
Abstract
Background
TKIs are the first-line treatment for patients with Ph-positive (Ph+) leukemia. However, drug resistance is frequently observed, mainly due to mutations within the breakpoint cluster region-Abelson leukemia virus (BCR-ABL) kinase domain. The T315I substitution confers complete resistance to TKIs. The aim of this study was to analyze the clinical characteristics of 17 patients with T315I mutation after TKI treatment and provide a basis for prognosis.
Patients and methods
The clinical data of 17 TKI-resistant Ph+ leukemia patients who were found to have a ABL kinase domain mutation from September 2008 to January 2017 were collected. Karyotypes and BCR-ABL fusion gene were analyzed by R-banding and fluorescence in situ hybridization, respectively. Total RNA was extracted by TRIzol reagent, and the ABL kinase domain mutation was detected by direct sequencing.
Results
A total of 17 patients reached effective remission including major molecular response and complete cytogenetic response. However, all the patients subsequently developed a T315I mutation after treatment with TKIs. The rate of the BCR-ABL fusion gene in most of the patients who developed the T315I mutation was significantly higher than that before the mutation. At initial diagnosis, patients average platelet count was 149.7×109/L, whereas the average platelet count was only 53.88×109/L after the T315I mutation (P<0.01). The results also showed that the survival time of patients with a high proportion of blast cells or a high number of white blood cells was obviously shortened.
Conclusion
Patients platelet count decreased when detected with the T315I mutation compared with the initial diagnosis. Combined use of different TKIs and complex chromosomal karyotypes may promote the development of the T315I mutation. When the ratio of blast cells was >50% and the number of white blood cells was >20×109/L, poor survival prognosis was observed.
Introduction
For adult patients with acute lymphoblastic leukemia (ALL) and chronic myeloid leukemia (CML), the Philadelphia chromosome (Ph) resulting from t(9;22) (q34;q11) translocation between the Abelson leukemia virus (ABL) oncogene on chromosome 9 and the breakpoint cluster region (BCR) gene on chromosome 22 is the most common cytogenetic abnormality.Citation1 The BCR-ABL fusion oncogene encoded by Ph can activate tyrosine kinase activity, which causes the proliferation of tumor cells by multiple signaling pathways, such as MAPK/ERK cascades, PI3K/AKT/mTOR, and STAT pathway.Citation2–Citation4 Therefore, the BCR-ABL fusion oncogene plays an essential role in the pathogenesis of Ph-positive (Ph+) leukemia and is considered the underlying mechanism of treatment.
Imatinib, the first tyrosine kinase inhibitor (TKI) approved by the Food and Drug Administration in 2001 is currently the first-line therapy for Ph+ leukemia. However, some patients still have primary or acquired drug resistance, of whom the BCR-ABL kinase domain point mutation is the most common reason for acquired resistance. Second-generation drugs, such as nilotinib and dasatinib, were developed to benefit patients who are unable to achieve effective remission with imatinib or who have BCR-ABL kinase domain mutations other than the T315I mutation.Citation5 Third-generation TKIs, such as ponatinib, are designed to overcome all BCR-ABL kinase domain mutations, including the T315I mutation.Citation6 However, recent research revealed that the T315I mutation still occurs to patients even when second- and third-generation TKIs are used. For example, the T315I gatekeeper mutation appeared in 5 of 12 patients receiving dasatinib.Citation7 These molecular mutations have driven the development and standardization of sophisticated molecular monitoring methods to identify therapeutic failures.Citation8
So far, no consensus about the reasons for mutations has been achieved. Thus, we collected the clinical data of 17 Ph+ ALL and CML cases treated with TKIs in Nanjing Drum Tower Hospital. We analyzed the possible reasons of the T315I mutation to provide a theoretical basis for timely prediction of prognosis and improve therapeutic regimen.
