
Abstract
Introduction
Vasculogenic mimicry (VM) describes the formation of an epithelial-independent tumor microcirculation system that differs from traditional angiogenesis. Angiogenesis and the formation of VM are closely related through the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) pathway and the epithelial–mesenchymal transition (EMT) process.
Materials and methods
In this study, 8-Br-cAMP, a cAMP analog and PKA activator, was used to activate the cAMP/PKA pathway to evaluate the effects of cAMP/PKA on angiogenesis and VM in colorectal cancer (CRC) cells. We used a syngeneic model of CRC in BALB/c mice.
Results
We discovered that treatment with 8-Br-cAMP significantly reduced tumor number compared to control mice after the 7th, 14th, and 28th days of treatment. VM was evaluated by periodic acid–schiff (PAS)–CD31 staining, and we found that VM was inhibited by 8-Br-cAMP treatment in vivo. Immunohistochemistry confirmed the inhibition of vascular endothelial growth factor (VEGF) and cAMP and the activation of PKA by 8-Br-cAMP; quantitative real-time-PCR (qRT-PCR) demonstrated that 8-Br-cAMP regulated the expression of vascular endothelial (VE)-cadherin, matrix metalloproteinase 2 (MMP2), ephrin type-A receptor 2 (EphA2), and VEGF in vivo. Experiments in vitro revealed that treatment with 8-Br-cAMP and U0126 decreased VEGF expression through PKA–ERK in CT26 cells by qRT-PCR. We further confirmed that tube formation of human umbilical vein endothelial cells was inhibited by 8-Br-cAMP in vitro.
Discussion
This study demonstrates that angiogenesis and VM are inhibited by 8-Br-cAMP treatment. Our data indicate that 8-Br-cAMP acts through the cAMP/PKA–ERK pathway and through EMT processes in CRC. These findings provide an insight into mechanisms of CRC and suggest that the cAMP/PKA–ERK pathway is a novel potential therapeutic target for the treatment of CRC.
Introduction
Colorectal cancer (CRC) has the third highest incidenceCitation1 and the second highest mortality among all cancers worldwide.Citation2 Liver metastasis is the leading cause of mortality in patients diagnosed with CRC. It is estimated that ~50% of patients with CRC die from distant metastases.
Angiogenesis is a key event in the malignant progression of CRC and enables CRC tumor growth by providing the growing tumor with access to the blood supply. This process not only enhances cell proliferation but can also facilitate dissemination of CRC cells. Vascular endothelial growth factor (VEGF) is a central player in angiogenesis and is one of the best characterized angiogenic factors.Citation3 VEGF recruits and stimulates the proliferation of endothelial cells, directly leading to angiogenesis.Citation4
It has been reported that there is a significant difference in VEGF expression between primary CRC tissue and metastatic liver lesions from CRC patients with liver metastases.Citation5 We hypothesize that there are additional mechanisms underlying the establishment of tumor microcirculation in CRC beyond VEGF-driven angiogenesis.
Vasculogenic mimicry (VM), first described in melanoma by Maniotis et al,Citation6 is the establishment of an epithelial-independent tumor microcirculation system that differs from traditional angiogenesis. In VM, vascular channels are composed of periodic acid–schiff (PAS)-positive basement membrane and CD31-negative epithelial-like colorectal cell. These vessels allow red blood cells to pass through, providing direct exposure of tumor cells to a blood supply and potentially increasing opportunities for metastatic dissemination. Since VM was first described in 1999, the occurrence of VM has been confirmed in ovarian cancer, breast cancer, glioblastoma, and liver cancer.Citation7–Citation9 We have reported that VM occurs in CRC and is associated with tumor node metastasis staging. Additionally, VM has been associated with metastasis and reduced survival in CRC patients.Citation10
Cyclic adenosine monophosphate (cAMP) acts to convey extracellular signals inside cells, and the activity of protein kinase A (PKA) is dependent on cellular levels of cAMP. The cAMP/PKA pathway is a classical G-protein-coupled receptor-regulated pathway. In addition, the activation of the second messenger cAMP is correlated with VEGF expression. Amano et alCitation11 found that in a rat sponge implanting model, the activation of cAMP promoted VEGF expression, resulting in neoplastic vascularization. A study by Namkoong et alCitation12 revealed that adenylate cyclase-activating forskolin promoted human vascular epithelial cells to secret VEGF and form vasculature. Lissitzky et alCitation13 reported that cAMP affects the formation of VM in cultured melanoma cells. Collectively, these studies indicate that angiogenesis and formation of VM are closely related through the cAMP/PKA pathway. Therefore, we hypothesized that the cAMP/PKA pathway may be a potential common pathway of angiogenesis and VM and that this axis may be a potential target to suppress the neovascularization of primary CRC and liver metastasis, thereby restricting CRC growth and improving survival outcomes for patients with CRC.
