
Abstract
Hutchinson–Gilford progeria syndrome (HGPS) is a rare congenital disease caused by mutations in the LMNA gene. Children with HGPS are phenotypically characterized by lipodystrophy, short height, low body weight, scleroderma, reduced joint mobility, osteolysis, senile facial features, and cardiovascular compromise that usually lead to death. We aimed to describe the case of a patient who reached above-average age expectancy for children with HGPS in Latin America and describe the clinical and molecular characteristics of the patient. A 14-year-old female patient was presented with progeria-compatible phenotypic characteristics. HGPS was confirmed via LMNA gene sequencing that detected a heterozygous c.1824C>T (p.Gly608Gly) mutation. The primary aim is to describe the HGPS case, the molecular gene mutation finding, and make a short review of the limited available treatment options for children with HGPS. Such as the farnesyl transferase inhibitors in conjunction with other pharmacological therapies that have insinuated improvement in health, and survival rate.
Introduction
Hutchinson–Gilford progeria syndrome (HGPS) is a rare sporadic autosomal dominant segmental premature aging disease, with a prevalence of 1 in 20 million births in the United States.Citation1 Associated with de novo missense heterozygous mutations of the LMNA gene in most cases.Citation2,Citation3
Little is known of the prevalence of HGPS in middle-income-countries, but in 2013, there was a report of 16 cases in Central and South AmericaCitation4 that described a life expectancy of 13 years of age. Taking this into account we will describe the clinical and molecular characterization of a female patient with HGPS that reached above-average age expectancy in Latin America, and review some available treatment options.
Case Report
A 14-year-old female previously diagnosed with HGPS was the firstborn child of non-related, healthy parents, with no previous family genetic disorders and a healthy 9-year-old sibling. The mother was aged 20 years during conception, and the father was aged 26 years. Normal weight and height data were recorded throughout pregnancy. According to the parents, the patient appeared to be a healthy newborn, and the patient’s development and growth was normal until her second year of age.
Subsequently, she had trouble gaining weight, even with an adequate diet, started losing hair, and her skin thickened and hardened. Her cognitive development was normal until she was 13 years old, when she had to stop school owing to Chikungunya viral infection that triggered secondary medical conditions.
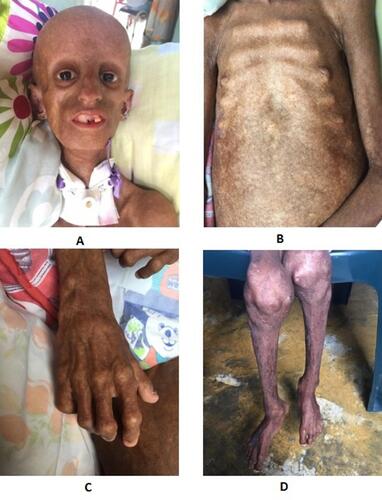
The physical examination of our patient was compatible with a classic progeria phenotype she had alopecia, posterior low hair implantation with prominent scalp veins and eyes, beaked nose, micrognathia, partial anodontia, and senile facial features. Her thorax had “rosary” costal grating, and abdominal outpouching, with the absence of subcutaneous fat. Genitals were normal. Skin showed altered skin pigment, with scleroderma. Her extremities presented with tufting of fingers, osteoarthritis, and joint fibrosis ().
Figure 1 Alopecia, posterior low hair implantation with prominent scalp veins and eyes, beaked nose, micrognathia, partial anodontia, and senile facial features (A). Her thorax had “rosary” costal grating, and abdominal outpouching, with absence of subcutaneous fat (B). Her extremities presented with tufting of fingers (C), osteoarthritis, and joint fibrosis (D).
