
Abstract
Lynch syndrome is the most common cause of hereditary colon cancer, and accounts for as much as 3% of all colon and endometrial cancers. The identification and management of individuals with Lynch syndrome have evolved over the past 20 years, yet the syndrome remains vastly underdiagnosed. It is important for clinicians to recognize individuals and families who are at risk in order to be able to manage them appropriately and reduce their morbidity and mortality from this condition. This review will touch on the history of Lynch syndrome, the current knowledge of genotype–phenotype correlations, the cancers associated with Lynch syndrome, and management of individuals who are gene carriers.
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Overview and genetic basis
Lynch syndrome is a dominantly inherited cancer syndrome in which predisposition to colorectal, endometrial, and other cancers occurs due to an underlying defect in the cellular mismatch repair (MMR) system. MMR proteins form a complex that detects and corrects replication errors. A compromised MMR system leads to accelerated accumulation of somatic mutations, often resulting in carcinogenesis.
MLH1, MSH2, MSH6, and PMS2 are among the genes that produce MMR proteins. Lynch syndrome is caused by a heritable mutation in one copy of an MMR gene. At a phenotypic level, Lynch syndrome is dominant with variable expressivity. Secondary, somatic loss of the corresponding normal allele compromises the function of the entire MMR complex; Lynch syndrome is therefore recessive at the cellular level. An estimated 70%–90% of Lynch syndrome is attributable to deleterious mutations in MLH1 and MSH2, with the remaining 10%–30% distributed approximately equally between MSH6 and PMS2.Citation1–Citation3 Up to 3% of Lynch syndrome is due to mutations in the EPCAM gene, which is involved in epithelial cell adhesion, cell signaling, and proliferation. EPCAM is directly upstream of MSH2, and deletions of the 3′ end of EPCAM result in epigenetic hypermethylation of the MSH2 promoter, causing Lynch syndrome.Citation4
Lynch syndrome exhibits characteristic features of cancer predisposition syndromes, including substantially elevated risks for specific cancers, earlier onset, high rates of multiple primary cancers, and the absence of typical risk factors. Cancers associated with Lynch syndrome include colorectal, endometrial, ovarian, stomach, hepatobiliary, urinary, small bowel, brain/central nervous system, and sebaceous tumors. Cancer risks are strongly influenced by which MMR gene mutation is present but may also vary substantially between and within families, due to broader influences of the genome and gene–environment interaction.
Historical perspective and evolution of descriptive terms
The first colorectal cancer syndrome to be well characterized was called familial adenomatous polyposis (FAP) and was characterized by very early onset, massively prolific development of colorectal polyps. Later, when high rates of colorectal cancer were observed in some families in the absence of florid polyposis, the term “hereditary nonpolyposis colorectal cancer” (HNPCC) was used to describe this new clinical entity, distinguishing it from the previously recognized FAP. Based on clinical observations, the association of colorectal cancers with brain tumors was named Turcot syndrome, and colorectal cancers associated with sebaceous neoplasms and keratoacanthomas were termed Muir–Torre syndrome. Identifying the underlying molecular etiology led to the realization that Turcot syndrome with colorectal cancer and glioblastoma is due to an MMR deficit. Muir–Torre is also caused by underlying MMR defects, and both conditions are now recognized as part of the broader clinical spectrum of Lynch syndrome.
HNPCC became defined by an evolving series of criteria. The first was published in 1991 after an international meeting of researchers and clinicians (the International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer) in Amsterdam, the Netherlands.Citation5 The Amsterdam criteria, which can be remembered using a “3–2–1” mnemonic, were intended to more precisely define a homogeneous population for research purposes. The Amsterdam criteria describe families who do not have FAP and in which three closely related individuals spanning at least two generations have had colorectal cancer, with at least one diagnosis occurring prior to age 50 years. “Closely related” is defined as one of the affected trio being a first degree relative of the other two. With increasing recognition of the extracolonic manifestations of Lynch syndrome over the next decade, criteria were revised in 1999 to include extracolonic cancers.Citation6
The Amsterdam and revised Amsterdam criteria were developed with emphasis on specificity rather than sensitivity, and were intended for use as research criteria. Nevertheless, they became widely used clinically to identify high-risk families, with an estimated sensitivity and specificity of 60% and 70%.Citation7,Citation8 Authors of these criteria were careful to point out that these criteria should not be used to exclude individuals or families with features of Lynch syndrome from mutation analysis. Nevertheless, these criteria continue to be utilized in ways that were not intended, and it is unfortunate that some payers still utilize these criteria to determine eligibility for coverage of genetic testing.