Patients and methods
Patient selection
Among 17 patients with T315I mutation, 10 and 7 cases had ALL and CML, respectively. All 17 patients were diagnosed by means of clinical manifestations, laboratory tests, cell morphology, and immunology. They were followed up at intervals of 2–3 months. Patients with the primary T315I mutation who detected through direct sequencing were excluded from the observation group. Each patient was treated with TKIs, and the ABL kinase domain mutation was detected by direct sequencing when TKI resistance was observed during treatment. We used a coding system to ensure the privacy of patients participating in our study. All the patients provided informed consent for genetic analysis based on the Declaration of Helsinki. They were informed of the existence of other treatment options according to the ethics committee of the Affiliated Drum Tower Hospital, School of Medicine, Nanjing University. The ethics committee of the Affiliated Drum Tower Hospital, School of Medicine, Nanjing University approved this study.
Detection method
All the patients underwent bone marrow puncture, chromosome examination, immunofluorescence in situ hybridization, and nested polymerase chain reaction (PCR) for surveillance of disease. Karyotypes and BCR-ABL fusion gene were analyzed by R-banding and fluorescence in situ hybridization, respectively. Identification and description of anomalous karyotypes were according to the International System of Human Cytogenetic Nomenclature (2009). The specific method and Ph+ cell standard were presented in the literature.Citation9 Total RNA was extracted by TRIzol reagent and reverse transcribed into cDNA via reverse transcriptase. After amplification and electrophoresis, the PCR products were sequenced by Sanger sequencing to detect ABL kinase mutation.Citation10 The patients were divided into two groups to draw the survival curve, and it depended on whether the ratio of blast cells is >50% and whether the number of white blood cells in the mutation is >20×109/L. Meanwhile, we collected the platelet count at relapse in patients with T315I mutation and patients without T315I mutation to observe the differences between them.
Follow-up
The median follow-up time was 10 months (range: 4–84 months) from the time of diagnosis to death or the end of follow-up, as of January 30, 2017.
Statistical analysis
SPSS 22.0 software (IBM Corporation, Armonk, NY, USA) was used for statistical analysis. Paired-samples t-test and independent-samples t-test were used to compare the platelet count. P<0.05 was considered statistically significant. Survival function was analyzed by life tables.
Results
Patient characteristics and treatment
The study population consisted of 17 patients (10 ALL and seven CML; 10 males and 7 females), and the median age was 41 years (20–74 years). A total of 12 patients presented with splenomegaly and lymphadenopathy at diagnosis, suggesting that the disease was in the terminal stage. Karyotype analysis showed that 6 patients had complex karyotypes (patients 5, 7, 8, 11, 15, and 17), and the remaining 11 patients had t(9;22) at initial diagnosis. However, 8 patients had complex karyotypes (patients 5, 7, 8, 11, 12, 14, 15, and 17) at relapse because the karyotype of 2 patients changed from t(9;22) to complex karyotype ().
Table 1 Karyotypes at initial diagnosis and at relapse
Treatment adherence and resistance of these patients were also assessed. Only six (35.3%) patients showed adequate treatment adherence to first- and second-generation TKI therapy, and the rest (64.7%) showed poor treatment adherence. The duration of TKI therapy per patient demonstrated that the minimum duration of TKI therapy before the T315I mutation was 3 months (patient 6), and the maximum duration was 2 years (patient 11), showing high variability in the duration of TKI therapy. Seven patients treated with dasatinib were given a maximum dose of 140 mg/day (patients 1, 2, 5, 6, 7, 12, and 14), and patient 11 who used nilotinib was given a maximum dose of 800 mg/day. Patient 15 received first-generation TKI (imatinib) with a maximum dose of 600 mg/day and did not receive any second-generation TKI thereafter. However, eight patients were treated with two kinds of TKIs (patients 3, 4, 8, 9, 10, 13, 16, and 17), seven of whom were administered imatinib at the beginning and given a second-generation TKI (dasatinib) after they had the Y253H, E255k, Q252H, F317L, M351T, and D276G mutations. Patient 8 developed the T315I mutation after taking dasatinib for 6 months and then switched to ponatinib ().
Table 2 Clinical information of patients with T315I mutation
After TKI treatment, 17 patients had effective remission, of whom 11 patients achieved major molecular response and 6 patients achieved complete cytogenetic response. However, all the patients subsequently developed the T315I mutation; even patients 7 and 9 had allogeneic hematopoietic stem cell transplantation (allo-HSCT). The median time of T315I mutation was 10 months.
