
Abstract
Primary aldosteronism is the most common cause of secondary hypertension. The syndrome accounts for 10% of all cases of hypertension and is primarily caused by bilateral adrenal hyperplasia or aldosterone-producing adenoma. Over the last few years, the use of exome sequencing has significantly improved our understanding of this syndrome. Somatic mutations in the KCNJ5, ATP1A1, ATP2B3 or CACNA1D genes are present in more than half of all cases of aldosterone-producing adenoma (~40%, ~6%, ~1% and ~8%, respectively). Germline gain-of-function mutations in KCNJ5 are now known to cause familial hyperaldosteronism type III, and an additional form of genetic hyperaldosteronism has been reported in patients with germline mutations in CACNA1D. These genes code for channels that control ion homeostasis across the plasma membrane of zona glomerulosa cells. Moreover, all these mutations modulate the same pathway, in which elevated intracellular calcium levels lead to aldosterone hyperproduction and (in some cases) adrenal cell proliferation. From a clinical standpoint, the discovery of these mutations has potential implications for patient management. The mutated channels could be targeted by drugs, in order to control hormonal and overgrowth-related manifestations. Furthermore, some of these mutations are associated with high cell turnover and may be amenable to diagnosis via the sequencing of cell-free (circulating) DNA. However, genotype-phenotype correlations in patients harboring these mutations have yet to be characterized. Despite this recent progress, much remains to be done to elucidate the yet unknown mechanisms underlying sporadic bilateral adrenal hyperplasia.
Introduction
Primary aldosteronism (PA) represents the autonomous hypersecretion of aldosterone that is relatively independent of the reninangiotensin system, adrenocorticotropic hormone (ACTH) and plasma [K+].Citation1 The disease is mainly caused by bilateral adrenal hyperplasia or aldosterone-producing adenoma (APA),Citation2,Citation3 which together account for over 90% of cases. Other rare causes (such as primary unilateral adrenal hyperplasia, aldosterone-producing adrenocortical carcinoma, and ectopic aldosterone-producing tumor and a number of familial forms) are responsible for less than 10% of cases.
In 1955, Jerome W Conn described PA for the first time.Citation4 A few years later, he suggested that PA could account for as many as 10% of all cases of essential hypertension. Nevertheless, PA continued to be considered as a rare cause of hypertension until a number of studies confirmed the presence of PA in between 5% and 13% of hypertensive patientsCitation5–Citation8 and in as many as one fifth of patients with resistant hypertension.Citation9,Citation10 Hence, PA is undoubtedly the most common cause of secondary hypertension. Moreover, research also confirmed that patients with PA have an elevated risk of cardiovascular complications (stroke, nonfatal myocardial infarction and atrial fibrillation) when compared with matched controls with essential hypertension and the same degree of blood pressure control.Citation11–Citation13 William F Young called this rediscovery of PA’s prevalence and morbidity as the “renaissance of a syndrome.”Citation14 However, the pathophysiology of PA remained unclear until Choi et al made an important breakthrough by reporting the presence of mutations in the gene encoding the inwardly rectifying potassium channel Kir3.4 (KCNJ5) in patients with aldosterone-producing adenoma.Citation15 In the last two years, the use of whole genome sequencing has led to a revolution in PA diagnosis and research. Mutations in three further membrane proteins (the Na+/K+ ATPase, the Ca2+ ATPase and Cav1.3) have been described. Genetic variances in these genes are associated with elevated intracellular calcium levels and/or activated calcium signaling pathways – the end result of which is aldosterone hyperproduction plus (in some cases) adrenal cell proliferation (adenoma formation or hyperplasia). Moreover, the molecular basis for a recently reported form of familial hyperaldosteronism has been elucidated and a further form had been described. These major steps have significantly improved our knowledge of PA and its pathogenesis.
In the present article, we review the genetics of PA. By way of an introduction, we provide a summary of aldoster-one biosynthesis. Lastly, we comment on applications and perspectives.
Aldosterone biosynthesis
Aldosterone is secreted by zona glomerulosa (ZG) cells located in the subcapsular layer of the adrenal gland cortex. It is produced in a series of specific enzymatic reactions, the final steps of which are catalyzed by aldosterone synthase (encoded by CYP11B2). The CYP11B2 gene shares a high degree of homology (95%) with the gene for 11 β-hydroxylase (CYP11B1), which catalyzes the final steps in cortisol biosynthesis in the zona fasciculata (ZF). Although both genes are located in tandem on chromosome 8q21–22, each is expressed in its respective adrenal zone only.Citation16
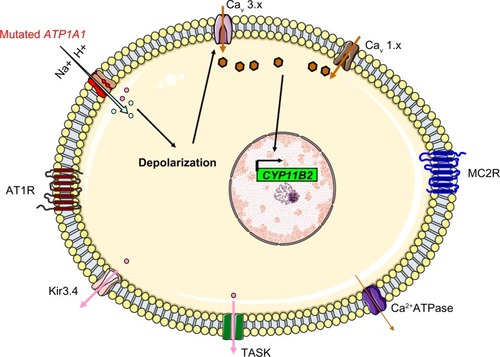
The main aldosterone secretagogs are angiotensin II, potassium and (to a lesser extent) ACTH. In the basal state (), ZG cells are hyperpolarized (–78 to –90 mV) because of background K “leakage” essentially through TWIK (tandem of P domains in a weak inward rectifying K+ channel)-related acid sensitive K (TASK) channels 1 and 3.Citation17,Citation18 Binding of angiotensin II to the type 1 angiotensin II receptor (AT1R) blocks these leaky channels as well as other potassium channels (such as Kir3.4) as well as the Na+/K+ ATPase and thus leads to glomerulosa membrane depolarization, activation of voltage-gated calcium channels (essentially low-voltage activated channels but also high-voltage activated channels) and calcium influx (). Calcium is also released from intracellular stores via the activation of phospholipase C.Citation19 Changes in extracellular potassium can also directly cause membrane depolarization and thus calcium entry.Citation20 Calcium stimulates aldosterone production in several actions at different steps,Citation21 by:
Figure 1 A simplified, schematic diagram of ZG cells at rest, when the cells are hyperpolarized due to leaky K+ channels (TASK). Na+/K+ ATPase and Kir3.4 contribute to the maintenance of membrane hyperpolarization. The voltage-gated Ca2+ channels are closed.
