- GenoveseMCInhibition of p38: has the fat lady sung?Arthritis Rheum200960231732019180514
- DarnellJKerrIStarkGJak-STAT pathways and transcriptional activation in responseScience19942645164141514218197455
- WardACTouwIYoshimuraAThe Jak-Stat pathway in normal and perturbed hematopoiesisBlood2000951192910607680
- IgazPTóthSFalusABiological and clinical significance of the JAK-STAT pathway: lessons from knockout miceInflamm Res200150943544111603847
- O’SheaJJGadinaMSchreiberRDCytokine signaling in 2002: new surprises in the Jak/Stat pathwayCell2002109SupplS121S13111983158
- TaniguchiTCytokine signaling through nonreceptor protein-tyrosine kinasesScience199526852082512557716517
- IhleJNSignaling by the cytokine receptor superfamily in normal and transformed hematopoietic cellsAdv Cancer Res19966823658712070
- JatianiSSBakerSJSilvermanLRReddyEPJak/STAT pathways in cytokine signaling and myeloproliferative disorders: approaches for targeted therapiesGenes Cancer201011097999321442038
- ShuaiKLiaoJSongMMEnhancement of antiproliferative activity of gamma interferon by the specific inhibition of tyrosine dephosphorylation of Stat1Mol Cell Biol1996169493249418756652
- KisselevaTBhattacharyaSBraunsteinJSignaling through the JAK/STAT pathway, recent advances and future challengesGene20022851–212412039028
- SchindlerCCytokines and JAK-STAT signalingExp Cell Res1999253171410579906
- DarnellJESTATs and gene regulationScience19972775332163016359287210
- LevyDEDarnellJEJrSTATs: transcriptional control and biological impactNat Rev Mol Cell Biol20023965166212209125
- NiwaHBurdonTChambersISmithASelf-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3Genes Dev19981213204820609649508
- HughesKWatsonCJThe spectrum of STAT functions in mammary gland developmentJAKSTAT20121315115824058764
- AbellKBilancioAClarksonRWStat3-induced apoptosis requires a molecular switch in PI(3)K subunit compositionNat Cell Biol20057439239815793565
- HaricharanSLiYSTAT signaling in mammary gland differentiation, cell survival and tumorigenesisMol Cell Endocrinol2014382156056923541951
- ClarksonRWBolandMPKritikouEAThe genes induced by signal transducer and activators of transcription (STAT)3 and STAT5 in mammary epithelial cells define the roles of these STATs in mammary developmentMol Endocrinol200620367568516293640
- O’SheaJJSchwartzDMVillarinoAVGadinaMMcInnesIBLaurenceAThe JAK-STAT pathway: impact on human disease and therapeutic interventionAnnu Rev Med201566131132825587654
- GreenmanCStephensPRBignellGPatterns of somatic mutation in human cancer genomesNature2007446713215315817344846
- JeongEGKimMSNamHKSomatic mutations of JAK1 and JAK3 in acute leukemias and solid cancersClin Cancer Res200814123716372118559588
- CaffarelMMZaragozaRPensaSLiJGreenARWatsonCJConstitutive activation of JAK2 in mammary epithelium elevates Stat5 signalling, promotes alveologenesis and resistance to cell death, and contributes to tumourigenesisCell Death Differ201219351152221941370
- ZhangYToyKAKleerCGMetaplastic breast carcinomas are enriched in markers of tumor-initiating cells and epithelial to mesenchymal transitionMod Pathol201225217818422080057
- ChanSRVermiWLuoJSTAT1-deficient mice spontaneously develop estrogen receptor α-positive luminal mammary carcinomasBreast Cancer Res2012141R1622264274
- WeichselbaumRRIshwaranHYoonTAn interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancerProc Natl Acad Sci U S A200810547184901849519001271
- WatsonCJMillerWRElevated levels of members of the STAT family of transcription factors in breast carcinoma nuclear extractsBr J Cancer19957148408447710952
- DiazNMintonSCoxCActivation of Stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated Src and survivin expressionClin Cancer Res2006121202816397019