A total of 12 patients discontinued TKIs and switched to high-intensity chemotherapy regimens, such as hyper-CVAD (cyclophosphamide, vincristine, adriamycin and dexamethasone), VDCP (vincristine, daunorubicin, cyclophosphamide, and dexamethasone), HAG (homoharringtonine, cytarabine and G-CSF), and HD-MTX (high-dose mitoxantrone) with VP (vincristine and dexamethasone). However, they did not achieve good clinical results, and four of them died due to complications, such as respiratory failure (one), heart failure (one), and sepsis (two), whereas others chose to discharge automatically. Patients 8 and 11 underwent allo-HSCT involving haploid sibling donors, and the conditioning regimen contained modified Bu/Cy. However, the second T315I mutation still occurred (patient 8), and subsequent lymphocyte infusion failed to improve the disease state. Finally, patient 8 died due to viremia. Fortunately, patient 11 did not undergo any mutations until the end of follow-up.
Mutation analysis
We used immunofluorescence in situ hybridization to dynamically monitor the BCR-ABL fusion rate in 17 patients. The fusion rate of BCR-ABL decreased in all the patients after taking TKIs, indicating that the disease status is improved. However, the rate of the BCR-ABL fusion gene in most patients was obviously higher than that before they had the T315I mutation (). When the T315I mutation occurred, most of the patients discontinued TKIs and switched to other chemotherapy regimens, but the disease was not effectively alleviated.
Figure 1 BCR-ABL fusion rate of 17 patients.
Abbreviations: BCR-ABL, breakpoint cluster region-Abelson leukemia virus; HSCT, hematopoietic stem cell transplantation; VDP, vincristine, daunorubicin, dexamethasone; Hyper-CVAD, cyclophosphamide, vincristine, adriamycin and dexamethasone; VDCP, vincristine, daunorubicin, cyclophosphamide and dexamethasone; Hyper-CVAD-B, mitoxantrone, cytarabine; VP, vincristine and dexamethasone; HD-MTX, high-dose mitoxantrone; VDLP, vincristine, daunorubicin, L-asparaginase and dexamethasone; Hyper-CVAD-A, cyclophosphamide, pirarubicin, vincristine and dexamethasone; VDCP, vincristine, daunorubicin, cyclophosphamide and dexamethasone.
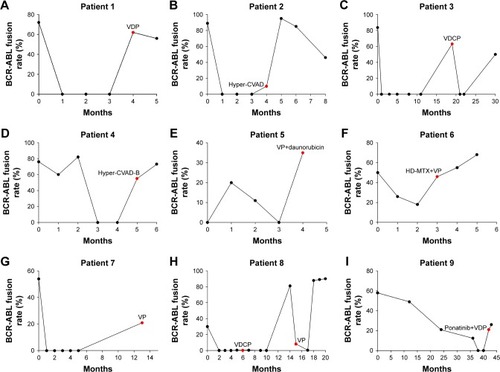
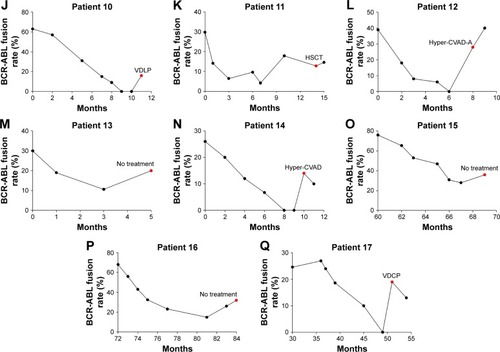
We analyzed the platelet count of patients at initial diagnosis and after developing the T315I mutation. At initial diagnosis, the average platelet count of 17 patients was 149.7×109/L, which was still within the normal range. In the T315I mutation, the average platelet count of patients was only 53.88×109/L, which was a statistically significant difference compared with the former (P<0.01). A notable linear change was observed between the platelet count in the mutation and at initial diagnosis (P=0.028; and ). The platelet count at relapse in patients with T315I mutation and patients without the T315I mutation was compared, but there was no significant difference (P>0.05; ).
Figure 2 Platelets in different stages (P<0.01).