“Lynch syndrome”, named for Dr Henry T Lynch, who was among the first to recognize and describe families with hereditary cancer predisposition, is now the accepted and preferred term to describe a hereditary syndrome caused by germline mutations that disrupt the function of an MMR gene. Although “HNPCC” is still used somewhat interchangeably with “Lynch syndrome”, it fails to recognize the associated extracolonic features and is less specific, as not all family history-defined HNPCC has underlying MMR defects. The eponymous Dr Henry Lynch is internationally recognized for his contributions to the discovery of the syndrome, his descriptions of the natural history, raising awareness by publishing and speaking, and his graciousness and support for organizations that work directly with individuals and families with Lynch syndrome.
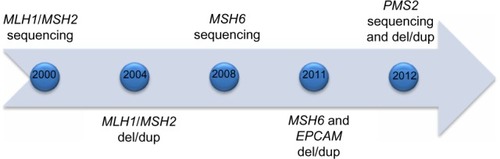
Not long after discovery in the mid-1990s, commercial testing became available for MLH1 and MSH2 around the turn of the decade, with genetic testing for four MMR genes plus EPCAM available within 12 years (). Families who meet Amsterdam I criteria but do not have an MMR deficit as the underlying etiology (so-called familial colorectal cancer type X) have been described and characterized.Citation9 These families have elevated colorectal cancer risk compared with a general population but not the same magnitude of risk as Lynch syndrome, and do not appear to have elevated risk for extracolonic cancers. The underlying genetic causes remain undefined, although with the recent advent of next-generation sequencing panels, additional genes will likely be implicated in some cases.
Figure 1 Approximate time line for availability of mismatch repair gene tests.
Clinical spectrum of Lynch syndrome
The risks of developing Lynch syndrome-associated cancers are gene and sex influenced. Initial studies tended to overestimate penetrance, due to the purposeful selection bias of the Amsterdam criteria. Penetrance data and cancer risk estimates have continued to evolve as genetic testing becomes more widespread. Generally, cancer risk estimates have trended downward, and the lower penetrance genes PMS2 and MSH6 have been found to account for a higher proportion of Lynch syndrome than previously recognized.Citation10 As gene and age-specific data evolve, it becomes important from a clinical standpoint to review recent literature for the most accurate risk estimates associated with a particular gene. Specific cancer risks associated with Lynch syndrome are reviewed regularly and displayed in tabular format in the National Comprehensive Cancer Network (NCCN) colorectal screening guidelines.Citation11
Individuals with an MLH1 or MSH2 gene mutation have the highest risks and the widest array of cancers attributable to Lynch syndrome. In particular, men with an MSH2 mutation have the highest risk for several types of cancers.Citation12–Citation14 MSH6 carriers have lower colorectal cancer risks but substantial gynecologic cancer risks.Citation15,Citation16 PMS2 carriers have lower colorectal and gynecologic cancer risks.Citation17 Data on extracolonic, nongynecologic cancers specific to MSH6 and PMS2 are sparse. Risks associated with EPCAM deletions are being elucidated. Deletions may occur in the 3′ end of EPCAM, or may span both EPCAM and MSH2. In cases where the deletion is in EPCAM only, the epigenetic silencing of MSH2 occurs only in cells that express EPCAM, and therefore creates a mosaic pattern of MSH2 inactivation. It appears that in people with this cause for Lynch syndrome the risk of colorectal cancer remains high, but endometrial cancer risk is low. Individuals with deletions that span both EPCAM and MSH2 have cancer risks similar to those with MSH2 mutations.Citation4,Citation18
Colorectal cancer
Features of colorectal cancer associated with Lynch syndrome include earlier average age at onset, right-sided predominance, elevated risk of synchronous and metachronous cancers, and rapid adenoma to carcinoma progression compared with sporadic adenomas.Citation19–Citation21 Histologic characteristics of Lynch syndrome-related colon cancers have been observed to be poorly differentiated, with tumor-infiltrating lymphocytes, mucin containing, and with signet ring or cribriform histology.Citation22,Citation23 There appears to be a survival advantage when matched stage for stage with non-Lynch syndrome colorectal cancers.Citation24–Citation26