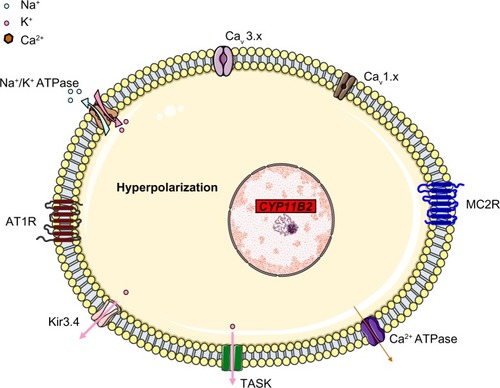
Figure 2 Regulation of aldosterone secretion in the physiologic state. Binding of angiotensin II to the AT1R blocks Kir3.4 potassium channels, TASK and Na+/K+ ATPase (1), this depolarizes the membrane (2) and results in the opening of voltage-gated Ca2+ channels (3). The resulting increase in intracellular levels provides the signal for increased expression of aldosterone synthase (4) through many different actions (including increased transcription of steroidogenic acute regulator protein).
Increasing the activity of cholesterol ester hydrolase, which de-esterifies cholesterol and thus releases it from its cytoplasmic storage sites.
Stimulating the delivery of cholesterol to the outer mitochondrial membrane.
Promoting the transcription and translation of steroidogenic acute regulator protein (StAR), which thus increases the intramitochondrial transfer of cholesterol to the inner membrane (a rate-limiting step in aldosterone synthesis).
Increasing mitochondrial oxidative metabolism and the formation of certain cofactors required for CYP11B2 activity.
Activating the calcium/calmodulin-dependent protein kinase I and IV, which activate many transcription factors such as Nur-related factor 1 (NURR1), nerve growth factor induced clone B (NGFIB), activating transcription factor 1 (ATF1), and cyclic AMP (cAMP) response element binding protein 1 (CREB1). These transcription factors in turn enhance the transcription of aldosterone synthase.Citation22
Somatic mutations in aldosterone-producing adenoma
Aldosterone-producing adenoma is responsible for 30%–50% of cases of PA.Citation23 Indeed, its prevalence in PA might have been underestimated because of failure to systematically perform adrenal vein sampling in the diagnostic workup. The identification of unilateral APA is very important in the management of PA, since it implies that curative surgical treatment is possible.
Somatic mutations in KCNJ5
The KCNJ5 gene (located on 11q24.3) codes for Kir3.4, a member of the G-protein-activated inwardly rectifying potassium channel family (subfamily J, member 5). Kir3.4 is expressed in the ZG, where it forms a highly-active heterotetramer with Kir3.1 (KCNJ3) that contributes to maintenance of the K+ equilibrium potential across the cell membrane. It also can form a less active homotetramer. These channels normally draw potassium out of the cell and thus help to maintain the hyperpolarization of the ZG membrane at rest. Selective passage of potassium through the channel pore is conferred by a GYG motif at the narrowest part of the pore. This selectivity filter is very well conserved among different species and among the other channel in the family. A loss-of-function mutation in KCNJ5 reportedly causes a rare form of long QT syndrome (type 13).Citation24
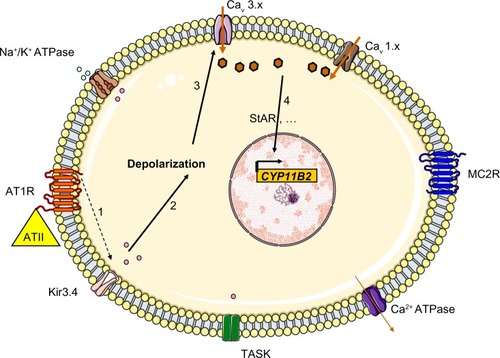
By using whole-genome sequencing, Choi et al searched for somatic mutations in a cohort of 22 patients with APA and found two recurrent mutations in eight cases (38%): p.Gly151Arg (G151R) or p.Leu168Arg (L168R). These mutations occurred within or near the Kir3.4’s selectivity filter and thus altered the channel’s selectivity. G151 is the first glycine in the GYG motif, whereas L168’s side chain abuts the side chain of the tyrosine in the same motif (Y152). These amino acid substitutions are associated with a diminished K+/Na+ permeability ratio (1 for the G151R mutant and 1.3 for the L168R mutant, relative to a value of 25.3 in the wild type channel). This loss of channel selectivity is responsible for increased Na+ conductance. In turn, Na+ entry triggers cell depolarization and thus subsequent calcium entry. Elevation of intracellular calcium levels is a key step in the aldosterone hyperproduction process (). Functional studies have confirmed that overexpression of the mutant KCNJ5 in the HAC15 human adrenocortical carcinoma cell line results in increased aldosterone production. CYP11B2 messenger RNA (mRNA) levels were found to be upregulated in APAs with KCNJ5 mutations (by a factor of 3.1, relative to APAs without KCNJ5 mutations).Citation25 However, the mechanism by which chronic Ca2+ stimulation promotes increased proliferation in the ZG has not been fully elucidated, since calcium overload is known to be cytotoxic. Visinin-like 1 (VSNL1, a neuronal calcium-sensor protein which participates in the transduction of calcium signaling) is found to be overex-pressed by a factor of 8.1 in APAs harboring mutations in KCNJ5 (when compared with APAs expressing wild-type KCNJ5). Increased expression of VSNL1 is thought to protect cells from calcium-induced death (ie, it has an anti-apoptotic effect).Citation26
Figure 3 The mechanism thought to underlie hyperaldosteronism in patients with KCNJ5 mutations that cause Kir3.4 to lose its potassium selectivity. The mutated channels allow sodium entry, which triggers depolarization.