VM is regulated by vascular endothelial (VE)-cadherin, matrix metalloproteinases (MMPs), and ephrin type A receptor 2 (EphA2), which play roles in epithelial–mesenchymal transition (EMT)Citation14 and can contribute to extracellular matrix remodeling and the formation of VM.Citation15 EMT is a dynamic process by which epithelial cancer cells are converted into dedifferentiated cells with additional mesenchymal propertiesCitation16 and can promote VM in cancer. VE-cadherin is an important adhesion protein that regulates vasculogenic activities. EphA2 is a member of the ephrin receptor family of PTKs and is of importance in angiogenesis. VE-cadherin regulates the expression and phosphorylation of EphA2 on the cell membrane. Phosphorylation of EphA2 in turn promotes the phosphorylation of VE-cadherin, resulting in the eventual loss of adhesion between cells and subsequently leading to cell migration and infiltration into VM. Hess et alCitation17 reported that EphA2 promotes the progression of VM by dephosphorylating focal adhesion kinase and remodeling the extracellular matrix in melanoma. Wang et alCitation3 reported that VEGF induces EphA2 to increase the expression of MMPs and remodel the extracellular matrix, leading to the regulation of VM. These findings also suggest that angiogenesis and VM may be correlated with EMT processes.
In the current study, we used 8-Br-cAMP, a cAMP analog and PKA activator, to activate the cAMP/PKA pathway to evaluate the impact on angiogenesis and VM in CRC cells in vivo and in vitro and to explore the potential of targeting the cAMP/PKA axis in the treatment of CRC.
Material and methods
Cell line and syngeneic tumor formation
Our cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The human umbilical vein endothelial cells (HUVEC) originates from normal neonatal human umbilical vein endothelial cells, and the CT26 cell line was derived from colon carcinoma tissues of BALB/c mice according to ATCC websites. The CT26 CRC cell line, derived from a BALB/c murine adenocarcinoma, was maintained in Roswell Park Memorial Institute-1640 medium supplemented with 10% fetal bovine serum at 37°C and 5% CO2. For passaging or the preparation of CT26 cells to inject into BALB/c mice, CT26 cells were dissociated with 0.25% trypsin and centrifuged for 5 min at 1,000 rpm. Following aspiration of the supernatant, phosphate buffer saline (PBS) was added and cell concentration was adjusted to 1×106 cells/mL.
Forty-two 4- to 5-week-old male BALB/c mice, weighing from 18 to 22 g, were purchased from the Changzhou Kawensi Biotech Company (Changzhou, China). Six mice were injected subcutaneously in the back with 0.4 mL of the CT26 cell suspension (4×105 cells/mouse). Tumors measuring 1 cm in diameter emerged 10 days later in these mice. Pathological examination of tissue confirmed the presence of carcinoma. All tumor tissues were removed and sliced into pieces for serial implantation into the cecum of BALB/c mice.
Thirty-six mice received the implantation of CT26 carcinoma tissue in their cecum. After general anesthesia and sterilization, a 2 cm vertical incision was made at the right lower quadrant of the abdomen. The cecum was then pulled out of the abdomen. The serosa of the cecum that was exposed out was scratched, and a 2 mm diameter tumor tissue was attached with fibrin glue. Finally, the cecum was put back into place and the skin was sealed. After tumor implantation, mice were randomly divided into a control group and an experimental group. In the experimental group, the intraperitoneal injection of 8-Br-cAMP (60 mg/kg/day) was performed for 7 days, while control mice received injection of normal saline. Mice were sacrificed on the 7th, 14th, and 28th days, and tumor tissue was harvested for the evaluation of gene expression. However, due to a high mortality rate, the number of mice for sacrifice at each time point was adjusted in order to guarantee that mice were available for culling on the 28th day. This study was approved by the Animal Ethics Committee of Northern Jiangsu People’s Hospital, Yangzhou Clinical School of Nanjing Medical University. This study was also conducted following the guidelines and regulations of Animal Ethics Committee of Northern Jiangsu People’s Hospital, Yangzhou Clinical School of Nanjing Medical University.
