
Abstract
We aimed to detect the pathogenic gene mutations in a patient with lamellar ichthyosis (LI). The genomic DNA of the patient was examined using high-throughput whole-exome sequencing to identify the causative mutations. Compound heterozygous mutations of c.1187G>T (p.Arg396Leu) and c.607C>T (p.Gln203*) were found in the transglutaminase-1 gene (TGM1) on chromosome 14 of the proband. The mutations stated above have been reported to impair the function of TGM1 protein and to be pathogenic. Our data suggest that the proband carried compound heterozygous mutations of c.1187G>T(p.Arg396Leu) and c.607C>T(p.Gln203*) in TGM1, which were in the trans position and the cause of his disease. We also found some dermoscopic in this patient which may be specific in LI.
Introduction
Lamellar ichthyosis (LI) is a rare autosomal-recessive inherited and genetically heterogeneous disease. It is one of the main skin phenotypes of autosomal-recessive congenital ichthyosis (ARCI).Citation1 There are eight genes reported to be related with LI: TGM1, ABCA12, ALOXE3, ALOX12B, CERS3, CYP4F22, NIPAL4/ICHTHYIN and PNPLA1.Citation2 TGM1 mutations are not only the most common cause of LI, but also the main cause of ARCI.Citation3,Citation4 The genotype–phenotype correlation of ARCI is not clear. Nevertheless, researchers have discovered that ARCI caused by TGM1 mutations is linked significantly to the presence of collodion membrane at birth, ectropion, plate-like scales and alopecia.Citation3 Dermoscopic findings of LI are rare.Citation5 Thus, we investigated the genetical cause and dermoscopic features of the proband.
Case Report
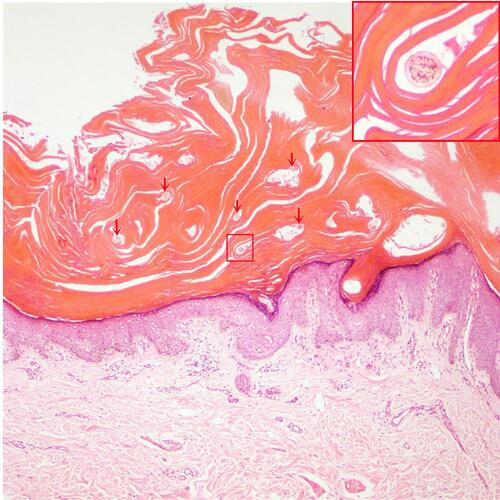
A 19-year-old male was admitted to our hospital with generalized squamous keratosis. He had been diagnosed as a collodion baby at birth, and later developed the lamellar form of ichthyosis gradually all over his body without systemic treatment. Following the ichthyosis, he had light-to-dark-brown ichthyotic keratotic scales with hair loss, ectropion, and incomplete eye closure (). We magnified the scales on different areas of the proband with a dermoscope. Dermoscopy showed alopecia and pustules on his head (), greasy light-brown scales on his face (), “sheet-like” scales on his hand (), pre-thoracic skin () and “bark-like” thickened scales with deep fissures between scales on the abdominal area () and on his thigh (). The degree of hyperkeratosis differed in different parts of the skin. In general, scales on areas with thicker subcutaneous fat tended to be thicker. Histopathology () showed severe lamellar hyperkeratosis, mild hypertrophy of the spinous layer, and vacuolated degeneration of some spinous cells. In the stratum corneum, there were some cross-sections of hair shaft (arrows in ). One of those structures was highlighted and magnified in the upper right corner (red box in ). The proband’s father denied consanguineous marriage or a similar family history.
Figure 1 Clinical features of the patient: dark, thickened and tightened lamellar scales spread over almost all the skin surface with hair loss and ectropion.

Figure 2 Dermoscopic features of the patient. Dermoscopy (×50 magnification) shows alopecia and pustules on his head (A), greasy light-brown scales on his face (B), “sheet-like” scales on his hand (C), pre-thoracic skin (D) as well as “bark-like” thickened scales with deep fissures between scales on the abdominal area (E) and on his thigh (F).
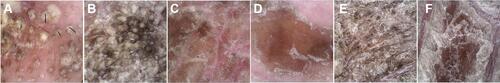
Figure 3 Histology (hematoxylin and eosin, ×100 magnification) shows severe lamellar hyperkeratosis, mild hypertrophy of the spinous layer, and vacuolated degeneration of some spinous cells. In the stratum corneum, there were some cross-sections of hair shaft (arrows). One of those structures was highlighted and magnified in the upper right corner (red box).
The clinical diagnosis was severe LI. High-throughput whole-exome sequencing was requested to further identify the cause.