Mulatero et al discovered a somatic p.Thr158Ala (T158A) mutation in an Italian patient with APA and a family history of PA.Citation27 The same mutation had been reported in the original article by Choi et al but as a germline mutation. Threonine in position 158 is also a well-conserved amino acid that forms hydrogen bonds with two conserved residues (P128 and C129) in the loop between the first transmembrane domain and the pore helix, and thus constrains the structure. The T158A mutation eliminates these hydrogen bonds and thus is expected to modify channel selectivity. To date, only four other somatic mutations in KCNJ5 have been described in patients with APAs. Each mutation has been reported only once: a deletion mutation affecting isoleucine 157 (p.Ile157del), which is presumed to induce a conformational change near the selectivity filter;Citation28 two substitutions, both of which affect the same amino acid (glutamic acid 145), p.Glu145Gln (E145Q)Citation29 and p.Glu145Lys (E145K);Citation30 and a p.Trp126Arg (W126R) mutation.Citation31 Either (G151R) or (L168R) is present in 99% of KCNJ5-mutated APAs, whereas the five other mutations are extremely rare.
It has now been established in a number of studies that somatic mutations of KCNJ5 are present in approximately 40% (range: 13%–65%) of APAs.Citation32,Citation33 The proportion of APAs with a KCNJ5 mutation differs considerably from one center to another, which might reflect disparities in the criteria used to differentiate between unilateral PA and bilateral PA and establish the diagnosis of APA. Only one study found a very low prevalence (12.5%).Citation34 The highest prevalence (65.2%) was found in a Japanese study. It is noteworthy that when compared with studies in Western countries, studies from Japan report a much higher prevalence of APAs (approximately 80%) in patients with PA.Citation35 However, the results of almost all studies confirm that KCNJ5 mutations are more frequent in women than in men. Only studies in Japan describe a high prevalence in men – again suggesting the presence of epidemiological differences between Japanese and Western populations. Furthermore, KCNJ5 mutation carriers are generally younger than nonmutated APA patients, which suggests earlier disease onset and/or more severe disease and thus earlier diagnosis. Some of the abovementioned studies have found evidence of more severe disease in KCNJ5 mutation carriers, including higher aldosterone levels at diagnosis and lower potassium levels.Citation36 Only a few studies have found that patients with KCNJ5 mutations have larger tumors (on average) than patients without KCNJ5 mutations.Citation37
Interestingly, some researchers suggest that there are at least two subtypes of APA, with one subtype containing clear-type cells with a ZF-like morphology and another containing predominantly compact cells with a ZG-like morphology. To test the relationship between APA subtypes and KCNJ5 mutations, Azizan et al analyzed CYP17A1 expression in 22 APAs. CYP17A1 was chosen as a biomarker because it codes for an enzyme that is normally not expressed in the ZG. Nine of the 22 APAs showed relatively high levels of CYP17A1 and CYP11B1 expression. In histological analyses, these APAs appeared to contain a majority of clear cells with a high cytoplasm-to-nucleus ratio. Hence, Azizan et al referred to this subtype as a “ZF-like” APA. Another nine cases showed low expression of CYP17A1 and were referred to as “ZG-like” APAs, which contained compact cells with a low cytoplasm-to-nucleus ratio. CYP17A1 expression was higher in APAs with a KCNJ5 mutation than in those lacking a mutation (ie, the wild type). Furthermore, the proportion of ZG-like cells was lower in APAs with a KCNJ5 mutation than in wild-type APAs.Citation38 Azizan et al suggested that APAs with a somatic KCNJ5 mutation arise from ZF cells, even though the possibility that cytological features and modifications were due to the effect of the mutation on ZG-derived cells could not be ruled out. One can hypothesize that the occurrence of a somatic mutation in the activated adrenal stem cell/progenitor cells could cause autonomous secretion of aldosterone and morphological changes that would make mutated cells resemble ZF cells. However, there is no valid evidence to support any of these three hypotheses. Furthermore, the abovementioned report about a relationship between ZF-like APAs and KCNJ5 mutations has not been confirmed by any other group so far. On the contrary, Boulkroun et al investigated the cell composition of 31 APAs and found that 26 APAs were mainly composed of ZF-like cells and five were mainly composed of ZG-like cells; however, there was no significant intergroup difference in terms of the prevalence of KCNJ5 mutations.Citation39
Somatic mutations in ATP1A1
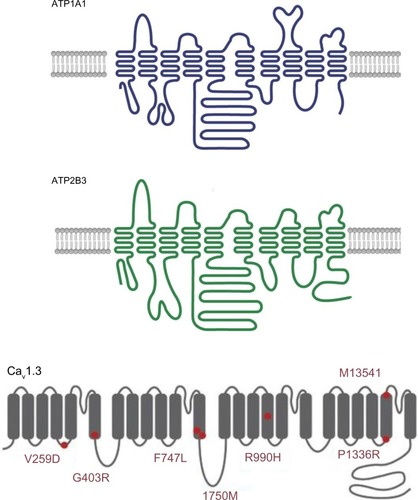
The ATP1A1 gene encodes the alpha-1 (catalytic) subunit of the Na+/K+ ATPase, a member of the P-type ATPase family. It maps to chromosome 1p13.1 and comprises 10 transmembrane segments (from M1 to M10, with M1 being the most N-terminal segment). There are two functionally important cytoplasmic loops: a large loop between M4 and M5 (which binds ATP) and a small loop between M2 and M3 (which has a key role in energy transduction) (). Upon activation, the transmembrane segment M1 interacts and cooperates with segment M4 to facilitate ion binding. For each ATP hydrolyzed, the Na+/K+ ATPase transports two K+ ions into the cell and three Na+ ions out of the cell. The resulting potassium and sodium gradients contribute to the resting membrane potential and action potentials. ATP1A1 is strongly expressed in the ZG and (to a lesser degree) in the ZF.