- CharpinCSecqVGiusianoSA signature predictive of disease outcome in breast carcinomas, identified by quantitative immunocytochemical assaysInt J Cancer200912492124213419142869
- HynesNEWatsonCJMammary gland growth factors: roles in normal development and in cancerCold Spring Harb Perspect Biol201028a00318620554705
- HenryMDTriplettAAOhKBSmithGHWagnerKParity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic miceOncogene200423416980698515286714
- MeydanNGrunbergerTDadiHInhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitorNature199637965666456488628398
- ThompsonJECubbonRMCummingsRTPhotochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitorBioorganic Med Chem Lett200212812191223
- LucetISFantinoEStylesMThe structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitorBlood2006107117618316174768
- BuchertMBurnsCJErnstMTargeting JAK kinase in solid tumors: emerging opportunities and challengesOncogene201535893995125982279
- GarciaRBowmanTLNiuGConstitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cellsOncogene200120202499251311420660
- KumarJFraserFWRileyCAhmedNMcCullochDRWardACGranulocyte colony-stimulating factor receptor signalling via Janus kinase 2/signal transducer and activator of transcription 3 in ovarian cancerBr J Cancer2014110113314524220695
- HaddadBRGuLMirttiTSTAT5A/B gene locus undergoes amplification during human prostate cancer progressionAm J Pathol201318262264227523660011
- Catlett-FalconeRLandowskiTHOshiroMMConstitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cellsImmunity199910110511510023775
- BanerjeeSBiehlAGadinaMHasniSSchwartzDMJAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospectsDrugs201777552154628255960
- Costa-PereiraAPTinininiSStroblBMutational switch of an IL-6 response to an interferon-γ-like responseProc Natl Acad Sci U S A200299128043804712060750
- FurqanMAkinleyeAMukhiNMittalVChenYLiuDSTAT inhibitors for cancer therapyJ Hematol Oncol201369024308725
- ChenJBaiLBernardDStructure-based design of conformationally constrained, cell-permeable STAT3 inhibitorsACS Med Chem Lett201012858920596242
- LiuALiuYJinZXZH-5 inhibits STAT3 phosphorylation and enhances the cytotoxicity of chemotherapeutic drugs in human breast and pancreatic cancer cellsPLoS One2012710e4662423056374
- SiddiqueeKZhangSGuidaWCSelective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activityProc Natl Acad Sci U S A2007104187391739617463090
- TurksonJZhangSMoraLBBurnsASebtiSJoveRA novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cellsJ Biol Chem200528038329793298816046414
- JingNLiYXiongWShaWJingLTweardyDJG-quartet oligonucleotides: a new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosisCancer Res200464186603660915374974
- KunigalSLakkaSSSodadasuPKEstesNRaoJSStat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancerInt J Oncol20093451209122019360334
- YangZCaiJHXieSJTherapeutic effects of signal transducer and activator of transcription 3 siRNA on human breast cancer in xenograft miceChin Med J (Engl)2011124121854186121740845
- YuQCVerheyenEMZengYAMammary development and breast cancer: a Wnt perspectiveCancers (Basel)201687126
- TanakaKKitagawaYKadowakiTDrosophila segment polarity gene product porcupine stimulates the posttranslational N-glycosylation of Wingless in the endoplasmic reticulumJ Biol Chem200227715128161282311821428
- KimYMKahnMThe role of the Wnt signaling pathway in cancer stem cells: prospects for drug developmentRes Rep Biochem2014411226566491
- PohlSGBrookNAgostinoMArfusoFKumarAPDharmarajanAWnt signaling in triple-negative breast cancerOncogenesis201764e31028368389
- KomiyaYHabasRWnt signal transduction pathwaysOrganogenesis200842687519279717