Hematoxylin and eosin staining
Paraffin-embedded sections of murine tumor tissues were prepared and stained with hematoxylin and eosin. Slices were cut into 5 μm sections and stained with hematoxylin–eosin for histological evaluation under a microscope (Olympus Corporation, Tokyo, Japan) by two independent pathologists.
Observation of VM
Immunohistochemical staining with CD31/CD34 and PAS is used to identify VM in tumor tissues.Citation18–Citation20 VM is recognized as vascular-like patterns of tumor cells gathered together without necrosis, hemorrhage, or infiltration of inflammatory cells. CD31/CD34 and PAS staining demonstrated that VM is composed of extracellular basement membrane (PAS positive) with red blood cells inside. Tumor cells were CD31 negative, while typical vessels showed CD31 positivity and PAS positivity. Therefore, the double staining could validate VM as it is PAS positive and CD31 negative. Five VM-rich areas were evaluated under 100× magnification in each section, and the quantity of VM in each area was calculated under 400× magnification. The mean value of all five areas is defined as VM density (VMD).
Quantitative real-time-PCR (qRT-PCR)
Total RNA was isolated from cells using the TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA). cDNAs were generated using the PrimeScript RT Master Mix kit (Takara, Dalian, China). Primers for qRT-PCR were designed using online tools available at the Primer3 website. qRT-PCR was performed on a StepOnePlus Real-Time PCR system (Thermo Fisher Scientific) using the SYBR Green PCR kit (Hoffman-La Roche Ltd., Basel, Switzerland) in a final volume of 10 μL. Relative quantification was performed for genes listed in . Each reaction was performed and repeated at least three times. The data were analyzed using the 2−ΔΔCt method.
Table 1 Primer sequence used for qRT-PCR
Immunohistochemistry analysis
After animal sacrifice, tumor tissues obtained from CT26 tumor-bearing subjects were prepared in formalin-fixed, paraffin-embedded sections. Samples were cut into 4-μm-thick serial sections, deparaffinized with xylene, and rehydrated in alcohol. Sections were subsequently submerged in antigen retrieval buffer and microwaved for antigen fixation. Sections were treated with hydrogen peroxide to block endogenous nonspecific binding activity and were incubated overnight at 4°C with diluted primary antibodies. Slides were incubated with the appropriate Horseradish Peroxidase-polymer-conjugated secondary antibodies at 37°C for 1 h. The primary antibodies that were used are as follows: mouse anti-cAMP antibody (sc73761; Santa Cruz Biotechnology Inc., Dallas, TX, USA), mouse anti-PKA antibody (sc28315; Santa Cruz Biotechnology Inc.), rabbit anti-CD31 antibody (sc-1506; Santa Cruz Biotechnology Inc.), and rabbit anti-VEGF antibody (sc-507; Santa Cruz Biotechnology Inc.). Negative control slides followed the same protocol, with the exception that primary antibodies were replaced with PBS. Subsequently, we stained sections with 3,3-diamin-obenzidine solution for 3 min and counterstained the nuclei with hematoxylin. Stained tissue slices were reviewed and independently scored by two pathologists.
Drug treatments
Because we hypothesized that the cAMP/PKA–ERK angiogenesis axis plays an important role in the regulation of VM, the ERK inhibitor U0126 was used to inhibit the cAMP/PKA-ERK axis. CT26 and HUVEC cell cultures were divided into three groups: treatment with 10 μM 8-Br-cAMP, treatment with 10 μM U0126 treatment, and untreated control. Treatment duration lasted 1 h, after which total RNA was extracted.
Tube formation assay
We used HUVEC cells to process the tube formation assay. Matrigel (BD, Franklin Lakes, NJ, USA) and pipette tips were stored 4°C overnight prior to preparation of the tube formation assays. Fifty microliters of matrigel was added to pre-chilled 96-well plates on ice. When cells reached 70%–80% confluence, they were digested and then resuspended in serum-free dulbecco’s modified eagle medium (DMEM) to a concentration of 2×105 cells/mL. A total of 50 μL of cell suspension was added onto the matrigel of each well. 8-Br-cAMP was added into serum-free DMEM. After overnight incubation at 37°C, tube network formation was observed under the microscope and photographed, and the area and connectivity of the networks were calculated.