Figure 4 A schematic diagram of the structure of Na+/K+ ATPase subunit alpha 1 (upper panel, ATP1A1), Ca2+ ATPase 3 (middle panel, ATP2B3) and the alpha subunit of low-voltage activated calcium channel Cav1.3, with known mutations indicated by red circles (lower panel).
Recently, Beuschlein et al performed exome sequencing of tumor and matched control tissue samples from nine males affected with hypokalemic PA and bearing wild-type Kir3.4. The researchers found that three patients had somatic ATP1A1 mutations: substitution of the leucine in position 104 (in M1) p.Leu104Arg (L104R) in two cases and substitution of the valine in the position 332 (in segment M4) p.Val332Gly (V332G) in the third case. Both of these amino acids are highly conserved among species and among different members of the P-type ATPase family. Beuschlein et al then sequenced the entire coding region of ATP1A1 in an additional 100 APAs and detected six cases with somatic mutations: four had the L104R mutation, while the other two featured a five-amino-acid-deletion encompassing the same leucine (p.Phe100_Leu104del). Finally, the researchers performed targeted sequencing of the affected genomic locations in 199 additional APAs and confirmed that ATP1A1 mutations are present in 5.2% of patients with APA.Citation40 The sequencing data indicated that both the wild-type and mutated alleles were present in tumor tissues – suggesting that the mutation was heterozygous. All the abovementioned alterations in the ATP1A1 disrupt the glutamic acid in position 334 (site II), which is crucial for sodium and potassium ion binding and gating of the binding pocket. The leucine104 is located only 4 Å from glutamic acid 334; replacing it by a large, positively charged arginine would alter the position of site II. The V332G mutation would be associated with greater flexibility (affecting the position of site II) because glycine does not have a side chain.
Beuschlein et al also examined the functional impact of the ATP1A1 mutations and found that mutated ATP1A1 have impaired activity. When compared with the wild type, mutated ATP1A1 proteins showed very low affinity for potassium (0.2% for the L104R mutant and 2% for the V332G mutant). The impaired activity did not result from underexpression, since expression levels of the mutants were normal. When cultured, adenoma cells with an ATP1A1 mutation showed substantially higher levels of depolarization. The cells hyperpolarized when extracellular sodium was removed – thus suggesting higher sodium conductance and perhaps a disturbance of the intracellular ion composition.
Another group selected 10 ZG-like APAs (>50% compact cells and low CYP17A1 expression) for genome sequencing and identified four mutations in ATP1A1: two L104R mutations, one with the (p.Phe100_Leu104del) deletion-mutation, and a substitution of residues 960–963 by serine (p.GluGluThrAla963Ser). The latter mutation includes the glutamic acid 961 (in segment M9), which is thought to be important for the Na+-specific site III. Functional studies showed that all mutants were associated with an inward current. By varying the extracellular pH and replacing Na+ with N-methyl-D-glucamine, Azizan et al found that protons carry the current in the case of the L104R mutation, whereas Na+ is the main carrier in the case of the p.Phe100_Leu104del mutation. Hence, the researchers suggested that mutations in ATP1A1 change the transport mode from active pumping (three Na+ outwards and two K+ inwards) to passive conduction (inward current due to protons or Na+ ions, depending on the mutation) (). This impairment in conduction leads to membrane depolarization, opening of voltage-gated calcium channels, increased intracellular calcium and thus increased CYP11B2 expression and aldosterone hyperproduction.Citation30
Figure 5 The molecular mechanism thought to underlie hyperaldosteronism in patients with ATP1A1 mutations. The Na+/K+ ATPase changes from actively pumping (three Na+ outwards and two K+ inwards) to passive conduction of an inward current (protons or Na+ ions, depending on the mutation).
A further report from Italy found that ATP1A1 mutations are present in 6.3% of patients with APAs. A new somatic mutation was described (p.Gly99Arg, G99R). Glycine 99 interacts with isoleucine 292 to mediate the opening of the gate at the entrance of the binding pocket. Arginine in the position 99 may alter the surrounding structure (including glutamic acid at position 334).Citation31
Somatic mutations in ATP2B3
The ATP2B3 gene encodes the plasma membrane calcium transport ATPase 3 (PMAC3), another member of the P-type ATPase family. Its structure is similar to that of ATP1A1, with 10 transmembrane segments and two functionally important cytoplasmic loops (). ATP2B3 is required for clearing calcium from the cytoplasm of eukaryotic cells and thereby has an important role in intracellular calcium homeostasis. ATP2B3 is located on the X chromosome (Xq28) and is expressed throughout the adrenal cortex.
Mutations in ATP2B3 were discovered at the same time as those in ATP1A1; Beuschlein et al reported three different in-frame deletions that resulted in either p.Leu425_Val426del or p.Val426_Val427del. These deletions affect a highly conserved residue and thus alter the M4 transmembrane helix in a region that is thought to be crucial for calcium ion binding. Again, a functional study confirmed that ATP2B3 mutations impair activity and inactivate pump function. When cultured, adenoma cells with an ATP2B3 mutation showed substantially higher levels of depolarization.Citation40 To date, only one other deletion mutation (c.1281_1286delGGCTGT) had been reported,Citation41 suggesting that ATP2B3 mutations have a low prevalence in patients with APAs (0.9%–1.6%).
According to the results of some (but not all) studies, ATPase mutations are, 1) associated with more severe forms of PA and, 2) are more common in men. It has also been suggested that APAs with ATPase mutations are smaller than APAs with KCNJ5 mutations. However, these associations need to be confirmed in larger studies. It is noteworthy that KCNJ5 mutations have not been found with ATP1A1 or ATP2B3 mutations in the same tumor.