Colorectal cancer risks are reported to be as high as 75%, with median ages reported from 44 years to 61 years in those with Lynch syndrome.Citation27 These vary according to which gene is involved and are well documented in other publications and summarized in .Citation12,Citation16,Citation17,Citation28–Citation34 Although 10% of colorectal cancers in the general population occur prior to age 50 years, in Lynch syndrome approximately 50% occur prior to age 50 years, before routine colorectal screening would typically commence.Citation35,Citation36 The rate of synchronous and metachronous colorectal cancers is dramatically elevated in colon and rectal cancer survivors with Lynch syndrome, with approximately 15%–20% developing a second colorectal cancer within 10 years, 40%–50% within 20 years, and >60% within 30 years.Citation37 Finally, the average dwell time from onset of a polyp to onset of carcinoma is much shorter in Lynch syndrome. Polyps may progress to carcinoma within 2–3 years among individuals with Lynch syndrome, compared with from 4 years to >10 years in the general population.Citation38,Citation39
Table 1 Colorectal and gynecologic cancer risks for people with Lynch syndrome compared with the general US population
Endometrial cancer
Endometrial cancer is at least as likely as colorectal cancer to be the initial cancer diagnosis in women with Lynch syndrome, and synchronous endometrial/ovarian cancers are more likely.Citation40–Citation42 As many as 26% of female survivors of colorectal cancer due to Lynch syndrome will develop endometrial cancer within 10 years of initial diagnosis.Citation42 Individuals with an MSH2 or MSH6 mutation have the highest risk for endometrial cancer, with a lifetime risk of up to 44% ().Citation15,Citation33
Lynch syndrome-associated endometrial cancers have primarily endometrioid histology, but other types, including clear cell, are observed.Citation43 MMR-deficient endometrial cancers are more likely to exhibit specific morphological features, including peritumoral lymphocytes, prominent tumor-infiltrating lymphocytes, and heterogeneous tumors displaying two morphologically distinct tumor cell populations.Citation44 The reported average ages of onset vary significantly and, in general, are younger, but there is evidence that the average age of onset may not be as early as previously thought, with the advent of universal screening for Lynch syndrome among individuals with endometrial cancer.Citation16,Citation45 Lynch syndrome is present in 8%–9% of women with early onset endometrial cancers, and 7%–21% of women with synchronous endometrial and ovarian cancers.Citation41,Citation44,Citation46–Citation49 Features of lower uterine segment endometrial cancers, which account for 3.5% of endometrial cancers overall, were observed in 42% of women with Lynch syndrome.Citation50 As many as 30% of women with endometrial cancer of the lower uterine segment may have Lynch syndrome.Citation51,Citation52
Ovarian cancer
Approximately 2% of ovarian cancers are due to Lynch syndrome.Citation53 When selected for early age of onset, specifically those diagnosed under the age of 40 years, the association with Lynch syndrome may be closer to 4%.Citation54
Reported lifetime risks for ovarian cancer in women with Lynch syndrome fall primarily within the range of 6.7%–12% and appear highest for carriers of MSH2 mutations, followed by MSH6 and MLH1 ().Citation13,Citation16,Citation55,Citation56 Synchronous endometrial cancer is reported in ∼22%, and 55% have a synchronous or metachronous Lynch syndrome-related cancer.Citation49 Mean ages for diagnosis of ovarian cancer in Lynch syndrome are primarily in the 40 to 50-year range, with up to 30% of Lynch syndrome-related ovarian cancers diagnosed prior to age 35 years.Citation13,Citation49,Citation53 Although Lynch syndrome-associated ovarian cancers are predominantly epithelial, unlike BRCA-related ovarian cancers, which are characteristically high grade serous, Lynch syndrome-associated ovarian cancers tend to display a higher proportion of endometrioid, clear cell, and mucinous cancers.Citation49,Citation57–Citation60
Gastric cancer