Western blot
Protein was extracted from CRC cells and tissues by using a RIPA kit (Beyotime, Shanghai, China) according to the manufacturer’s protocol. Different molecular weights of protein were separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels in the running buffer and transferred to polyvinylidene fluoride membranes (EMD Millipore, Bedford, MA, USA) in the transfer buffer. Then, the membranes were blocked in 5% nonfat milk for >2 h and incubated in specific primary antibodies at 4°C overnight. After washing in tris-buffered saline and Tweens for three times (10 min each time), the membranes were transferred to secondary antibodies (rabbit or mouse) at room temperature for 2 h. Protein bands could be exposed by using ECL Plus (EMD Millipore) with a Bio-Imaging System. The specific primary antibodies were as follows: E-cadherin, VE-cadherin, EphA2, MMP2, PKA, VEGF, and Tubulin, which were used as an internal control.
Results
8-Br-cAMP significantly inhibits the growth of CT26 tumors in mice
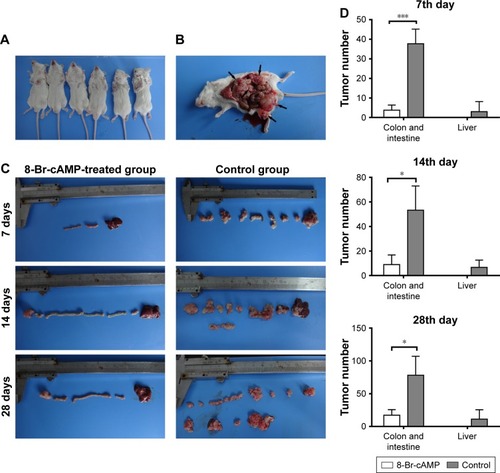
Initially, subcutaneous CT26 tumors were established in six male BALB/c mice (). Afterward, 36 mice received serial transplantation of tumor fragments into their cecum and were randomized to treatment with 8-Br-cAMP or control. Subjects in both groups were sacrificed for the observation of tumor progression on the 7th, 14th, and 28th days (). Primary tumor nodules and abdominal metastatic lesions were counted and compared between both groups. Seven days after implantation, the mean amounts of primary CRC tumor nodules and liver metastases in the control group were 49.33±26.86 and 3.00±5.20, respectively, while in the 8-Br-cAMP treatment group, the mean amounts of primary CRC tumor nodules and liver metastases were 3.75±2.63 and 0, respectively. On the 14th day, the mean amounts of primary tumor nodules in the control and 8-Br-cAMP groups were 53.25±12.58 and 9.00±7.81, respectively; the mean amounts of liver metastases in the control and 8-Br-cAMP groups were 6.75±5.91 and 0, respectively. Ascites with pyaemia and macrosopic liver metastases were observed in mice from control group on the 14th day. Subjects from the 8-Br-cAMP treatment group did not show signs of liver metastases, metastasis to other organs, or ascites on the 7th and 14th days. On the 28th day, an increased mean number of primary tumors and metastatic lesions were observed in the control group (78±29.03 and 11.25±14.41, respectively). In these mice, massive liver metastases were observed, and tumors were diffusively distributed throughout the abdomen. The abdominal cavity was enlarged and had massive pyemic ascites. In contrast, three mice from the 8-Br-cAMP-treated group had no metastatic lesions and a mean count of colorectal and intestinal tumors of 17.33±8.50 (). Counts of colorectal and intestinal tumors in mice from the 8-Br-cAMP group were significantly lower than the control group on the 7th, 14th, and 28th days (P<0.05). However, no significant difference was observed in liver metastasis between the two groups. These results demonstrate that 8-Br-cAMP has significant inhibitory effects on primary CRC growth.
Figure 1 Syngeneic tumor formation in mice treated with 8-Br-cAMP vs control.
Abbreviation: cAMP, cyclic adenosine monophosphate.
Treatment with 8-Br-cAMP inhibits VM
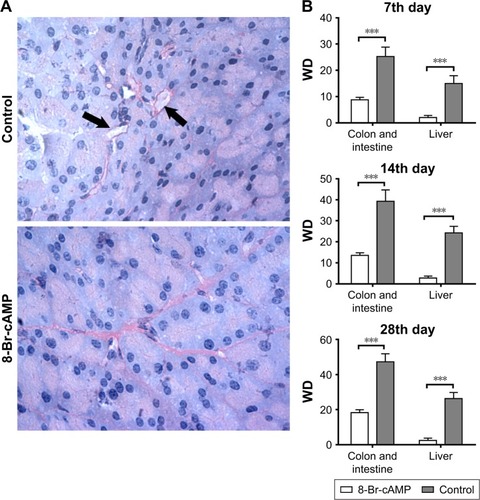
VM was evaluated by PAS–CD31 staining of colorectal and intestinal tumors from control and experimental groups. VM is recognized as PAS positive (membrane matrix stained pink) and CD31 negative (membrane matrix lacking brown stain). Less VM was observed in the 8-Br-cAMP-treated group than in the control group (). VMD, calculated as the mean VM of five areas in a sample, was significantly lower in the 8-Br-cAMP group than in the control group throughout the experiment (P<0.05; ). These results demonstrate that, in addition to inhibiting tumor growth, disruption of the cAMP/PKA axis with 8-Br-cAMP regulates the formation of VM.