Somatic mutations in CACNA1D
The CACNA1D gene encodes Cav1.3, the alpha subunit of an L-type voltage-gated calcium channel. This family of channels (also known as high-voltage activated calcium channels) is characterized by sensitivity to dihydropyridine calcium channel blockers, which act as weak antagonists of Cav1.3. High-voltage-gated calcium channels differ from low-voltage-gated calcium channels (T-type channels) in terms of the voltage dependence of opening and inactivation and the kinetics of closure and inactivation. Cav1.3 is composed of four homologous repeats (I–IV), each of which has six transmembrane segments (S1–S6) and a membrane-associated loop between S5 and S6 (). The channel’s pore is formed by the S5, S6 and the loop. The CACNA1D gene is located on 3p14.3. Alternative splicing gives rise to many different isoforms. CACNA1D is expressed in the ZG and in many other tissues (heart, neurons, cochlear hair cells, etc,). Loss-of-function mutations in this gene have been recently discovered in sinoatrial node dysfunction and deafness (SANDD) syndrome.Citation42
By performing exome sequencing, Scholl et al found two somatic mutations in CACNA1D in 18 cases of APA. Both mutations affected well-conserved amino acids (p.Gly403Arg and p.Ile770Met) located at the cytoplasm ends of the S6 segment of repeats I and II (which line the inner pore). Interestingly, the mutation reported in SANDD syndrome is a homozygous three-base insertion that results in the insertion of glycine at position 404 and thus highlights the importance of this region in channel function. When expressed in HEK293T cells, both mutant genes are associated with peak current amplitudes at less depolarized potentials (with half-maximal activation voltages of −9.2, −25.6, and −31.7 mV for wild type, Gly403Arg, and Ile770Met, respectively). This finding indicates that mutations lead to channel opening at lower potentials (ie, closer to the resting membrane potential in ZG cells). The p.Gly403Arg mutation was also associated with sustained activation. When Scholl et al sequenced all S6 segments in an additional 46 APAs, they identified three additional somatic mutations (corresponding to a prevalence of 7.8%). Easier activation of the mutant channels results in increased Ca2+ entry and greater aldosterone production. Even spontaneous membrane oscillations may activate these mutant channels. In the same study, Scholl et al identified another two somatic mutations (p.Phe767Val and p.Val1373Met).Citation43
Another group studied ten ZG-like APAs and identified five somatic mutations in CACNA1D including two previously undescribed mutations. The authors then screened three additional cohorts (from three different countries) and found seven somatic mutations (three of which were previously undescribed) in 142 patients (giving an overall prevalence of 7%–8%). All the mutations affected conserved sites and were located in domains that are important for channel function. These mutations generally occur in males and are more frequent in small APAs. It has been suggested that these tumors are small because they are composed of ZG-type cells, which are lipid-poor and thus smaller than lipid-rich ZF-type cells.Citation44
Inherited genetic alterations in familial hyperaldosteronism
Four forms of genetic hyperaldosteronism have been described to date, including two reported very recently. These forms are rare and account for only 1%–10% of patients with PA in total.Citation45
Familial hyperaldosteronism type I
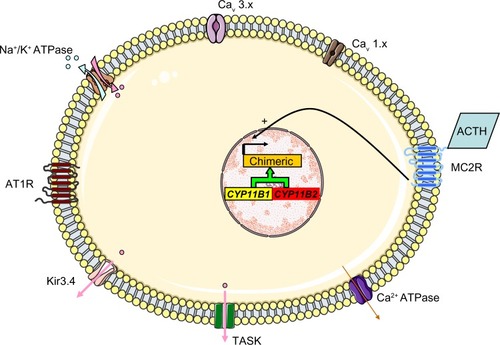
In 1966, Sutherland el alCitation46 described the case of a father and son with hypertension due to hyperaldosteronism. The father’s condition persisted after left adrenalectomy but was relieved by dexamethasone treatment. Familial hyperaldosteronism type I (FHI) is also known as glucocorticoid-remediable aldosteronism (GRA) and is characterized by severe, early-onset hypertension. Although GRA accounts for less that 1% of patients with PA, it is considered to be the most common form of monogenic hypertension.Citation47 Patients with GRA have an elevated risk of early hemorrhagic stroke and they secrete high levels of hybrid steroids (18-hydroxycortisol and 18-oxocortisol). Glucocorticoid-remediable aldosteronism is inherited as an autosomal, dominant trait. The molecular etiology of GRA was characterized in 1992;Citation48 unequal crossing-over between the 11 β-hydroxylase (CYP11B1) and aldosterone synthase (CYP11B2) genes results in a chimeric gene that combines the 5′ promoter region of the 11 β-hydroxylase with the coding region of aldosterone synthase (). As a result, aldosterone synthesis is regulated by ACTH (rather than by the renin-angiotensin system) and has a circadian rhythm similar to that of cortisol. This aberrant regulation explains why dexamethasone administration reduces aldosterone levels and normalizes hypertension. Nowadays, GRA is diagnosed with a polymerase chain reaction-based assay and so a dexamethasone suppression test or hybrid steroid assays are no longer required. The Endocrine Society recommends genetic testing in patients with confirmed PA before the age of 20 and in those who have a family history of PA or strokes at a young age.Citation1
Figure 6 The molecular mechanism thought to underlie GRA. Unequal crossing-over between the 11 β-hydroxylase (CYP11B1) and aldosterone synthase (CYP11B2) genes results in a chimeric gene and the control of aldosterone secretion by ACTH.