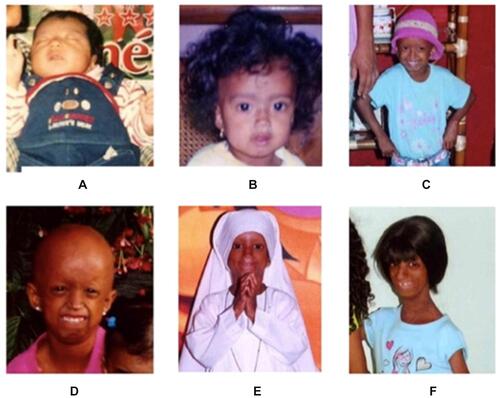
Additionally, she had dilated cardiomyopathy, severe aortic and mitral valve insufficiency, congestive heart failure, severe insulin resistance and, altered lipid metabolism. Owing to these complications, she was receiving congestive heart failure treatment: furosemide, digoxin, and propranolol, and had concomitant follow-ups with pediatric gastroenterology, endocrinology, and pediatric palliative care. Patients’ physical alterations through time were documented in a picture time-line ().
Figure 2 Patient at one month-of-age (A), six months-of-age (B), five years-of-age (C), six years-of-age (D), seven years-of-age (E), and thirteen years-of-age (F).
Via a blood test, we sequenced the LMNA gene. A heterozygous mutation detected in exon 11 of the LMNA gene at c.1824C>T (p.Gly608Gly) confirmed the molecular diagnosis of classic HGPS. The present study was previously approved by the institutional Internal Review Board, Comite de etica en investigacion biomedica. A written informed consent was signed by the parents authorizing to perform genetic test, use case details, pictures and publish the case.
Discussion
HGPS follows an autosomal dominant inheritance pattern.Citation2,Citation3 But, most patients with HGPS have de novo missense mutation in the LMNA gene leading to activation of a cryptic splice site, which means children do not inherit the disease from their parents.Citation5 Although at birth, these patients appear healthy, the symptoms begin to appear after the first year of life, and the average age of diagnosis is 2.9 years.Citation2 These children have a life expectancy of 13.4 yearsCitation6 and experience accelerated atherosclerosis usually resulting in early death associated with myocardial infarction or less commonly, stroke.Citation7–Citation9 Therefore, children with this life-threatening condition must be followed up by a pediatric palliative care team.
In 2003, two independent studies reported on the mutation c.1824C>T (p.Gly608Gly) within exon 11 of the LMNA gene, now referred to as the “classic” mutation that occurs in ~90% of HGPS patients,Citation6,Citation10,Citation11 including the patient in the present study.
Although this mutation is usually silent, it activates a cryptic splice that deletes 150 nucleotides, extending to the beginning of exon 12.Citation2,Citation3,Citation10,Citation11 Therefore, the final post-translational process of prelamin A (suppression of the 15C-terminal amino acid) is halted, resulting in abnormal farnesylation, and a mutant lamin A called progerin. Scaffidi and MistelliCitation12 evaluated fibroblasts of patients with classic HGPS and concluded that the presence of progerin and not the absence of lamina A causes the phenotype.Citation12 Additionally, with the insertion of a modified oligonucleotide that targets the cryptic splice and causes mutation of p.G608G, fibroblasts recover nuclear distribution of the studied proteins and normal morphology.Citation13 Other authors like Fong et alCitation14 demonstrated that toxicity caused by progerin is responsible for the abnormal HGPS phenotype.