- YostCTorresMMillerJRHuangEKimelmanDMoonRTThe axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3Genes Dev19961012144314548666229
- JangGKimJChoSBlockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotypeSci Rep201551246526202299
- PolakisPWnt signaling and cancerGenes Dev200014151837185110921899
- Boras-GranicKWysolmerskiJJWnt signaling in breast organogenesisOrganogenesis20084211612219279723
- VeltmaatJMVan VeelenWThieryJPBellusciSIdentification of the mammary line in mouse by Wnt10b expressionDev Dyn2004229234935614745960
- BriskenCHeinemanAChavarriaTEssential function of Wnt-4 in mammary gland development downstream of progesterone signaling serviceGenes Dev200061765065410733525
- HsuWShakyaRCostantiniFImpaired mammary gland and lymphoid development caused by inducible expression of Axin in transgenic miceJ Cell Biol200115561055106411739413
- IncassatiAChandramouliAEelkemaRCowinPKey signaling nodes in mammary gland development and cancer: β-cateninBreast Cancer Res201012621321067528
- RoartyKSerraRWnt5a is required for proper mammary gland development and TGF-β-mediated inhibition of ductal growthDevelopment2007134213929393917898001
- CleversHNusseRWnt/β-catenin signaling and diseaseCell201214961192120522682243
- HoweLRBrownAMWnt signaling and breast cancerCancer Biol Ther200431364114739782
- KhramtsovAIKhramtsovaGFTretiakovaMHuoDOlopadeOIGossKHWnt/β-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcomeAm J Pathol201017662911292020395444
- YangWYanHXChenLWnt/β-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cellsCancer Res200868114287429518519688
- LinSYXiaWWangJCβ-Catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progressionProc Natl Acad Sci U S A20009784262426610759547
- MacDonaldBTTamaiKHeXWnt/β-catenin signaling: components, mechanisms, and diseasesDev Cell200917192619619488
- SmidMWangYZhangYSubtypes of breast cancer show preferential site of relapseCancer Res20086893108311418451135
- PolakisPThe many ways of Wnt in cancerCurr Opin Genet Dev2007171455117208432
- CleversHWnt/β-catenin signaling in development and diseaseCell2006127346948017081971
- BrownAMWnt signaling in breast cancer: have we come full circle?Breast Cancer Res20013635135511737884
- NagahataTShimadaTHaradaAAmplification, up-regulation and over-expression of DVL-1, the human counterpart of the Drosophila disheveled gene, in primary breast cancersCancer Sci200394651551812824876
- PrasadCPGuptaSDRathGRalhanRWnt signaling pathway in invasive ductal carcinoma of the breast: relationship between β-catenin, dishevelled and cyclin D1 expressionOncology2007731–211211718337623
- KlemmFBleckmannASiamLβ-Catenin-independent WNT signaling in basal-like breast cancer and brain metastasisCarcinogenesis201132343444221173432
- NishitaMEnomotoMYamagataKMinamiYCell/tissue-tropic functions of Wnt5a signaling in normal and cancer cellsTrends Cell Biol201020634635420359892
- HenryCQuadirAHawkinsNJExpression of the novel Wnt receptor ROR2 is increased in breast cancer and may regulate both β-catenin dependent and independent Wnt signallingJ Cancer Res Clin Oncol2014141224325425209439
- ZengRHuangJZhongMZMultiple roles of WNT5A in breast cancerMed Sci Monit2016225058506728005837
- ZhongZShanMWangJLiuTShiQPangDDecreased Wnt5a expression is a poor prognostic factor in triple-negative breast cancerMed Sci Monit2016221726721633
- BjörklundPSvedlundJOlssonAKAkerströmGWestinGThe internally truncated LRP5 receptor presents a therapeutic target in breast cancerPLoS One200941e424319158955
- ZhanTRindtorffNBoutrosMWnt signaling in cancerOncogene201736111461147327617575
- WangYBuFRoyerCASPP2 controls epithelial plasticity and inhibits metastasis through β-catenin-dependent regulation of ZEB1Nat Cell Biol201416111092110425344754