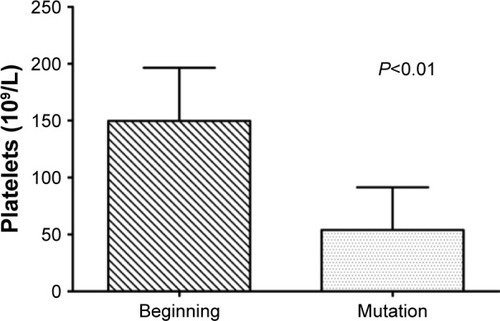
Table 3 Laboratory data of patients with T315I mutation
Table 4 Comparison of platelet count at relapse in patients with T315I mutation and patients without T315I mutation
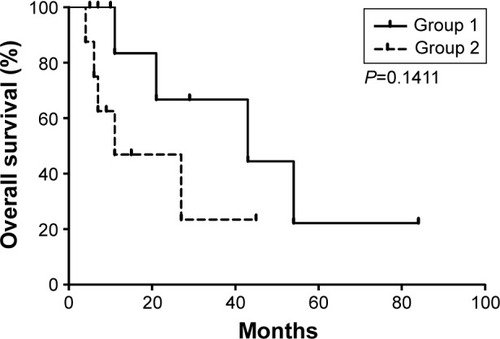
We divided the patients into two groups to draw the survival curve depending on whether the bone marrow aspirate contains >50% blast cells. We concluded that the median survival time of patients was 43 months when the rate of blast cells was <50% (group 1). However, it reduced to only 11 months when the rate of blast cells was >50% (group 2; ).
Figure 3 Total survival time demonstrated by life tables.
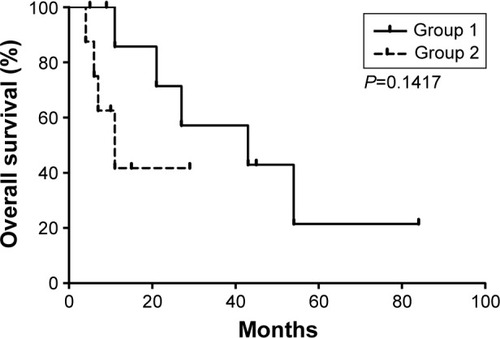
We also dynamically tested the blood routine of patients and found that the median survival time was 43 months for patients whose white blood cells were <20×109/L in the T315I mutation (group 1), whereas it was only 11 months for patients whose white blood cells were more than 20×109/L (group 2; ).
Figure 4 Total survival time demonstrated by life tables.
Discussion
The BCR-ABL fusion gene is an oncogene of CML and 30%–50% cases of adult ALL.Citation11 The BCR-ABL kinase domain has three parts, namely P-loop (P), catalytic domain (C), and activation loop (A). More than 70 types of mutations, involving more than 50 amino acid changes, can occur in the P-ring, A-ring, catalytic region or contact points of the two parts (such as T315 and F317). Point mutations cause changes in the ABL kinase amino acid by interfering directly with the binding of the TKI to the ABL kinase or by inhibiting inactivation of ABL kinase, thereby interfering with the binding of the drug to the target site and eventually leading to drug resistance.Citation12 The most frequent detectable mutation in Ph+ ALL is T315I (detected in 37% of all patients positive for mutations), followed by E255K and Y253H.Citation1
The median time from diagnosis to the T315I mutation in 17 patients was 10 months, suggesting that treatment with TKIs did not postpone disease progression. Ten patients in the course of treatment failed to undergo regular treatment and timely monitoring due to various reasons, which may also be an important reason for the deterioration of the disease. Johansson et alCitation13 and Pfeifer et alCitation14 reported that additional cytogenetic and molecular genetic aberrations are likely to promote disease progression because of increased genomic instability. In the current study, eight patients with complex chromosomal karyotypes developed the T315I mutation; such results were similar to a related study. Aggoune et alCitation15 showed that the combined use of different TKIs may inadvertently promote the development or selection of mutations in the BCR-ABL kinase domain. Seven patients with the T315I mutation in our cohort had combined with Y253H, E255k, and other mutations, because imatinib was used before dasatinib, which may contribute to the T315I mutation.