Gastric cancer is reported to occur in approximately 5%–13% of individuals with Lynch syndrome, with considerable variability based on country of origin.Citation13,Citation60 Risks are reported to be higher in MLH1 and MSH2 than other mutation carriers, and higher in males than in females, with a mean age of onset of 55 years ().Citation13,Citation55,Citation60,Citation61 Lynch syndrome-related gastric cancer is primarily, but not exclusively, the intestinal type, with diffuse-type gastric cancers representing 12.5%–23%.Citation60–Citation62
Table 2 Extracolonic, nongynecologic cancer risks for MLH1/MSH2 carriers compared with the general US population
Small bowel cancer
Up to 6% of individuals with Lynch syndrome develop small bowel cancer at a median age of <50 years for carriers of MLH1 and MSH2 mutations, and 54 years for MSH6 carriers ().Citation55,Citation63 As with several Lynch syndrome-associated extracolonic cancers, the risk appears highest in men with MSH2 mutations and lowest in carriers of MSH6 mutations, with scant data available for carriers of PMS2 mutations.Citation55
Urinary tract cancer
Renal pelvis and urothelial (transitional cell) cancers are exceedingly rare in the general population.Citation64 In contrast, people with Lynch syndrome have up to an 8% risk of developing upper urothelial cancers by age 70 years, at a median age of 58–62 years, with the highest risk occurring in men with MSH2 mutations ().Citation13,Citation65–Citation67 Recent data suggest a two- to four-fold elevated risk of bladder cancer as well, such that for men with MSH2 mutations the risk of developing a urinary tract cancer by age 70 years may approach or exceed 20%.Citation65,Citation68
Sebaceous neoplasms
The presence of sebaceous neoplasms in individuals and families with other internal malignancies was referred to as Muir–Torre syndrome before molecular genetic testing demonstrated a common underlying etiology. Sebaceous neoplasms, particularly carcinomas, are exceedingly rare.Citation69 Sebaceous neoplasms in people with Lynch syndrome are more likely than sporadic neoplasms to occur prior to age 60 years (median age 56 years), be multiple rather than isolated, and occur in the context of a personal or family history of Lynch syndrome-related cancer(s).Citation70 The incidence of sebaceous neoplasms among individuals with Lynch syndrome has been reported to be as high as 9% ().Citation71,Citation72
Other rare tumors associated with Lynch syndrome
The spectrum of Lynch syndrome-associated tumors is wide, and several very rare cancers in the general population are seen more frequently in Lynch syndrome ().Citation55 Although the risks for these rare tumors are greatly increased above the general population risks, the absolute risks are low. Individuals with Lynch syndrome have a risk of up to nearly 4% to develop pancreatic cancer by age 70 years. Pancreatic cancers appear most frequently in families with MSH2 mutations, followed by MLH1 and MSH6.Citation73 Up to 4% of people with Lynch syndrome develop hepatobiliary cancer by age 70 years (median 50–57 years), another rare cancer in the general population.Citation13 Finally, individuals with Lynch syndrome have up to a 3% lifetime risk of developing cancers of the brain and central nervous system, particularly glioblastoma.Citation74
Evolving spectrum of Lynch syndrome- associated cancers
Prostate cancer has recently been associated with Lynch syndrome, and data are beginning to emerge regarding the risks and ages of onset. Several studies have found the lifetime risk for prostate cancer in Lynch syndrome to be increased by two-to five-fold.Citation75–Citation77 Additional studies are needed to determine whether Lynch syndrome-associated prostate cancers occur at an earlier average age or are more aggressive.Citation75,Citation76
The relationship between breast cancer and Lynch syndrome remains unresolved. Studies have not consistently demonstrated a higher than expected incidence of breast cancer among individuals with Lynch syndrome.Citation78,Citation79 Several studies have demonstrated evidence of MMR with loss of immunohistochemical staining in breast cancers found among known carriers of a mismatch gene mutation.Citation80–Citation82 As breast cancer is fairly common in the general population, larger studies are needed to determine whether breast cancer is indeed part of the Lynch syndrome cancer spectrum.