- GujralTSChanMPeshkinLSorgerPKKirschnerMWMacBeathGA noncanonical Frizzled 2 pathway regulates epithelial-mesenchymal transition and metastasisCell2014159484485625417160
- WuZQLiXYHuCYFordMKleerCGWeissSJCanonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic breast cancer 1, early onset (BRCA1) repressionProc Natl Acad Sci U S A201210941166541665923011797
- PishvaianMJByersSWBiomarkers of WNT signalingCancer Biomark200734–526327417917155
- SatoNYamabukiTTakanoAWnt inhibitor Dickkopf-1 as a target for passive cancer immunotherapyCancer Res201070135326533620551066
- NuñezFBravoSCruzatFMontecinoMde FerrariGVWnt/β-catenin signaling enhances cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer cellsPLoS One201164e1856221494638
- WangZZhangHHouJClinical implications of β-catenin protein expression in breast cancerInt J Clin Exp Pathol2015811149891499426823833
- LiuJPanSHsiehMHTargeting Wnt-driven cancer through the inhibition of Porcupine by LGK974Proc Natl Acad Sci U S A201311050202242022924277854
- AnzanoMARobertsABMeyersCASynergistic interaction of two classes of transforming growth factors from murine sarcoma cellsCancer Res19824211477647786290046
- MosesHLRobertsABDerynckRThe discovery and early days of TGF-β: a historical perspectiveCold Spring Harb Perspect Biol201687a02186527328871
- TuckerRFShipleyGDMosesHLHolleyRWGrowth inhibitor from BSC-1 cells closely related to platelet type β transforming growth factorScience198422646757057076093254
- RobertsABWakefieldLMThe two faces of transforming growth factor β in carcinogenesisProc Natl Acad Sci U S A2003100158621862312861075
- MassaguéJTGFβ signalling in contextNat Rev Mol Cell Biol2012131061663022992590
- MassaguéJSeoaneJWottonDSmad transcription factorsGenes Dev200519232783281016322555
- LamouilleSXuJDerynckRMolecular mechanisms of epithelial-mesenchymal transitionNat Rev Mol Cell Biol201415317819624556840
- DanielCWRobinsonSDRegulation of mammary growth and function by TGF-βMol Reprod Dev19923221451511637552
- SilbersteinGBDanielCWReversible inhibition of mammary gland growth by transforming growth factor-βScience198723748122912933474783
- PierceDFJrJohnsonMDMatsuiYInhibition of mammary duct development but not alveolar outgrowth during pregnancy in transgenic mice expressing active TGF-β1Genes Dev1993712A230823178253379
- BottingerEPJakubczakJLHainesDCBagnallKWakefieldLMTransgenic mice overexpressing a dominant-negative mutant type II transforming growth factor β receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz-[a]-anthraceneCancer Res19975724556455709407968
- EwanKBShyamalaGRavaniSALatent transforming growth factor-β activation in mammary gland: regulation by ovarian hormones affects ductal and alveolar proliferationAm J Pathol200216062081209312057913
- JhappanCGeiserAGKordonECTargeting expression of a transforming growth factor β1 transgene to the pregnant mammary gland inhibits alveolar development and lactationEMBO J1993125183518458491177
- KordonECMcKnightRAJhappanCHennighausenLMerlinoGSmithGHEctopic TGFβ1 expression in the secretory mammary epithelium induces early senescence of the epithelial stem cell populationDev Biol1995168147617883078
- RobinsonSDRobertsABDanielCWTGFβ suppresses casein synthesis in mouse mammary explants and may play a role in controlling milk levels during pregnancyJ Cell Biol199312012452518416990
- StrangeRLiFSaurerSBurkhardtAFriisRRApoptotic cell death and tissue remodelling during mouse mammary gland involutionDevelopment1992115149581638991
- BierieBGorskaAEStoverDGMosesHLTGF-β promotes cell death and suppresses lactation during the second stage of mammary involutionJ Cell Physiol20092191576819086032