Familial hyperaldosteronism type II
In 1992, Stowasser et alCitation49 described a second form of hereditary hyperaldosteronism in 13 patients with PA from five families. This form of PA (designated as familial hyperaldosteronism type II [FHII]) cannot be suppressed by dexa methasone. Patients with FHII are biochemically and clinically indistinguishable from patients with sporadic PA.Citation50 Hence, FHII is defined as the occurrence of PA in two or more members of the same family. The true prevalence of this form is not known but might be as high as 6% in patients with PA.Citation51 The syndrome is probably heterogeneous. Although the molecular basis for FHII has yet to be determined, a link with the chromosomal region 7p22 has been established in some families. It is noteworthy that sequencing has not identified any candidate genes in and around the 7p22 region. Interestingly, Mulatero et al searched for mutations in KCNJ5 in 46 patients with familial hyperaldosteronism (presumed to be FHII) from 21 families and identified two patients from the same family with a germline mutation in KCNJ5 (ie, with familial hyperaldosteronism type III). This raises the possibility that some of the cases classified as FHII are caused by another form of familial hyperaldosteronism. Moreover, the same authors found three APAs with somatic mutations in KCNJ5.Citation27 This finding suggests that some cases of FHII might result from the familial aggregation of cases of sporadic PA. With regard to recently discovered mutations in patients with APA, Beuschlein et al did not find any germline mutations in ATP1A1 or ATP2B3 in 18 patients with FHII.Citation40
Familial hyperaldosteronism type III
In 2008, a third form of familial hyperaldosteronism was described. In fact, FHIII is characterized by severe hypertension in childhood (before the age of 7), severe hypokalemia, and PA with very high levels of hybrid steroids (18-oxocortisol and 18-hydroxycortisol). Glucocorticoid-remediable aldosteronism was suspected but dexamethasone treatment doubled aldosterone levels instead of normalizing them. Negative genetic tests for GRA definitively ruled out GRA as a cause of this clinical picture. The hypertension was refractory to combination therapy (meaning that adrenalectomy was required) in all three members of the described family (a father and his two daughters). Interestingly, the excised adrenals were markedly enlarged and microscopic examination revealed a thin, atrophic ZG, a slightly decreased zona reticularis, and diffuse hyperplasia of the ZF with transitional features on electron microscopy.Citation52 The genetic basis of this syndrome was discovered three years later, when a germline heterozygous T158A mutation in KCNJ5 was found in all affected members. Oki et al confirmed that expression of the T158A mutant in HAC15 cells resulted in increased Na+ inflow, a higher plasma membrane voltage, increased intracellular calcium levels and a 5.3-fold increase in aldosterone production (relative to HAC cells expressing wild-type KCNJ5). Nifedipine and calmodulin inhibitor decreased aldosterone production in the T158A mutant, which further emphasizes the role of intracellular calcium in the physiopathology of this syndrome. However, cell proliferation was lower in the T158A mutant than in the wild type.Citation53
Scholl et al reported on four additional families with FHIII and a surprising genotype-phenotype correlation. Two of the four families had a clinical course similar to that of the patients with the T158A mutation. Upon genetic testing, both families were found to have a germline mutation identical to one of the recurrent somatic mutations found in APAs (G151R). Interestingly, the other two families had a germline mutation that affected the same amino acid but resulted in a glycine to glutamic acid substitution (G151E). The clinical course of the families with the G151E mutation was also characterized by early-onset hypertension that was nevertheless easily controlled. No adrenal hypertrophy was evident. Most interestingly, functional studies confirmed that the G151E mutation was the most severe (with greater Na+ conductance than the G151R mutation) and was associated with poorer survival. Scholl et al concluded that much greater Na+ conductance is responsible for increased cell death, which hinders the formation of adenoma and/or hyperplasia.Citation54
Other inherited mutations in KCNJ5 have been reported to cause FHIII.Citation55,Citation56 However, in view of the limited number of families with FHIII it seems too early to speak about genotype/phenotype correlation or to divide FHIII into two or more subtypes (for example, with and without adrenal hyperplasia).
Germline mutations in CACNA1D cause primary aldosteronism
After discovering CACNA1D somatic mutations in APAs and by analogy with the situation for KCNJ5, Scholl et alCitation43 posited that germline mutations in CACNA1D could occur in PA. They screened 100 unrelated patients with early-onset PA and found two de novo mutations, both of which affected amino acids known to be subject to somatic mutations in APA.
The first subject had suffered from hypertension since birth, as well as congenital heart disease. The hypertension was refractory to treatment until amlodipine normalized the blood pressure and reduced biventricular hypertrophy. The subject had other clinical manifestations, including seizures, apparent cerebral palsy, cortical blindness, and complex neuromuscular abnormalities. He was found to harbor a p.Gly403Asp mutation in CACNA1D.
The second subject had cerebral palsy, spastic quadriplegia, mild athetosis, severe intellectual disability and seizures. By the age of 5, he displayed hypertension and hypokalemia. Genetic testing revealed a p.Ile770Met mutation in CACNA1D.
Only these two such cases have been reported to date and so it appears to be premature to define a phenotype for this association between germline mutations in CACNA1D and PA; the clinical picture seems to be very complex and affects many systems (in accordance with the expression of CACNA1D in a wide range of tissues).
Applications and perspectives
KCNJ5
A highly selective inhibitor of mutant KCNJ5 might suppress aldosterone secretion, stop cellular proliferation or perhaps even lead to tumor regression. This type of inhibitor would be of great use in patients with FHIII and patients with sporadic APA, since 40% harbor one of two somatic mutations (G151R or L168R) in KCNJ5. Furthermore, a specific inhibitor could be used in a diagnostic test to distinguish between patients with and without mutations.Citation57 Alternatively, the channel itself (Kir3.4) could become a drug target in hypertension.
Several studies have indicated that solid tumors (especially those with a high turnover, as in cases with KCNJ5 mutations) release DNA into the circulation. This circulating DNA might be detectable in plasma and would thus provide us with an easy, simple, rapid and relatively inexpensive diagnostic test for APA with a KCNJ5 mutation. However, this technology is still in its infancy.Citation58
KCNJ5 mutations (leading to abnormally high intracellular calcium, hormone hypersecretion and cellular overgrowth) might serve as a model for other endocrinopathies in which a hormonal syndrome and benign adenoma coexist.
CACNA1D
At low doses, dihydropyridine calcium channel blockers are weak inhibitors of Cav1.3. Selective, potent Cav1.3 inhibitors have already been described and might serve as a specific treatment for CACNA1D-associated PA. This type of compound might be useful in patients with KCNJ5 mutations, since we know that the latter lead to aldosterone production through increased calcium influx.