Surveillance
A major reason to identify individuals with Lynch syndrome is to optimize surveillance, which ultimately minimizes morbidity and mortality. Surveillance recommendations for individuals with Lynch syndrome differ substantially from those of the general population, due to the accelerated progression from colorectal adenoma to carcinoma and the increased incidence of cancers that can be avoided with prophylactic surgery, such as endometrial and ovarian cancers.Citation83–Citation85 The elevated risk for colorectal cancer to occur at young ages justifies the initiation of surveillance as young as age 20–25 years, depending on the family history and genotype.Citation11 The right-sided predominance of colon cancers with Lynch syndrome necessitates a colonoscopy rather than a sigmoidoscopy.
The NCCN has published guidelines for management of individuals with Lynch syndrome that are regularly reviewed and updated, and there are several other publications that outline recommendations for surveillance.Citation11,Citation86,Citation87 It is notable that the most recent version of the NCCN guidelines reflects evolving evidence that individuals with a PMS2 or MSH6 mutation may have reduced penetrance and thus may not require the same intensity of surveillance.
Surveillance for colon cancer should include annual or biannual colonoscopy and begin around the age of 25 years.Citation86,Citation88,Citation89 Individuals with family members who were diagnosed at very young ages may consider colonoscopy earlier, typically 5–10 years before the earliest age of onset in the family. Recent evidence suggests that individuals with MSH6 or PMS2 mutations may be able to delay initiation of colonoscopy until as late as 30 years, due to the reduced penetrance.Citation11,Citation16,Citation17 Fewer colon cancers are identified when surveillance with polypectomy is performed at 1 to 2-year intervals and cancers are identified at earlier stages with overall improved survival rates.Citation89,Citation90 Studies comparing regular light colonoscopy with use of indigo carmine dye have not noted improved overall survival.Citation91 However, there was documented improvement in detection of very small polyps, and it remains to be determined whether this benefit may ultimately translate into better outcomes.
Endometrial cancer symptoms include abnormal uterine bleeding and pain, which are usually early indicators easily recognized by patients. Patients should be educated to seek medical evaluation if they experience abnormal bleeding. There is no clear management recommendation regarding endometrial biopsy for surveillance, as it is invasive, and there appears to be no evidence to suggest that outcomes are improved.Citation92
Surveillance for other cancers is widely debated due to the lack of evidenced-based improvement of outcomes. Therefore, most groups do not make any specific recommendations for extracolonic cancer surveillance. Some practitioners may consider small bowel X-ray and/or upper endoscopy to screen for cancers of the upper gastrointestinal tract, and urinalysis with cytology to screen for urothelial cancer in individual cases, but there are no guidelines to direct these surveillance methods. Finally, the issue of dermatology screening has been raised, based on a single study that found that almost 10% of individuals with Lynch syndrome had sebaceous adenomas or the Muir–Torre variant of Lynch syndrome.Citation71
Surgical considerations
There continues to be a debate about colectomy versus subtotal colectomy at the time of colon cancer treatment for individuals known to have Lynch syndrome. Surgeons may opt for a subtotal colectomy at the time of a colon cancer diagnosis in an individual with Lynch syndrome, despite lack of evidence demonstrating survival benefit.Citation93,Citation94 Quality of life issues following total abdominal colectomy should be carefully considered, as should access and adherence to surveillance, and management should be tailored on an individual basis.Citation95,Citation96 Few physicians would recommend prophylactic colectomy today, although it was considered early on in the chronicle of Lynch syndrome.