- WelchDRFabraANakajimaMTransforming growth factor β stimulates mammary adenocarcinoma cell invasion and metastatic potentialProc Natl Acad Sci U S A19908719767876822217201
- DalalBIKeownPAGreenbergAHImmunocytochemical localization of secreted transforming growth factor-β1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinomaAm J Pathol199314323813898393616
- XieWMertensJCReissDJAlterations of Smad signaling in human breast carcinoma are associated with poor outcome: a tissue microarray studyCancer Res200262249750511809701
- OftMPeliJRudazCSchwarzHBeugHReichmannETGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cellsGenes Dev19961019246224778843198
- Seton-RogersSELuYHinesLMCooperation of the ErbB2 receptor and transforming growth factor β in induction of migration and invasion in mammary epithelial cellsProc Natl Acad Sci U S A200410151257126214739340
- MuraokaRSDumontNRitterCABlockade of TGF-β inhibits mammary tumor cell viability, migration, and metastasesJ Clin Invest2002109121551155912070302
- MuraokaRSKohYRoebuckLRIncreased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor β1Mol Cell Biol200323238691870314612410
- SiegelPMShuWCardiffRDMullerWJMassaguéJTransforming growth factor β signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasisProc Natl Acad Sci U S A2003100148430843512808151
- TanEJThuaultSCajaLCarlettiTHeldinCHMoustakasARegulation of transcription factor Twist expression by the DNA architectural protein high mobility group A2 during epithelial-to-mesenchymal transitionJ Biol Chem2012287107134714522241470
- ThuaultSTanEJPeinadoHCanoAHeldinCHMoustakasAHMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transitionJ Biol Chem200828348334373344618832382
- BlancoMJMoreno-BuenoGSarrioDCorrelation of Snail expression with histological grade and lymph node status in breast carcinomasOncogene200221203241324612082640
- YangJManiSADonaherJLTwist, a master regulator of morphogenesis, plays an essential role in tumor metastasisCell2004117792793915210113
- GregoryPABertAGPatersonELThe miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1Nat Cell Biol200810559360118376396
- GuleiDMehterovNLingHStantaGBraicuCBerindan-NeagoeIThe “good-cop bad-cop” TGF-β role in breast cancer modulated by non-coding RNAsBiochim Biophys Acta2017186171661167528411077
- BrackenCPGregoryPAKolesnikoffNA double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transitionCancer Res200868197846785418829540
- KorpalMLeeESHuGKangYThe miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2J Biol Chem200828322149101491418411277
- ParkSMGaurABLengyelEPeterMEThe miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2Genes Dev200822789490718381893
- Perdigao-HenriquesRPetroccaFAltschulerGmiR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexesOncogene201635215817225798844
- KimNHKimHSLiXYA p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transitionJ Cell Biol2011195341743322024162
- NietoMAHuangRYJacksonRAThieryJPEMT: 2016Cell20161661214527368099
- Al-HajjMWichaMSBenito-HernandezAMorrisonSJClarkeMFProspective identification of tumorigenic breast cancer cellsProc Natl Acad Sci U S A200310073983398812629218
- ShipitsinMCampbellLLArganiPMolecular definition of breast tumor heterogeneityCancer Cell200711325927317349583
- ManiSAGuoWLiaoMJThe epithelial-mesenchymal transition generates cells with properties of stem cellsCell2008133470471518485877
- ScheelCEatonENLiSHParacrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breastCell2011145692694021663795
- BrunaAGreenwoodWLe QuesneJTGFβ induces the formation of tumour-initiating cells in claudinlow breast cancerNat Commun20123105522968701