Perspectives
Although great progress has been made over the last two years, much work is still to be done. In particular, the molecular basis for bilateral adrenal hyperplasia (the most common type of PA in Western countries) is still obscure. It is noteworthy, that Boulkroun et al did not find any germline KCNJ5 mutations in 174 patients with bilateral adrenal hyperplasia.Citation32 Furthermore, screening for germline mutations in ATP1A1 and ATP2B3 was negative in 91 patients with bilateral adrenal hyperplasia.Citation40
More research is needed in order to understand why KCNJ5 mutations lead to adenoma formation or hyperplasia in vivo but are associated with decreased cellular proliferation in vitro.
It remains to be established whether KCNJ5 mutations arise in ZG cells, ZF cells or transitional cells that have yet to differentiate.
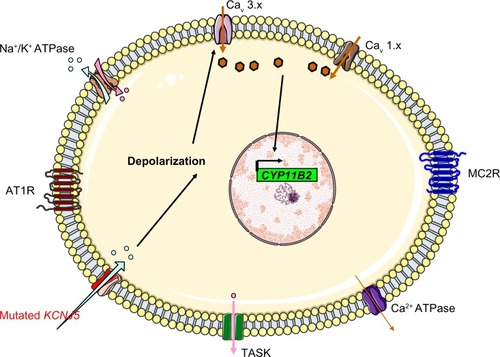
Do mutations outside the selectivity filter cause PA? And if so, by which mechanism? In a recent report Murthy et al resequenced the flanking and coding region of KCNJ5 in 251 patients with sporadic PA to search for rare variants (either germline mutations or single nucleotide polymorphisms). The authors identified three heterozygous missense mutations that affected well-conserved amino acids outside the selectivity filter (R52H, E246K and G247R). The R52H and E246K substitutions resulted in loss of channel selectivity while the third one (G247R) did not alter the channel behaviour. In the first family (R52H kinship) two sons of the index case carry the mutation without clinical manifestations of PA while only the index case was sequenced in the E246K family. So it is not possible at the present time to rule out a familial form (FHIII) with incomplete penetrance (due to age-related penetrance or to another factor). No clear mechanism exists to explain how mutations distant to selectivity filter affect the channel selectivity.Citation59
The role of Single Nucleotide Polymorphism (SNP) in the KCNJ5 gene is to be determined. Murthy et alCitation59 found that the (E282Q) SNP was present in 12 of 251 patients with PA while its presumed population frequency is about 2%. The E282Q showed a substantial loss of channel selectivity, modest increase in angiotensin II-induced aldosterone release and reduced cell viability when expressed in the H295R cells. The authors proposed that the E282Q is associated with PA but this association needs to be confirmed in larger studies in a population in which the frequency of E282Q carriers is well-known. Another report from the People’s Republic of China proposed that a SNP in the 3′ untranslated region of the KCNJ5 is associated with PA in Chinese males.Citation60
There is no current explanation for sex differences: KCNJ5 mutations are more frequent in women and ATPase mutations are more frequent in men. Do sex steroids have a role?
All the mutations described to date result in increased calcium influx and thus increased intracellular calcium levels. However, the status of other sites in the calcium signaling pathway is not known. Are there any downstream mutations that lead to increased intracellular calcium release or enhanced downstream pathways?
More cases are necessary if we are to define the phenotypes associated with familial hyperaldosteronism.
Acknowledgments
We are grateful to Felix Beuschlein and Hanne Poulsen for providing the images in .
Disclosure
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector. All authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- FunderJWCareyRMFardellaCEndocrine SocietyCase detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guidelineJ Clin Endocrinol Metab20089393266328118552288
- RossiGPSecciaTMPessinaACPrimary aldosteronism: part II: subtype differentiation and treatmentJ Nephrol200821445546218651533
- FunderJWThe genetics of primary aldosteronism: chapter twoHypertension201259353753822275537
- ConnJWPresidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndromeJ Lab Clin Med195545131713233623
- RossiGPBerniniGCaliumiCPAPY Study InvestigatorsA prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patientsJ Am Coll Cardiol200648112293230017161262
- RossiGPSecciaTMPessinaACPrimary aldosteronism – part I: prevalence, screening, and selection of cases for adrenal vein samplingJ Nephrol200821444745418651532
- FagugliRMTaglioniCChanges in the perceived epidemiology of primary hyperaldosteronismInt J Hypertens2011201116280421837271
- AmarLPlouinPFSteichenOAldosterone-producing adenoma and other surgically correctable forms of primary aldosteronismOrphanet J Rare Dis20105920482833
- MossoLCarvajalCGonzálezAPrimary aldosteronism and hypertensive diseaseHypertension200342216116512796282
- ZennaroMCPrimary aldosteronism takes (KCNJ) five!Endocrinology201215341575157722447911
- MilliezPGirerdXPlouinPFBlacherJSafarMEMouradJJEvidence for an increased rate of cardiovascular events in patients with primary aldosteronismJ Am Coll Cardiol20054581243124815837256
- StowasserMUpdate in primary aldosteronismJ Clin Endocrinol Metab200994103623363019737921
- MulateroPMonticoneSBertelloCLong-term cardio- and cerebrovascular events in patients with primary aldosteronismJ Clin Endocrinol Metab201398124826483324057288
- YoungWFPrimary aldosteronism: renaissance of a syndromeClin Endocrinol (Oxf)200766560761817492946
- ChoiMSchollUIYuePK+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertensionScience2011331601876877221311022
- McMahonGTDluhyRGGlucocorticoid-remediable aldosteronismArq Bras Endocrinol Metabol200448568268615761539
- CareyRMRole of K(+) channels in the pathophysiology of primary aldosteronismHypertension201259353453622252399
- Velarde-MirandaCGomez-SanchezEPGomez-SanchezCERegulation of aldosterone biosynthesis by the Kir3.4 (KCNJ5) potassium channelClin Exp Pharmacol Physiol2013401289590123829355
- HattangadyNGOlalaLOBollagWBRaineyWEAcute and chronic regulation of aldosterone productionMol Cell Endocrinol2012350215116221839803