Although currently, there are no Food and Drug Administration-approved treatments for HGPS, some clinical trials have been directed to test farnesyl transferase inhibitors such as lonafarnib.Citation3,Citation8,Citation10,Citation15 Gordon et al conducted two single-arm non-randomized, age and gender-matched clinical trials on farnesyltransferase inhibitors. In the first trial, they administered lonafarnib as monotherapyCitation15 with no concurrent control group. The primary outcome was an improvement in weight gain rate, followed by cardiovascular distensibility, increase in bone rigidity and sensorineural hearing, but the duration of the trial was insufficient to demonstrate an improvement in survival. In the second trail, they compared triple-therapyCitation16 (farnesyltransferase inhibitors, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor and a bisphosphonate) with historic and concurrent untreated control participants enrolled in the prior lonafarnib monotherapy treatment trial. Concluding that triple-therapy did not provide any additional benefit compared to lonafarnib monotherapy.Citation16 There was a third observational cohort study, age, gender and continent matched derived from the previous two treatment trials conducted to compare contemporaneous treated patients with lonafarnib vs untreated patients. Showing lower mortality rate after 2.2 years.Citation17 Despite these results, it is important to highlight that farnesylation inhibitors do not reverse the disease and therefore are not curative.Citation15
Currently, CRISPR/Cas9 gene editing seems a promising strategy for the treatment of genetic diseases, including HGPS.Citation18 Another important scenario to keep in mind with HGPS children is to prevent secondary complications; therefore, some authors recommend the use of aspirin (2–3 mg/kg per day) as a cardio-cerebrovascular protector.Citation9
Although most HGPS cases are associated with a new mutation, Wuyts et alCitation19 described an affected individual whose mutation was transmitted by his asymptomatic mother, who presented somatic and germline mosaicism to the classic mutation. Therefore, promoting genetic counseling to parents of children with HGPS is essential because prenatal tests are available, and the risk of recurrence is 1 in 500 siblings.Citation9
Differential diagnoses for HGPS include the following autosomal recessive syndromes: Wiedemann-Rautenstrauch syndrome, neonatal progeroid disorder characterized by lipodystrophy, growth retardation, triangular face, and dental anomalies suggested to be caused by biallelic variants in POLR3A;Citation20 Rothmund–Thomson syndrome that compromises the RECQL4 gene and is associated with baldness, short stature, skin pigmentation, cataracts, and abnormalities of bones, nails, and teeth; Cockayne syndrome, caused by mutations in the ERCC8 gene and usually presents with cutaneous photosensitivity, retinal degeneration, short height, large ears, long limbs and feet, and large hands; Werner syndrome, caused by mutations in the RECQL2 or WRN gene, that manifests as bilateral cataracts, thinning and graying of the hair, short stature, ankle sores, hyperkeratosis, subcutaneous atrophy, and “bird-like” facial features,Citation21 that appears at 20–30 years of age.
There are two more lipodystrophy syndromes linked to the LMNA gene. One of them is the mandibuloacral dysplasia type A (MADA) characterized for craniofacial, skeletal and cutaneous abnormalities, loss of subcutaneous fat from the extremities along with normal or excessive fat in the face and neck.Citation22,Citation23 The latter is associated with mutations that disrupt nuclear function and therefore premature cell death in many tissues.Citation24 The second one is the atypical progeroid syndrome caused by molecular defects in exon 1 through 6 of the LMNA gene. It presents with overlapping muscular symptoms, skin defects, cardiomyopathy and rhythm abnormalities, as well as variable progeroid features, and partial or generalized loss of subcutaneous fat.Citation22 Extremely rare genetic lipodystrophy syndromes are listed in .
Table 1 Extremely Rare Genetic Lipodystrophy Syndromes
This study suggests that patients with HGPS should be managed by a multidisciplinary health team that includes a geneticist, cardiologist, and pediatric palliative care, to meet all needs of children with this condition and their families.
Data Sharing Statement
Can be accessed contacting the corresponding author.
Ethics Approval
The present study was previously approved by the institutional Internal Review Board, Comite de etica en investigacion biomedica of Fundacion Valle Del Lili.
Consent for Publication
Written informed consent was signed by the parents authorizing to perform genetic tests, use case details, pictures and publish the case.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Acknowledgment
We thank the family and the patient for their participation in this study.
Disclosure
The authors report no conflicts of interest in this work.
Additional information
Funding
References
- La Fundación de Investigación Progeria. ¡Juntos, encontraremos la cura! Available from: https://www.progeriaresearch.org/es/. Accessed February 7, 2020.