After obtaining written informed consent, we extracted genomic DNA from the peripheral blood of the patient and his father. Mutation sites were identified by whole-exome sequencing with the genomic DNA of the proband. Whole-exome sequencing was based on a high-throughput sequencing platform with a paired-end strategy. We used the xGen Exome Research Panel from IDT (https://eu.idtdna.com/) for mutation capture. We screened for pathogenetic mutations based on American College of Medical Genetics and Genomics guidelines.Citation6 Pathogenic mutations that correlated with the patient’s clinical phenotype were verified using Sanger sequencing of genomic DNA of the proband and his parents.
Two known heterozygous variants on TGM1 gene were identified in the genomic DNA of the proband. One was c.1187G>T (p. Arg396Leu) (NM_000359.3; exon 8) on Chr14:24260600 (NC_000014.9), which was inherited from his father. The other was c.607C>T (p.Gln203*) (NM_000359.3; exon 4) on Chr14:24258646 (NC_000014.9) (). According to the American College of Medical Genetics and Genomics guidelines,Citation6 the proband carried two mutant loci (c.1187G>T and c.607C>T) and, although only the father was tested for carriage (the mother was not tested), we found that the father carried only the mutation at the c.1187G>T locus. Hence, we assumed that the two variant loci in the proband were in the trans position according to the Mendelian law.
Figure 4 TGM1 mutations. (A) and (B) TGM1 mutations in the proband. (C) TGM1 structure. TGM1 has 15 exons and the translation start is located in the second exon.Citation26 (D) Protein domains of TGM1. It consists of five chains (817 amino acids): anchoring domain (1–92), β-sandwich domain (94–246), catalytic domain (247–572), β-barrel 1 domain (573–688), and β-barrel-2 domain (689–817).Citation19
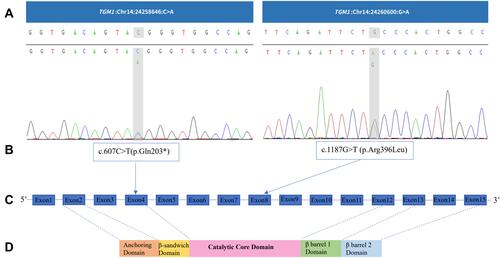
The TGM1 mutations in exon 8 and exon 4 have been reported to be pathogenic.Citation1,Citation7–Citation11 The former, c.1187G>T (p.Arg396Leu), located in the catalytic core domain of transglutaminase 1 (TGase-1), forms a compound heterozygous state with pathogenic variant (c.919C>T, c.160C>T, c.1135G>C) or suspected pathogenic variant (c.652G>A) and thus leads to ichthyosis.Citation1,Citation7,Citation8 In vitro study, it is reported that c.1187G>T (p.Arg396Leu) affects the structure of the enzyme and influences the catalytic reaction.Citation1 The latter, located in the β-sandwich domain of TGase-1, is reported to cause severe LI with ectropion and alopecia in addition to the typical thick lamellar scales on the trunk and extremities in a Chinese patient with c.607C>T (p.Gln203*) homozygous mutation. Moreover, in situ TGM1 assays have revealed a loss of TGase-1 cDNA and an absence of transglutaminase activity in the skin.Citation11 In another Chinese patient with LI in which this variant and the pathogenic variant c.944G>A formed a compound heterozygous state, in addition to erythema and scaling, external ear deformities were observed.Citation9
The proband was eventually diagnosed with severe LI caused by TGM1 compound heterozygous mutations. He was treated with acitretin (20 mg/day, P.O.) and topical agents (Vaseline™ and tazarotene). Unfortunately, the patient was lost during follow-up.