Our data revealed that platelet count in the T315I mutation was significantly less than that at initial diagnosis. Chui et alCitation16 found that PDGF/PDGFR plays an important role in promoting differentiation and anti-apoptosis of megakaryocyte. TKIs can block the initiation of the PDGF/PDGFR signaling pathway; interrupt several downstream signal cascades, such as PI3K/AKT phosphorylation; and weaken the inhibition of caspase-3 activation, eventually promoting megakaryocyte apoptosis and leading to thrombocytopenia.Citation17 Therefore, we hypothesized that patients with the T315I mutation were resistant to TKIs, so TKIs acted more on the PDGF/PDGFR signaling pathway and decreased the number of platelets. Whether the decreased platelet count can be used as a marker for predicting the T315I mutation is worthy of further study. Our experience showed that the survival time of patients with a high proportion of blast cells and high number of white blood cells was significantly shortened. The increase in the proportion of blast cells and the number of white blood cells showed a high degree of malignancy, which accelerated the progression of the disease when it was combined with the T315I mutation.
After taking TKIs, all the patients achieved effective remission, including complete cytogenetic response and major molecular response. However, the results were unsatisfactory. All the patients had the T315I mutation after a period of remission, indicating that the mutation in the BCR-ABL kinase domain was not related to the remission state. Therefore, the detection rate of gene mutation must be improved, and the therapeutic regimen must be adjusted in time. The mutation in the BCR-ABL kinase domain and chromosomal abnormalities can be detected in CD34+ leukemic cell subsets, including CD34+ CD38-LSCs.Citation18 In addition to standard karyotype analysis, real-time PCR and quantitative analysis of BCR-ABL transcripts by sequencing alone or in combination with denaturing high performance liquid chromatography have become tools for monitoring patients undergoing TKI therapy.Citation7 Baer et alCitation19 argued that mutation detection by conventional Sanger sequencing requires 10%–20% expansion of the mutated subclone, whereas mutations are detected at loads of 1%–2% by ultra-deep sequencing. Thus, early mutation detection by ultra-deep sequencing might allow treatment to be changed before clonal increase in cells with the T315I mutation.
The broad-spectrum kinase inhibitor, ponatinib, is a clinically available inhibitor that has shown efficacy against the T315I mutation. In a recent Phase III clinical trial with ponatinib, frequent severely adverse vascular effects were observed, leading to termination of the trial and temporary withdrawal from the market.Citation20 Meanwhile, axitinib shows significant therapeutic effects and relatively few side effects.Citation21 The most feasible approach to cure leukemia with the T315I mutation is allo-HSCT. However, our research showed that allo-HSCT still failed to achieve the desired results. The feasibility of allo-HSCT is remarkably reduced in clinical practice due to various reasons, such as the source of stem cells, human leukocyte antigen matching, and decreased immunity. Therefore, some scholars searched for other ways to treat Ph+ leukemia. The new MK-0457 is a small-molecule aurora kinase inhibitor with in vitro activity against cells expressing wild-type or mutated BCR-ABL, including the T315I BCR-ABL mutation; three patients with T315I mutation have achieved clinical responses to doses of MK-0457 that are not associated with adverse events.Citation22 Peng et alCitation23 used the mouse model experiment to develop an alternative approach to kinase inhibition, namely, the heat shock protein 90 inhibitor IPI-504, which resulted in BCR-ABL protein degradation and decreased number of leukemia stem cells.
Conclusion
TKIs remain the first-line treatment choice for Ph+ leukemia. However, drug resistance caused by gene mutation must also be considered. Close monitoring of the BCR-ABL fusion gene and researching on novel therapeutic agents are crucial for leukemia patients with the T315I mutation or other mutations.
Acknowledgments
We would like to thank all the volunteers who took part in this study. This work was supported by the Jiangsu Provincial Medical Innovation Team (CXTDA2017046), the Six Talent Peaks Project of Jiangsu Province (2015-WSN-075), the Jiangsu Provincial Medical Youth Talent (QNRC2016039), and the Technique Development Foundation of Nan Jing (Ykk14069&YKK16099). Peipei Xu and Dan Guo are the co-first authors.
Disclosure
The authors report no conflicts of interest in this work.