- Hennekam RCM. Hutchinson–Gilford progeria syndrome: review of the phenotype. Am J Med Genet Part A. 2006;140A(23):2603–2624. doi:10.1002/ajmg.a.31346
- Merideth MA, Gordon LB, Clauss S, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358(6):592–604. doi:10.1056/NEJMoa0706898
- Coppedè F. The epidemiology of premature aging and associated comorbidities. Clin Interv Aging. 2013;8:1023–1032. doi:10.2147/CIA.S37213
- González Morán MG. Síndrome de Progeria de Hutchinson-Gilford. Causas, investigación y tratamientos farmacológicos. Educ Quim. 2014;25(4):432–439. doi:10.1016/S0187-893X(14)70063-1
- DeBusk FL. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J Pediatr. 1972;80(4):697–724. doi:10.1016/s0022-3476(72)80229-4
- Gordon LB, McCarten KM, Giobbie-Hurder A, et al. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120(4):824–833. doi:10.1542/peds.2007-1357
- Gordon LB, Massaro J, D’Agostino RB Sr, et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130(1):27–34. doi:10.1161/CIRCULATIONAHA.113.008285
- Gordon LB, Brown WT, Collins FS. Hutchinson-Gilford progeria syndrome - geneReviews® - NCBI bookshelf. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1121/. Accessed February 4, 2020.
- Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–298. doi:10.1038/nature01629
- De Sandre-giovannoli A, Bernard R, Cau P, et al. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003;300(5628):2055. doi:10.1126/science.1084125
- Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11(4):440–445. doi:10.1038/nm1204
- Kieran MW, Gordon L, Kleinman M. New approaches to progeria. Pediatrics. 2007;120(4):834LP- 841. doi:10.1542/peds.2007-1356
- Fong LG, Ng JK, Lammerding J, et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest. 2006;116(3):743–752. doi:10.1172/JCI27125
- Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109(41):16666–16671. doi:10.1073/pnas.1202529109
- Gordon LB, Kleinman ME, Massaro J, et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation. 2016;134(2):114–125. doi:10.1161/CIRCULATIONAHA.116.022188
- Gordon LB, Shappell H, Massaro J, et al. Association of lonafarnib treatment vs no treatment with mortality rate in patients with Hutchinson-Gilford progeria syndrome. JAMA. 2018;319(16):1687–1695. doi:10.1001/jama.2018.3264
- Santiago-Fernández O, Osorio FG, Quesada V, et al. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat Med. 2019;25(3):423–426. doi:10.1038/s41591-018-0338-6
- Wuyts W, Biervliet M, Reyniers E, D’Apice M, Novelli G, Storm K. Somatic and gonadal mosaicism in Hutchinson-Gilford progeria. Am J Med Genet A. 2005;135:66–68. doi:10.1002/ajmg.a.30663
- Paolacci S, Bertola D, Franco J, et al. Wiedemann–Rautenstrauch syndrome: a phenotype analysis. Am J Med Genet Part A. 2017;173(7):1763–1772. doi:10.1002/ajmg.a.38246
- Kashyap S, Shanker V, Sharma N. Hutchinson - Gilford progeria syndrome: a rare case report. Indian Dermatol Online J. 2014;5(4):478. doi:10.4103/2229-5178.142507
- Akinci B, Meral R, Oral EA. Phenotypic and genetic characteristics of lipodystrophy: pathophysiology, metabolic abnormalities, and comorbidities. Curr Diab Rep. 2018;18(12):12. doi:10.1007/s11892-018-1099-9
- Simha V, Agarwal AK, Oral EA, Fryns J-P, Garg A. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin Endocrinol Metab. 2003;88(6):2821–2824. doi:10.1210/jc.2002-021575
- Hussain I, Garg A. Lipodystrophy syndromes. Endocrinol Metab Clin North Am. 2016;45(4):783–797. doi:10.1016/j.ecl.2016.06.012
- Agarwal AK, Fryns J-P, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12(16):1995–2001. doi:10.1093/hmg/ddg213
- Pelosini C, Martinelli S, Ceccarini G, et al. Identification of a novel mutation in the polymerase delta 1 (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism. 2014;63(11):1385–1389. doi:10.1016/j.metabol.2014.07.010
- Garg A, Kircher M, Del Campo M, Amato RS, Agarwal AK, Genomics U of WC for M. Whole exome sequencing identifies de novo heterozygous CAV1 mutations associated with a novel neonatal onset lipodystrophy syndrome. Am J Med Genet A. 2015;167(8):1796–1806. doi:10.1002/ajmg.a.37115