Figure 2 Formation of VM between 8-Br-cAMP-treated group and control group.
Abbreviations: cAMP, cyclic adenosine monophosphate; PAS, periodic acid–schiff; VM, vasculogenic mimicry; VMD, VM density.
Effects of 8-Br-cAMP treatment on VEGF, cAMP, and PKA
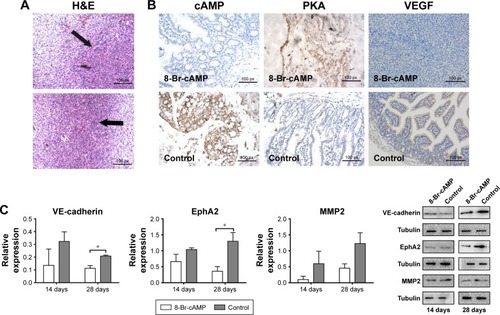
H&E staining demonstrated successful in situ tumor formation in the murine model (); the presence of carcinoma was confirmed independently by two pathologists. The expression of VEGF, cAMP, and PKA in tumor tissues from control and 8-Br-cAMP-treated groups was evaluated through immunohistochemistry (IHC), as previously described (). The expression of cAMP was significantly lower in 8-Br-cAMP-treated group than in the control group, indicating that the cAMP analog, 8-Br-cAMP, inhibits cAMP expression. The expression of PKA in the 8-Br-cAMP group was significantly higher than that in the control group, confirming that PKA was activated by treatment with 8-Br-cAMP. The expression of VEGF in CRC tissues was significantly lower in the 8-Br-cAMP-treated group than in the control group, demonstrating that this key factor of angiogenesis was inhibited by the PKA activator, 8-Br-cAMP. Lower expression of VEGF could lead to a decreased angiogenesis, diminishing neovasculature that promotes tumor growth. VEGF levels were positively correlated with VM and reduced following treatment with 8-Br-cAMP, indicating that VEGF and VM are regulated by 8-Br-cAMP and the cAMP/PKA axis.
Figure 3 Functions of 8-Br-cAMP treatment on VEGF, cAMP, and PKA.
Abbreviations: cAMP, cyclic adenosine monophosphate; EphA2, ephrin type A receptor 2; MMP2, matrix metalloproteinase 2; PKA, protein kinase A; qRT-PCR, quantitative RT-PCR; VE, vascular endothelial; VEGF, VE growth factor.
Regulation of EMT and VEGF expressions by 8-Br-cAMP in vivo evaluated by qRT-PCR and Western blot
qRT-PCR and Western blot were performed on CRC tissues obtained from BALB/c mice from the 8-Br-cAMP treatment and control groups after sacrifice to determine the expressions of EMT-associated genes, including VE-cadherin, EphA2, and MMP2. qRT-PCR, and Western blot were performed on tissues obtained on the 14th and 28th days (). On the 14th day, there was no significant difference in expression detected in these three genes between the 8-Br-cAMP-treated group and the control group (). However, on the 28th day, the expression of VE-cadherin and EphA2 was significantly lower in the 8-Br-cAMP-treated group than in the control group (P<0.05; ). This indicates that 8-Br-cAMP may have long-term effects on EMT processes, and the inhibitory influence may be exerted gradually. However, the MMP2 levels were not significantly different between the 8-Br-cAMP-treated group and the control group. Nevertheless, there was a trend that the expression of these genes was diminished over time in the 8-Br-cAMP-treated group by the 28th day, and lack of statistical significance at the 14th day may be due to the short follow-up time and small sample size. Moreover, the significant differences we observed may be more pronounced if we continued the observation for a longer follow-up time. These data demonstrate that 8-Br-cAMP regulates EMT processes, inhibiting the expression of VE-cadherin and EphA2; this regulation correlates with the inhibition of angiogenesis and VM.