- SatoMKadotaMTangBAn integrated genomic approach identifies persistent tumor suppressive effects of transforming growth factor-β in human breast cancerBreast Cancer Res2014163R5724890385
- VidakovicATRuedaOMVervoortSJContext-specific effects of TGF-β/SMAD3 in cancer are modulated by the epigenomeCell Rep201513112480249026686634
- AkhurstRJHataATargeting the TGFβ signalling pathway in diseaseNat Rev Drug Discov2012111079081123000686
- SalnikovAVRoswallPSundbergCGardnerHHeldinNERubinKInhibition of TGF-β modulates macrophages and vessel maturation in parallel to a lowering of interstitial fluid pressure in experimental carcinomaLab Invest200585451252115711566
- HerbertzSSawyerJSStauberAJClinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-β signaling pathwayDrug Des Devel Ther2015944794499
- AnidoJSaez-BorderiasAGonzalez-JuncaATGF-β receptor inhibitors target the CD44high/Id1high glioma-initiating cell population in human glioblastomaCancer Cell201018665566821156287
- BholaNEBalkoJMDuggerTCTGF-β inhibition enhances chemotherapy action against triple-negative breast cancerJ Clin Invest201312331348135823391723
- BandyopadhyayAWangLAgyinJDoxorubicin in combination with a small TGFβ inhibitor: a potential novel therapy for meta-static breast cancer in mouse modelsPLoS One201054e1036520442777
- BrunenDWillemsSMKellnerUMidgleyRSimonIBernardsRTGF-β: an emerging player in drug resistanceCell Cycle201312182960296823974105
- FischerKRDurransALeeSEpithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistanceNature2015527757947247626560033
- SenRBaltimoreDMultiple nuclear factors interact with the immunoglobulin enhancer sequencesCell19864657057163091258
- GilmoreTDThe Rel/NF-κB signal transduction pathway: introductionOncogene199918496842684410602459
- ModuleSHuxfordTGhoshGA structural guide to proteins of the NF-κB signaling moduleCold Spring Harb Perspect Biol200913a00007520066103
- GhoshGVan DuyneGGhoshSSiglerPBStructure of NF-κB p50 homodimer bound to a κB siteNature199537365123033107530332
- MüllerCWReyFASodeokaMVerdineGLHarrisonSCStructure of the NF-kappa B p50 homodimer bound to DNANature199537365123113177830764
- ChenFEHuangDBChenYQGhoshGCrystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNANature199839166654104139450761
- MayMGhoshSRel/NF-κB and IκB proteins: an overviewCancer Biol1997826373
- ChenFGhoshGRegulation of DNA binding by Rel/NF-κB transcription factors: structural viewsOncogene199918496845685210602460
- ChenZParentLManiatisTSite-specific phosphorylation of IκBα a by a novel ubiquitination-dependent protein kinase activityCell19968468538628601309
- YamaokaSCourtoisGBessiaCComplementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activationCell1998937123112409657155
- YaronAGonenHAlkalayIInhibition of NF-κB cellular function via specific targeting of the IκB-ubiquitin ligaseEMBO J19971621648664949351830
- YaronAHatzubaiADavisMIdentification of the receptor component of the IκBa-ubiquitin ligaseNature199839667115905949859996
- Ben-NeriahYRegulatory functions of ubiquitination in the immune systemNat Immunol200231202611753406
- SheppardKRoseDHaqueZTranscriptional activation by NF-κB requires multiple coactivatorsMol Cell Biol19991996367637810454583
- SunSCNon-canonical NF-κB signaling pathwayCell Res2010211718521173796
- XiaoGHarhajESunSCNF-κB-inducing kinase regulates the processing of NF-κB2 p100Mol Cell20017240140911239468
- MukherjeeSPBeharMBirnbaumHAHoffmannAWrightPEGhoshGAnalysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcriptionPLoS Biol2013119e100164724019758
- BrantleyDMYullFEMuraokaRSHicksDJCookCMKerrLDDynamic expression and activity of NF-κB during post-natal mammary gland morphogenesisMech Dev2000971–214915511025216