Discussion
TGM1 (NM_000359.3), located on chromosome 14 (NC_000014.9), is a gene that contains 15 exons encoding 817 amino acids and expresses TGase-1, which is a calcium-dependent, catalytic membrane-binding enzyme that plays an important part in formation of the epidermal keratinized envelope. TGase-1: (i) acts as a scaffold to organize secreted extracellular lipids into a continuous lamellar membrane that mediates the permeability barrier function and prevents water loss; (ii) constitutes a mechanical barrier to prevent infection.Citation12–Citation14 TGM1 mutations can lead to varying degrees of transglutaminase deficiency, which impairs protein crosslinking and esterification of epidermis-specific ceramides during formation of keratinocyte proteins and lipid envelopes, and disrupts the skin-barrier function.Citation15
Homozygous or compound heterozygous mutations on TGM1 lead to ARCI1, an autosomal recessive inherited disease which has two clinical phenotypes, LI and NCIE, and causes LI in most cases and NCIE in some cases.Citation16 LI presents as generalized brown or dark lamellar hyperkeratosis with or without erythema. NCIE presents as a distinct erythema covered with fine grayish-white scales. LI and NCIE may show scarring alopecia.Citation17 TGM1 mutations are associated with certain clinical manifestations: collodion membrane at birth, hair loss, hearing problems, eye problems, and skin odor.Citation18 However, clinical phenotypes cannot be inferred from genotypes, and even identical genotypes can lead to different clinical phenotypes.Citation19
A genetic diagnosis is the “gold standard” for the diagnosis of hereditary diseases. The common method is whole-exome sequencing of DNA extracted from peripheral blood and data analyses for possible pathogenic variants. Validation is undertaken by Sanger sequencing. In autosomal-recessive ichthyosis, novel (especially) missense mutations must be identified by enzyme-activity assays to determine their pathogenicity.Citation20 Alternatively, they can be verified by immunofluorescence blotting, which determines whether the mutated gene is pathogenic by comparing expression of the mutated gene with that of the wild-type protein.Citation21 Besides, computational analysis serves as a selective and attractive molecular approach. For instance, structural modeling has revealed that compound heterozygous missense mutations located in the catalytic core domain can lead to a significantly impaired structural stability of transglutaminase protein.Citation22
Researchers have discovered (through dermoscopy) many keratotic plugs in the cristae cutis where eccrine sweat pores are present. They believe these will block sweat glands and are the cause of hypohidrosis in LI.Citation4 In a study by Yang and colleagues, scales covered the cristae cutis but not the sulci cutis, in accordance with our findings. In our study, we also found the degree of hyperkeratosis differed in different parts of the skin and scales on areas with thicker subcutaneous fat tended to be thicker which may serve as another characteristic dermoscopic feature of LI. Yet further researches are needed.
Biopsy of ARCI-TGM1 cases often reveals non-specific features such as hyperkeratosis with focal parakeratosis, a slightly thickened granular layer, and mild-to-moderate psoriasiform hyperplasia.Citation23 In this case, except for those non-specific features, we also found some cross-sections of hair shaft in the stratum corneum in hematoxylin eosin stain of abdominal skin biopsy as a result of the over-thickened stratum corneum.
Acitretin is first-line treatment of LI in adults because it improves hyperkeratosis, hypohidrosis, hair regrowth, ectropion, hearing, and daily skin care. Emollients are recommended for patients with ichthyosis, and are used at least twice-daily, after bathing. Other topical therapies include lubricating agents, keratolytic agents, or topical retinoids depending on the overall lesions of the patient.Citation24 Patients with ectropion require regular eye examinations, long-term use of eye lubricants, and eyelid massage to reduce ocular symptoms. Eyelid skin grafts may be considered in patients with more severe ectropion, but only as third-line treatment.Citation25
Conclusions
The proband carried two pathogenic variants in TGM1 (c.1187G>T and c.607C>T). These compound heterozygous mutations were the causes of LI in the proband, and were in the trans position. Dermoscopy revealed keratotic scales anastomotic to the crista cutis, which led to a highlighted sulci cutis presenting like a fissure. The thickness of plate-like scales differed in different parts of the body, from sheets to bark-like. We found scales on areas with thicker subcutaneous fat tended to be thicker. Histology showed cross-sections of hair shaft in the stratum corneum, which is a sign of the over-thickened stratum corneum.
Ethics Approval
The patient’s informed consent for publication of the case details, including publication of the patient’s images, was obtained, and the study was approved by the Ethics Committee of the Second Hospital of Jilin University.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Acknowledgments
We thank the patient and his father for their participation in this study and consent of publication. We thank Dr. Jianxin Xia for assistance with the explanation of the histopathological result. We also thank Mr. Yanlong Wang and Miss Jingying Nie for assistance with the histopathological examination of the patient.
Disclosure
The authors declare no conflicts of interest in this work.