qRT-PCR and Western blot evaluation of VEGF expression after treatment with 8-Br-cAMP and U0126 in vitro
To further elucidate the pathways and mechanisms associated with VM in CRC cells, we conducted in vitro experiments on the CT26 cell line. CT26 cells were divided into the following three groups: 8-Br-cAMP treatment, U0126 treatment, and negative control. qRT-PCR and Western blot demonstrated that 8-Br-cAMP significantly enhanced PKA expression, corresponding to the results of the IHC analysis on in vivo tumors treated with 8-Br-cAMP (P<0.001; ). Importantly, the expressions of VEGF, VE-cadherin, MMP2, and EphA2 were significantly lower in CT26 cells treated with 8-Br-cAMP than in the control group (P<0.05; ), demonstrating that PKA can regulate EMT processes. Moreover, when CT26 cells were treated with U0126, the expression of VEGF, VE-cadherin, MMP2, and EphA2 was decreased relative to the control group. These findings demonstrated that 8-Br-cAMP is an analog of cAMP can activate PKA and further affect ERK, U0126 can function as a ERK inhibitor; thus, angiogenesis and EMT in CRC cells can be mediated through the PKA–ERK axis (P<0.05).
Figure 4 Expression of VEGF after treatment with 8-Br-cAMP and U0126 in vitro.
Abbreviations: cAMP, cyclic adenosine monophosphate; EphA2, ephrin type A receptor 2; MMP2, matrix metalloproteinase 2; PKA, protein kinase A; VE, vascular endothelial; VEGF, VE growth factor.
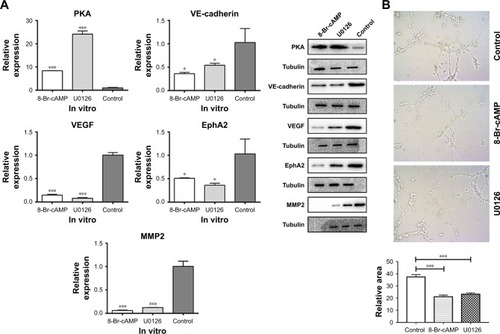
Inhibition of tube formation by 8-Br-cAMP
As described, HUVEC cells treated with 8-Br-cAMP form fewer polygonal networks from individual cell bodies than the control group, directly confirming that angiogenesis is inhibited by the 8-Br-cAMP. Also, cells treated with U0126 generate fewer polygonal networks compared to the control. This is consistent with the reduction in VEGF expression following 8-Br-cAMP and U0126 treatment, as VEGF is a critical mediator of angiogenesis. These data suggest that 8-Br-cAMP and U0126 significantly abolish angiogenesis through the cAMP/PKA–ERK pathway.
Discussion
VM is associated with poor prognosis in several malignant tumors, including glioblastoma, melanoma, and breast cancerCitation18 and VEGF is reported to be closely related to the formation of VM. We conducted a series of in vivo and in vitro experiments to determine the impact of treatment with 8-Br-cAMP, a cAMP analog, on angiogenesis and VM in CRC. In vivo, 8-Br-cAMP exerted potent antitumor effects in CRC tumor-bearing BALB/c mice, indicating that activation of the cAMP/PKA pathway has an impact on tumor growth. Levels of VM and VEGF expression were significantly lower in 8-Br-cAMP-treated mice than in nontreated mice. The reductions corresponded to observations of tumor number and size. This suggested that reduced blood supply is directly related to the reduced tumor burden resulting from treatment with 8-Br-cAMP. Other report indicates the antiproliferative effects of treating cells with 8-Br-cAMP,Citation21 indicating its strong antitumor potential.
While Lissitzky et alCitation13 mentioned that PKA is not involved in the VM process, our results demonstrate that the PKA activator, 8-Br-cAMP, inhibits the formation of VM, indicating that there may be a PKA-dependent regulation of VM and angiogenesis. It has also been reported that forskolin, a potent and specific activator of adenylyl cyclase, rather than PKA, significantly increases VEGF expression,Citation12 proliferation, and migration; this is in contrast to our results. However, it was also reported that forskolin inhibits cell growth, making the role of forskolin on VM and angiogenesis controversial.Citation22 The diverse actions of these drugs may be attributed to their different activation targets, and Namkoong et alCitation12 reported that forskolin not only increases intracellular cAMP but also participates in the PKA pathway. Forskolin increases levels of cAMP, leading to the activation of PKA and subsequent activation of associated downstream targets through PKA. Forskolin may also activate downstream targets through elevated cAMP levels, rather than PKA; this may partially contribute to the diverse outcomes observed with forskolin treatment. In terms of 8-Br-cAMP, forskolin can only activate PKA by competing with 8-Br-cAMP. However, the underlying mechanisms by which these two drugs lead to diverse outcomes are still unknown, and the diversity of the PKA and cAMP pathways is worth further study.