There is little debate with regard to prophylactic removal of the uterus and ovaries once childbearing is complete, due to the lack of effective surveillance of the ovaries and the significant decrease in the risk for both cancers following prophylactic surgery.Citation83 The average age of onset of endometrial cancer in Lynch syndrome is 55 years, and current recommendations suggest total abdominal hysterectomy and bilateral salpingo-oophorectomy by the age of 50 years.Citation97 Thorough pathological examination of surgical specimens is recommended, as gynecologic malignancies may be present already at the time of prophylactic surgery.Citation98,Citation99 Removing ovaries after or near the time of menopause eliminates some of the issues of early surgical menopause, as seen in BRCA1/2 carriers. However, 30% of Lynch syndrome-associated ovarian cancers occur prior to age 35 years, and there is no apparent contraindication for use of hormone replacement in this population. The likelihood of endometrial and ovarian cancer after prophylactic salpingo-oophorectomy is very low.Citation83
Chemoprevention
Trials of aspirin have shown promise in reducing polyp burden among individuals with Lynch syndrome. Use of aspirin for 4 years or longer is associated with a reduction in the risk for colon cancer, although the effect is not evident until at least 5 years after the intervention.Citation100 The optimal age to initiate the recommended dose and necessary duration of aspirin use has not been established, and studies are now ongoing. Birth control pills reduce the risk for both endometrial and ovarian cancer in the general population, and this effect was similar in a small cohort of women with Lynch syndrome.Citation101
Recommendations for treatment and surveillance for Lynch syndrome continue to evolve. NCCN guidelines are reviewed and updated regularly, incorporating new information as it arises. Coordination of care in Lynch syndrome is essential to ensure that patients are getting the most appropriate and up-to-date care. This often requires collaboration between many different specialists, such as gastroenterologists, gynecologic oncologists or gynecologists, primary care providers, and genetic counselors.Citation102
Psychosocial issues
Optimally, genetic testing should be done in a supportive setting with the expertise of a health care practitioner who is familiar with Lynch syndrome and some of the psychosocial issues that may be present. Genetic testing for Lynch syndrome may cause anxiety and distress, although studies have shown that the majority of individuals adapt to their results, and negative effects, if any, appear to be short term.Citation103,Citation104 Knowledge of one’s mutation and risk status may also provide individuals with a sense of control and optimism.Citation105
There may be psychological effects of living with the threat of cancer. This may be influenced by prior experience with illness or death from cancer in the family. Feelings of guilt with regard to passing on the gene mutation or the possibility of passing on the gene mutation may also be present.Citation105
Adherence to surveillance recommendations does not seem to be impacted by anxiety or cancer worry but may be impacted by an individual’s perceived barriers to screening.Citation106 Barriers that have been described include discomfort, embarrassment, and lack of awareness of surveillance recommendations.Citation106–Citation108 Recognizing these factors in individual patients is important so that health care practitioners can maximize compliance with surveillance recommendations.
Identification of Lynch syndrome: universal screening
Lynch syndrome accounts for ∼3% of all colorectal and endometrial cancers.Citation2,Citation97,Citation109,Citation110 Identification of Lynch syndrome has traditionally relied on multiple steps, including recognition of typical features and appropriate testing and/or referral to a genetics provider. Although there are some histological features within individual tumors that can indicate a likelihood of MMR deficit, and other clues, such as location within the body system (eg, lower uterine segment endometrial cancer or proximal colon cancer), Lynch syndrome-associated colon and endometrial cancers are not necessarily distinguishable from sporadic colon and endometrial cancers.Citation36,Citation111 Systematic collection, documentation, and assessment of family history are highly variable among health care providers, and rarely is this information readily available to pathologists who may recognize histological features of Lynch syndrome. Given these limitations and the compelling reasons to identify these individuals and their at-risk family members, universal screening has been proposed as a way to adequately identify individuals with Lynch syndrome.
Universal screening for Lynch syndrome is the evaluation of all colon and/or endometrial tumors at the time of diagnosis for evidence of MMR deficit. Microsatellite instability (MSI) and immunohistochemistry (IHC) are two screening methods used to identify affected individuals who may have Lynch syndrome. MSI is a measure of whether the MMR system is functioning. Loss of MMR function, which can be caused by Lynch syndrome or by epigenetic silencing of the MLH1 gene, results in MSI, which can be detected by polymerase chain reaction on a colon tumor specimen. Evidence of MSI suggests further workup to rule out Lynch syndrome. IHC is a demonstration of the presence or absence of MMR proteins in the tumor. Absence of a protein(s) as demonstrated by IHC staining suggests the possibility of a mutation in the corresponding gene.