Additional information
Funding
References
- Laiho E, Ignatius J, Mikkola H, et al. Transglutaminase 1 mutations in autosomal recessive congenital ichthyosis: private and recurrent mutations in an isolated population. Am J Hum Genet. 1997;61(3):529–538. doi:10.1086/515498
- Takeichi T, Akiyama M. Inherited ichthyosis: non-syndromic forms. J Dermatol. 2016;43(3):242–251. doi:10.1111/1346-8138.13243
- Farasat S, Wei M-H, Herman M, et al. Novel transglutaminase-1 mutations and genotype-phenotype investigations of 104 patients with autosomal recessive congenital ichthyosis in the USA. J Med Genet. 2009;46(2):103–111. doi:10.1136/jmg.2008.060905
- Fachal L, Rodríguez-Pazos L, Ginarte M, et al. Characterization of TGM1 c.984+1G>A mutation identified in a homozygous carrier of lamellar ichthyosis. Int J Dermatol. 2012;51(4):427–430. doi:10.1111/j.1365-4632.2011.05171.x
- Takeda M, Nomura T, Sugiyama T, et al. Compound heterozygous missense mutations p.Leu207Pro and p.Tyr544Cys in TGM1 cause a severe form of lamellar ichthyosis. J Dermatol. 2018;45(12):1463–1467. doi:10.1111/1346-8138.14675
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
- Diociaiuti A, El Hachem M, Pisaneschi E, et al. Role of molecular testing in the multidisciplinary diagnostic approach of ichthyosis. Orphanet J Rare Dis. 2016;11(1):4. doi:10.1186/s13023-016-0384-4
- Numata S, Teye K, Krol RP, et al. Mutation study for 9 genes in 23 unrelated patients with autosomal recessive congenital ichthyosis in Japan and Malaysia. J Dermatol Sci. 2015;78(1):82–85. doi:10.1016/j.jdermsci.2015.02.006
- Al-Naamani A, Al-Waily A, Al-Kindi M, et al. Transglutaminase-1 mutations in Omani families with lamellar ichthyosis. Med Princ Pract. 2013;22(5):438–443. doi:10.1159/000349914
- Cheng R, Liang J, Li Y, et al. Next-generation sequencing through multi-gene panel testing for diagnosis of hereditary ichthyosis in Chinese. Clin Genet. 2020;97(5):770–778. doi:10.1111/cge.13704
- Cao X, Lin Z, Yang H, et al. New mutations in the transglutaminase 1 gene in three families with lamellar ichthyosis. Clin Exp Dermatol. 2009;34(8):904–909. doi:10.1111/j.1365-2230.2009.03288.x
- Elias PM. Stratum corneum defensive functions: an integrated view. J Invest Dermatol. 2005;125(2):183–200. doi:10.1111/j.0022-202X.2005.23668.x
- Elias PM, Schmuth M, Uchida Y, et al. Basis for the permeability barrier abnormality in lamellar ichthyosis. Exp Dermatol. 2002;11(3):248–256. doi:10.1034/j.1600-0625.2001.110308.x
- Hohl D, Huber M, Frenk E, et al. Analysis of the cornified cell envelope in lamellar ichthyosis. Arch Dermatol. 1993;129(5):618–624. doi:10.1001/archderm.1993.01680260088013
- Richard G. Molecular genetics of the ichthyoses. Am J Med Genet C Semin Med Genet. 2004;131C(1):32–44. doi:10.1002/ajmg.c.30032
- Herman ML, Farasat S, Steinbach PJ, et al. Transglutaminase-1 gene mutations in autosomal recessive congenital ichthyosis: summary of mutations (including 23 novel) and modeling of TGase-1. Hum Mutat. 2009;30(4):537–547. doi:10.1002/humu.20952
- Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol. 2010;63(4):607–641. doi:10.1016/j.jaad.2009.11.020
- Sun Q, Burgren NM, Cheraghlou S, et al. The genomic and phenotypic landscape of ichthyosis: an analysis of 1000 kindreds. JAMA Dermatol. 2022;158(1):16–25. doi:10.1001/jamadermatol.2021.4242
- Hennies HC, Küster W, Wiebe V, et al. Genotype/phenotype correlation in autosomal recessive lamellar ichthyosis. Am J Hum Genet. 1998;62(5):1052–1061. doi:10.1086/301818
- Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol. 2009;129(6):1319–1321. doi:10.1038/jid.2009.57
- Numata S, Teye K, Karashima T, et al. Functional study of TGM1 missense mutations in autosomal recessive congenital ichthyosis. Exp Dermatol. 2016;25(8):657–659. doi:10.1111/exd.13000
- Nasser KK, Banaganapalli B, Shinawi T, et al. Molecular profiling of lamellar ichthyosis pathogenic missense mutations on the structural and stability aspects of TGM1 protein. J Biomol Struct Dyn. 2021;39(14):4962–4972. doi:10.1080/07391102.2020.1782770
- Yang CS, Pomerantz H, Mannava KA, et al. Comparing histopathology from patients with X-linked recessive ichthyosis and autosomal recessive congenital ichthyosis with transglutaminase 1 mutation: a report from the National Registry for Ichthyosis and Related Skin Disorders. J Am Acad Dermatol. 2016;74(5):1008–1010. doi:10.1016/j.jaad.2015.12.027
- Mazereeuw-Hautier J, Vahlquist A, Traupe H, et al. Management of congenital ichthyoses: European guidelines of care, part one. Br J Dermatol. 2019;180(2):272–281. doi:10.1111/bjd.17203
- Mazereeuw-Hautier J, Hernández‐Martín A, O’Toole EA, et al. Management of congenital ichthyoses: European guidelines of care, part two. Br J Dermatol. 2019;180(3):484–495. doi:10.1111/bjd.16882
- Yamanishi K, Inazawa J, Liew FM, et al. Structure of the gene for human transglutaminase 1. J Biol Chem. 1992;267(25):17858–17863. doi:10.1016/S0021-9258(19)37122-4