- GeymayerSDopplerWActivation of NF-κB p50/p65 is regulated in the developing mammary gland and inhibits STAT5-mediated β-casein gene expressionFASEB J20001491159117010834938
- ClarksonRWHeeleyJLChapmanRNF-κB inhibits apoptosis in murine mammary epitheliaJ Biol Chem200027517127371274210777569
- DemiccoEKavanaghKRomieuRRelB/p52 NF-κB complexes rescue an early delay in mammary gland development in transgenic mice with targeted superrepressor IκB-α expression and promote carcinogenesis of the mammary glandMol Cell Biol20052522101361014716260626
- ConnellyLRobinsonCChontMA transgenic model reveals important roles for the NF-κB alternative pathway (p100/p52) in mammary development and links to tumorigenesisJ Biol Chem200728213100281003517261585
- BrantleyDChenCMuraokaRNuclear factor-κB (NF-κB) regulates proliferation and branching in mouse mammary epitheliumMol Biol Cell20011251445145511359934
- CaoYBonizziGSeagrovesTIKKα provides an essential link between RANK signaling and cyclin D1 expression during mammary gland developmentCell2001107676377511747812
- BaxterFOIKK/2 induces TWEAK and apoptosis in mammary epithelial cellsDevelopment2006133173485349416887827
- ConnellyLBarhamWOnishkoHMInhibition of NF-κB activity in mammary epithelium increases tumor latency and decreases tumor burdenOncogene201130121402141221076466
- TorresLSernaEBoschANF-κB as node for signal amplification during weaningCell Physiol Biochem201128583384622178936
- ZaragozáRMirallesVJRusADWeaning induces NOS-2 expression through NF-κB modulation in the lactating mammary gland: importance of GSHBiochem J2005391Pt 358158815954866
- PerkinsNNF-κB: tumor promoter or suppressor?Trends Cell Biol2004142646915102437
- KarinMCaoYGretenFRLiZWNF-κB in cancer: from innocent bystander to major culpritNat Rev Cancer20022430131012001991
- RochaSMartinAMMeekDWNeilDPerkinsNDP53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-κB subunit with histone deacetylase 1Mol Cell Biol200323134713472712808109
- KimDWSovakMAZanieskiGActivation of NF-κB/Rel occurs early during neoplastic transformation of mammary cellsCarcinogenesis200021587187910783306
- PrattMATibboERobertsonSJThe canonical NF-κB pathway is required for formation of luminal mammary neoplasias and is activated in the mammary progenitor populationOncogene200928302710272219483731
- NakshatriHBhatPMartinDGoutletRSledgeGConstitutive activation of NF-κB during progression of breast cancer to hormone-independent growthMol Cell Biol1997177362936399199297
- SovakMBellasRKimDAberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancerJ Clin Invest199710012295229609399940
- CogswellPCGuttridgeDCFunkhouserWKSelective activation of NF-κB subunits in human breast cancer: potential roles for NF-κB2/p52 and for Bcl-3Oncogene20001991123113110713699
- ZhouYEppenberger-CastoriSEppenbergerUBenzCThe NFκB pathway and endocrine-resistant breast cancerEndocr Relat Cancer200512Suppl 1S37S4616113098
- HouMFLinSBYuanSSThe clinical significance between activation of nuclear factor κB transcription factor and overexpression of HER-2/neu oncoprotein in Taiwanese patients with breast cancerClin Chim Acta20033341–213714412867284
- BiswasDKShiQBailySNF-κB activation in human breast cancer specimens and its role in cell proliferation and apoptosisProc Natl Acad Sci U S A200410127101371014215220474
- LiuHLeeEGajdosCApoptotic action of 17β-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivoJ Natl Cancer Inst200395211586159714600091
- BoehmJZhaoJYaoJKimSIntegrative genomic approaches identify IKBKE as a breast cancer oncogeneCell200712961065107917574021
- HuttiJEShenRRAbbottDWPhosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKε promotes cell transformationMol Cell2010344461472