References
- SoveriniSDe BenedittisCPapayannidisCDrug resistance and BCR-ABL kinase domain mutations in Philadelphia chromosome-positive acute lymphoblastic leukemia from the imatinib to the second-generation tyrosine kinase inhibitor era: the main changes are in the type of mutations, but not in the frequency of mutation involvementCancer201412071002100924382642
- ModiHLiLChuSRossiJYeeJKBhatiaRInhibition of Grb2 expression demonstrates an important role in BCR-ABL-mediated MAPK activation and transformation of primary human hematopoietic cellsLeukemia201125230531221072043
- GesbertFSellersWRSignorettiSLodaMGriffinJDBCR/ABL regulates expression of the cyclin-dependent kinase inhibitor p27Kip1 through the phosphatidylinositol 3-kinase/AKT pathwayJ Biol Chem200027550392233923011010972
- DanialNNRothmanPJAK-STAT signaling activated by Abl oncogenesOncogene200019212523253110851051
- Liu-DumlaoTKantarjianHThomasDAO’BrienSRavandiFPhiladelphia-positive acute lymphoblastic leukemia: current treatment optionsCurr Oncol Rep201214538739422669492
- MillerGDBrunoBJLimCSResistant mutations in CML and Ph(+) ALL – role of ponatinibBiologics2014824325425349473
- Olsson-StrömbergUHermanssonMLundánTMolecular monitoring and mutation analysis of patients with advanced phase CML and Ph+ ALL receiving dasatinibEur J Haematol201085539940420659155
- MüllerMCCrossNCErbenPHarmonization of molecular monitoring of CML therapy in EuropeLeukemia200923111957196319710700
- ChenHYGeZWuYJImmunophenotypic analysis of Philadelphia chromosome positive acute lymphoblastic leukaemia in adultsZhongguo Shi Yan Xue Ye Xue Za Zhi2010183714717 Chinese20561435
- HochhausAKreilSCorbinASMolecular and chromosomal mechanisms of resistance to imatinib (STI571) therapyLeukemia200216112190219612399961
- KurzrockRKantarjianHMDrukerBJTalpazMPhiladelphia chromosome-positive leukemias: from basic mechanisms to molecular therapeuticsAnn Intern Med20031381081983012755554
- ShahNPNicollJMNagarBMultiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemiaCancer Cell20022211712512204532
- JohanssonBFioretosTMitelmanFCytogenetic and molecular genetic evolution of chronic myeloid leukemiaActa Haematol20021072769411919388
- PfeiferHLangeTWystubSPrevalence and dynamics of bcr-abl kinase domain mutations during imatinib treatment differ in patients with newly diagnosed and recurrent bcr-abl positive acute lymphoblastic leukemiaLeukemia20122671475148122230800
- AggouneDToscaLSorelNModeling the influence of stromal microenvironment in the selection of ENU-induced BCR-ABL1 mutants by tyrosine kinase inhibitorsOncoscience201411576825593988
- ChuiCMLiKYangMPlatelet-derived growth factor up-regulates the expression of transcription factors NF-E2, GATA-1 and c-Fos in megakaryocytic cell linesCytokine2003212516412670444
- ZhuangJHawkinsSFGlennMAAkt is activated in chronic lymphocytic leukemia cells and delivers a pro-survival signal: the therapeutic potential of Akt inhibitionHaematologica201095111011819713228
- PellicanoFSimaraPSinclairAThe MEK inhibitor PD184352 enhances BMS-214662-induced apoptosis in CD34+ CML stem/progenitor cellsLeukemia20112571159116721483442
- BaerCKernWKochSUltra-deep sequencing leads to earlier and more sensitive detection of the tyrosine kinase inhibitor resistance mutation T315I in chronic myeloid leukemiaHaematologica2016101783083827102501
- SeniorMFDA halts then allows sales of Ariad’s leukemia medicationNat Biotechnol201432191124406915
- PemovskaTJohnsonEKontroMAxitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformationNature2015519754110210525686603
- GilesFJCortesJJonesDBergstromDKantarjianHFreedmanSJMK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutationBlood2007109250050216990603
- PengCBrainJHuYInhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cellsBlood2007110267868517395781