Hyper-activation of the ERK pathway, which is the downstream of the cAMP/PKA pathway, is a major cause of human cancers.Citation23 In our research, 8-Br-cAMP reduced the expression of ERK through the cAMP/PKA pathway, which is concordant with previous report.Citation24 It is also reported that inhibition of ERK reduces the formation of VM and the expression of VEGF; our results are consistent with these reports.Citation13 However, the activation of downstream ERK may be caused not only by the activation of PKA but also by elevated intracellular cAMP levels,Citation3 as ERK is regulated by both pathways. While it is clear that ERK participates in the formation of VM and in the regulation of VEGF expression,Citation20 the specific upstream factors contributing to ERK activation require further elucidation.
A regulatory relationship between angiogenesis and VM has been described.Citation19,Citation25 During tumorigenesis, VEGF is released by proteases, such as MMP2, and induces angiogenesis.Citation26 Enhanced VEGF expression promotes the formation of VM, and EphA2 expression was also increased along with increased production of VEGF.Citation25 It has also been reported that EMT-associated genes are highly involved in VM, including VE-cadherin, EphA2, and MMPs.Citation15,Citation17,Citation27–Citation30 According to previous report and our own research, the expression of these EMT-associated genes is significantly decreased along with changes in VEGF and VM when cells are treated with 8-Br-cAMP, indicating that EMT is positively correlated with VM and VEGF expression.Citation31 Our data also revealed an association in the expression of ERK1/2 and VE-cadherin, a marker of VM formation, indicating that EMT is associated with the activity of the PKA–ERK axis, and this is consistent with previous reports.Citation14,Citation15
We selected the CT26-BALB/c syngeneic model of CRC as our in vivo model to simulate natural tumor growth in the presence of an intact immune system. However, serial implantation may have facilitated rapid tumor formation and growth, even in the presence of an immune system, resulting in more rapid mortality in subjects than we anticipated. In addition, the relatively small sample size in our study may have introduced bias into our results and was a limitation of our study.
Collectively, this study demonstrates that 8-Br-cAMP regulates angiogenesis and VM through the cAMP/PKA pathway and through downstream activation of the ERK signaling pathway in CRC in vitro and in vivo. We also confirmed that the expression of EMT markers (VE-cadherin and EphA2) and VEGF is associated with the formation of VM. To the best of our knowledge, this is the first study to evaluate the effect of activating the cAMP/PKA pathway with 8-Br-cAMP on the formation of VM and angiogenesis in CRC. This research indicates the potential importance of the cAMP/PKA pathway as a novel therapeutic target for the treatment of CRC.
Acknowledgments
This study was partially supported by the Jiangsu Key Medical Discipline (General Surgery) (ZDXKA2016005).
Disclosure
The authors report no conflicts of interest in this work.
References
- TanIBMalikSRamnarayananKHigh-depth sequencing of over 750 genes supports linear progression of primary tumors and metastases in most patients with liver-limited metastatic colorectal cancerGenome Biol2015163225808843
- HurKToiyamaYOkugawaYCirculating microRNA-203 predicts prognosis and metastasis in human colorectal cancerGut201766465466526701878
- WangJYSunTZhaoXLFunctional significance of VEGF-a in human ovarian carcinoma: role in vasculogenic mimicryCancer Biol Ther20087575876618376140
- YangJShiQDSongTBVasoactive intestinal peptide increases VEGF expression to promote proliferation of brain vascular endothelial cells via the cAMP/PKA pathway after ischemic insult in vitroPeptides20134210511123340020
- ChenJLiQWangCWuJZhaoGPrognostic significance of c-erbB-2 and vascular endothelial growth factor in colorectal liver metastasesAnn Surg Oncol20101761555156320069460
- ManiotisAJFolbergRHessAVascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicryAm J Pathol1999155373975210487832