- ParkBZhangHZengQNF-κB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSFNat Med2007131626917159986
- HuberMAAzoiteiNBaumannBNF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progressionJ Clin Invest2004114456958115314694
- TurnerNCReis-FilhoJSBasal-like breast cancer and the BRCA1 phenotypeOncogene200625435846585316998499
- ShehataMTeschendorffASharpGPhenotypic and functional characterisation of the luminal cell hierarchy of the mammary glandBreast Cancer Res2012145R13423088371
- SauALauRCabritaMAPersistent activation of NF-κB in BRCA1-deficient mammary progenitors drives aberrant proliferation and accumulation of DNA damageCell Stem Cell2016191526527292187
- ShostakKChariotANF-κB, stem cells and breast cancer: the links get strongerBreast Cancer Res201113421421867572
- SmithSMLyuYLCaiLNF-κB affects proliferation and invasiveness of breast cancer cells by regulating CD44 expressionPLoS One201499e10696625184276
- YeoSKFrenchRSpadaFClarksonROpposing roles of NFκB2 gene products p100 and p52 in the regulation of breast cancer stem cellsBreast Cancer Res Treat2017162346547728190248
- GodwinPBairdAMHeaveySBarrMPO’ByrneKJGatelyKTargeting nuclear factor-κB to overcome resistance to chemotherapyFront Oncol2013312023720710
- AhmedKMCaoNLiJJHER-2 and NF-κB as the targets for therapy-resistant breast cancerAnticancer Res2006266B4235424317201139
- JiangZLiuJCChungPEEganSEZacksenhausETargeting HER2+ breast cancer: the TBK1/IKKε axisOncoscience20141224
- TapiaMAGonzález-NavarreteIDalmasesAInhibition of the canonical IKK/NFκB pathway sensitizes human cancer cells to doxorubicinCell Cycle20076182284229217890907
- GilmoreTHerscovitchMInhibitors of NF-κB signaling: 785 and countingOncogene200625516887689917072334
- OrlowskiRZDeesECThe role of the ubiquitination-proteasome pathway in breast cancer: applying drugs that affect the ubiquitin-proteasome pathway to the therapy of breast cancerBreast Cancer Res2003511712559038
- AwadaAAlbanellJCanneyPABortezomib/docetaxel combination therapy in patients with anthracycline-pretreated advanced/metastatic breast cancer: a phase I/II dose-escalation studyBr J Cancer20089891500150718454159
- WakefieldARole of Bcl3 in the normal and neoplastic mammary gland2011 Available from: http://orca.cf.ac.uk/55135Accessed October 12, 2017
- BjörnströmLSjöbergMMechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genesMol Endocrinol200519483384215695368
- RoySSVadlamudiRKRole of estrogen receptor signaling in breast cancer metastasisInt J Breast Cancer2012201265469822295247
- LiYWuYAbbatielloTSlug contributes to cancer progression by direct regulation of ERα signaling pathwayInt J Oncol20154641461147225652255
- RobinsonMJCobbMHMitogen-activated protein kinase pathwaysCurr Opin Cell Biol1997921801869069255
- ChungJYParkYCYeHWuHAll TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transductionJ Cell Sci2002115Pt 467968811865024
- ParkSZhaoDHatanpaaKJRIP1 activates PI3K-Akt via a dual mechanism involving NF-κB-mediated inhibition of the mTOR-S6K-IRS1 negative feedback loop and down-regulation of PTENCancer Res200969104107411119435890
- LeeSAndrieuCSaltelFIκB kinase phosphorylates Dok1 serines in response to TNF, IL-1, or radiationProc Natl Acad Sci U S A200410150174161742115574499
- RawlingsJSRoslerKMHarrisonDAThe JAK/STAT signaling pathwayJ Cell Sci200411781281128315020666
- BandAMLaihoMCrosstalk of TGF-β and estrogen receptor signaling in breast cancerJ Mammary Gland Biol Neoplasia201116210911521390570
- KamdjeAHEtetPFVecchioLNew targeted therapies for breast cancer: a focus on tumor microenvironmental signals and chemoresistant breast cancersWorld J Clin cases201421276978625516852