- BeuschleinFRegulation of aldosterone secretion: from physiology to diseaseEur J Endocrinol20131686R85R9323568484
- GuagliardoNAYaoJHuCBarrettPQMinireview: aldosterone biosynthesis: electrically gated for our protectionEndocrinology201215383579358622689262
- BassettMHSuzukiTSasanoHWhitePCRaineyWEThe orphan nuclear receptors NURR1 and NGFIB regulate adrenal aldosterone productionMol Endocrinol200418227929014645496
- Gomez-SanchezCEGomez-SanchezEPMutations of the potassium channel KCNJ5 causing aldosterone-producing adenomas: one or two hits?Hypertension201259219619722203746
- YangYYangYLiangBIdentification of a Kir3.4 mutation in congenital long QT syndromeAm J Hum Genet201086687288020560207
- MonticoneSHattangadyNGNishimotoKEffect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cellsJ Clin Endocrinol Metab2012978E1567E157222628608
- WilliamsTAMonticoneSCrudoVWarthRVeglioFMulateroPVisinin-like 1 is upregulated in aldosterone-producing adenomas with KCNJ5 mutations and protects from calcium-induced apoptosisHypertension201259483383922331379
- MulateroPTauberPZennaroMCKCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronismHypertension201259223524022203740
- AzizanEAMurthyMStowasserMSomatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomasHypertension201259358759122252394
- ÅkerströmTCronaJDelgado VerdugoAComprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filterPLoS One201277e4192622848660
- AzizanEAPoulsenHTulucPSomatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertensionNat Genet20134591055106023913004
- WilliamsTAMonticoneSSchackVRSomatic ATP1A1, ATP2B3, and KCNJ5 Mutations in Aldosterone-Producing AdenomasHypertension201463118819524082052
- BoulkrounSBeuschleinFRossiGPPrevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronismHypertension201259359259822275527
- StowasserMPrimary aldosteronism and potassium channel mutationsCurr Opin Endocrinol Diabetes Obes201320317017923426162
- XekoukiPHatchMMLinLKCNJ5 mutations in the National Institutes of Health cohort of patients with primary hyperaldosteronism: an infrequent genetic cause of Conn’s syndromeEndocr Relat Cancer201219325526022323562
- TaguchiRYamadaMNakajimaYExpression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomasJ Clin Endocrinol Metab20129741311131922278422
- ZennaroMCJeunemaitreXBoulkrounSIntegrating genetics and genomics in primary aldosteronismHypertension201260358058822802222
- Al-SalamehACohenRChansonPPlouinPFUpdate on endocrine hypertensionAnn Endocrinol (Paris)201273Suppl 1S26S35 French23089379
- AzizanEALamBYNewhouseSJMicroarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumorsJ Clin Endocrinol Metab2012975E819E82922442279
- BoulkrounSGolib DzibJFSamson-CouterieBKCNJ5 mutations in aldosterone producing adenoma and relationship with adrenal cortex remodelingMol Cell Endocrinol20133711–222122723376008
- BeuschleinFBoulkrounSOsswaldASomatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertensionNat Genet2013454440444444e1–223416519
- Kumar DuttaRWelanderJBrauckhoffMComplementary somatic mutations in sporadic aldosterone producing adrenal adenomasEndocr Relat Cancer2014211L1L424179102
- BaigSMKoschakALiebALoss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafnessNat Neurosci2011141778421131953
- SchollUIGohGStöltingGSomatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronismNat Genet20134591050105423913001
- BrownMJIns and outs of aldosterone-producing adenomas of the adrenal: from channelopathy to common curable cause of hypertensionHypertension2014631242624082060
- ZennaroMCRickardAJBoulkrounSGenetics of mineralocorticoid excess: an update for cliniciansEur J Endocrinol20131691R15R2523610123
- SutherlandDJRuseJLLaidlawJCHypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasoneCan Med Assoc J19669522110911194288576
- McMahonGTDluhyRGGlucocorticoid-remediable aldosteronismCardiol Rev2004121444814667264
- LiftonRPDluhyRGPowersMA chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertensionNature199235563572622651731223
- StowasserMGordonRDTunnyTJKlemmSAFinnWLKrekALFamilial hyperaldosteronism type II: five families with a new variety of primary aldosteronismClin Exp Pharmacol Physiol19921953193221521363
- Hassan-SmithZStewartPMInherited forms of mineralo-corticoid hypertensionCurr Opin Endocrinol Diabetes Obes201118317718521494136
- MulateroPTizzaniDViolaAPrevalence and characteristics of familial hyperaldosteronism: the PATOGEN study (Primary Aldosteronism in TOrino-GENetic forms)Hypertension201158579780321876069
- GellerDSZhangJWisgerhofMVShackletonCKashgarianMLiftonRPA novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronismJ Clin Endocrinol Metab20089383117312318505761
- OkiKPlonczynskiMWLuis LamMGomez-SanchezEPGomez-SanchezCEPotassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesisEndocrinology201215341774178222315453
- SchollUINelson-WilliamsCYuePHypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5Proc Natl Acad Sci U S A201210972533253822308486
- CharmandariESertedakiAKinoTA novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertensionJ Clin Endocrinol Metab2012978E1532E153922628607
- MonticoneSHattangadyNGPentonDA Novel Y152C KCNJ5 Mutation Responsible for Familial Hyperaldosteronism Type IIIJ Clin Endocrinol Metab20139811E1861E186524037882
- SchollUILiftonRPNew insights into aldosterone-producing adenomas and hereditary aldosteronism: mutations in the K+ channel KCNJ5Curr Opin Nephrol Hypertens201322214114723318698
- GoetzLBethelKTopolEJRebooting cancer tissue handling in the sequencing era: toward routine use of frozen tumor tissueJAMA20133091373823280221
- MurthyMXuSMassimoGRole for germline mutations and a rare coding single nucleotide polymorphism within the KCNJ5 potassium channel in a large cohort of sporadic cases of primary aldosteronismHypertension201463478378924420545
- LiNFLiHJZhangDLGenetic variations in the KCNJ5 gene in primary aldosteronism patients from Xinjiang, ChinaPLoS One201381e5405123382865