- HendrixMJSeftorEAKirschmannDASeftorREMolecular biology of breast cancer metastasis. Molecular expression of vascular markers by aggressive breast cancer cellsBreast Cancer Res20002641742211250735
- SoodAKSeftorEAFletcherMSMolecular determinants of ovarian cancer plasticityAm J Pathol200115841279128811290546
- LiuXMZhangQPMuYGClinical significance of vasculogenic mimicry in human gliomasJ Neurooncol2011105217317921533525
- LiWZongSShiQLiHXuJHouFHypoxia-induced vasculogenic mimicry formation in human colorectal cancer cells: involvement of HIF-1a, Claudin-4, and E-cadherin and VimentinSci Rep201663753427869227
- AmanoHAndoKMinamidaSAdenylate cyclase/protein kinase A signaling pathway enhances angiogenesis through induction of vascular endothelial growth factor in vivoJpn J Pharmacol200187318118811885966
- NamkoongSKimCKChoYLForskolin increases angiogenesis through the coordinated cross-talk of PKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signalingCell Signal200921690691519385062
- LissitzkyJCParriauxDRistorcelliEVerineALombardoDVerrandoPCyclic AMP signaling as a mediator of vasculogenic mimicry in aggressive human melanoma cells in vitroCancer Res200969380280919176384
- MengJSunBZhaoXDoxycycline as an inhibitor of the epithelial-to-mesenchymal transition and vasculogenic mimicry in hepatocellular carcinomaMol Cancer Ther201413123107312225277383
- QiaoLLiangNZhangJAdvanced research on vasculogenic mimicry in cancerJ Cell Mol Med201519231532625598425
- LiuQQiaoLLiangNThe relationship between vasculogenic mimicry and epithelial–mesenchymal transitionsJ Cell Mol Med20162091761176927027258
- HessARSeftorEAGardnerLMMolecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: role of epithelial cell kinase (Eck/EphA2)Cancer Res20016183250325511309274
- YaoLZhangDZhaoXDickkopf-1-promoted vasculogenic mimicry in non-small cell lung cancer is associated with EMT and development of a cancer stem-like cell phenotypeJ Cell Mol Med20162091673168527240974
- ZhangSFuZWeiJGuoJLiuMDuKPeroxiredoxin 2 is involved in vasculogenic mimicry formation by targeting VEGFR2 activation in colorectal cancerMed Oncol201532141425471788
- LiuXWangJHLiSHistone deacetylase 3 expression correlates with vasculogenic mimicry through the phosphoinositide3-kinase/ERK-MMP-laminin5γ2 signaling pathwayCancer Sci2015106785786625940092
- LoKWKanHMGagnonKALaurencinCTOne-day treatment of small molecule 8-bromo-cyclic AMP analogue induces cell-based VEGF production for in vitro angiogenesis and osteoblastic differentiationJ Tissue Eng Regen Med2016101086787524493289
- SugimotoNMiwaSTsuchiyaHTargeted activation of PKA and Epac promotes glioblastoma regression in vitroMol Clin Oncol20131228128524649161
- IchikawaKKubotaYNakamuraTMCRIP1, an ERK substrate, mediates ERK-induced gene silencing during epithelial–mesenchymal transition by regulating the co-repressor CtBPMol Cell2015581354625728771
- KeranenTHommoTMoilanenEKorhonenRbeta2-receptor agonists salbutamol and terbutaline attenuated cytokine production by suppressing ERK pathway through cAMP in macrophagesCytokine2017941728162907
- ZhaoNSunBCZhaoXLRole of Bcl-2 and its associated miRNAs in vasculogenic mimicry of hepatocellular carcinomaInt J Clin Exp Pathol2015812157591576826884845
- VempatiPPopelASMac GabhannFExtracellular regulation of VEGF: isoforms, proteolysis, and vascular patterningCytokine Growth Factor Rev201425111924332926
- PaulisYWSoetekouwPMVerheulHMTjan-HeijnenVCGriffioenAWSignalling pathways in vasculogenic mimicryBiochim Biophys Acta201018061182820079807
- HuangBXiaoEHuangMMEK/ERK pathway is positively involved in hypoxia-induced vasculogenic mimicry formation in hepatocellular carcinoma which is regulated negatively by protein kinase AMed Oncol201532140825487444
- FanYLZhengMTangYLLiangXHA new perspective of vasculogenic mimicry: EMT and cancer stem cells (review)Oncol Lett2013651174118024179490
- KirschmannDASeftorEAHardyKMSeftorREHendrixMJMolecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implicationsClin Cancer Res201218102726273222474319
- TangNNZhuHZhangHJHIF-1alpha induces VE-cadherin expression and modulates vasculogenic mimicry in esophageal carcinoma cellsWorld J Gastroenterol20142047178941790425548487