The Bethesda criteria were developed to define populations for which colon tumor testing with MSI and/or IHC was indicated.Citation112 These evolved over time to include personal and family history of extracolonic cancers.Citation113 However, studies of universal screening of all colorectal and endometrial cancers suggest that as many as 70% of people with Lynch syndrome do not meet Amsterdam or Bethesda guidelines.Citation97,Citation114
Universal screening for Lynch syndrome has been demonstrated to be cost-effective, largely due to the identification of unaffected family members and subsequent prevention of colon and endometrial cancers.Citation1,Citation115–Citation118 The issue of whether or not consent should be obtained prior to screening has been debated. However, in practice, direct informed consent prior to tumor screening is rare.Citation119–Citation121
There are documented challenges to implementation of a universal screening program for Lynch syndrome.Citation122 One challenge has been to establish an effective process for notification and discussion with the patient, with subsequent patient uptake of genetic testing and notification of at-risk relatives.Citation123–Citation125 Individuals identified by universal screening for Lynch syndrome may not return for genetic counseling and testing if it is not apparent to them how it would impact their care and/or if their perception is that further evaluation is not warranted based on their family history.Citation126 There have been several studies attempting new approaches to remove barriers and improve compliance with follow-up genetic counseling and testing, with variable success.Citation127–Citation129 Cascade testing for at-risk relatives is complex and depends upon effective intrafamilial communication.Citation123,Citation130 Factors that correlate with how well information is transmitted among family members include the education of individuals with Lynch syndrome and recommendations by the health care professionals who care for them.Citation123,Citation126,Citation131 Most individuals with Lynch syndrome inform first-degree family members but are less likely to notify more distant relatives, due to lack of closeness and concerns that relatives may not understand the information shared.Citation132 Resources to assist health care providers and families in the process of notifying at-risk relatives include an online tool, informational brochures, and a searchable database to identify genetic counselors ().
Table 3 Lynch syndrome resources
Molecular testing does not always identify the underlying mutation in screen-positive individuals who are identified by tumor screening. Current technology may not detect all mutations, or there may be additional, yet-to-be-identified genes that cause Lynch syndrome. Biallelic somatic mutations may explain some cases of absent MMR proteins detected by IHC.Citation133 Personal and family history may be helpful in distinguishing sporadic biallelic somatic mutations from true Lynch syndrome with no identifiable mutation.
Finally, the clinical utility of evaluating nonendometrial, extracolonic tumors for evidence of MMR continues to be unclear. The accuracy of screening ovarian tumors for MMR with IHC or MSI is questionable, although abnormal findings certainly warrant further evaluation and consideration of germline testing.Citation134,Citation135 Rare sebaceous tumors (including sebaceous adenomas, epitheliomas, and carcinomas) are strongly associated with the Muir–Torre variant of Lynch syndrome, and 30%–60% of sebaceous tumors are related to an MMR defect.Citation136 However, there are differing opinions on the utility of screening all sebaceous tumors with IHC. Some authors have advocated for IHC screening of all sebaceous tumors.Citation137,Citation138 Others have argued that in the absence of a personal or family history of Lynch syndrome-associated cancers, the positive predictive value of IHC on sebaceous tumors is not high enough to warrant routine screening.Citation70 A recent publication describes a scoring system based on the number of sebaceous neoplasms, age at diagnosis, and personal and family history of Lynch syndrome-associated cancers, and concludes that IHC alone is a poor predictor for identifying people with Lynch syndrome.Citation139
Other Lynch syndrome-associated cancers may display evidence of MMR, but results tend to be less reliable than screening other tumors. There is incomplete concordance between MSI and IHC analysis in gastric tumors, so one group has recommended screening gastric tumors with both MSI and IHC when meeting revised Bethesda criteria.Citation62 Other tumors have much fewer data regarding reliability of MSI and IHC in assessing likelihood for Lynch syndrome. The one exception is small bowel tumors, in which the performance of MSI and IHC appears to be similar to that in colon cancers.Citation140
In conclusion, there are case reports of abnormal IHC among rare and unusual tumors in individuals with confirmed Lynch syndrome, but not enough evidence to routinely screen these tumors for evidence of MMR, nor enough evidence to be able to rule out suspected Lynch syndrome with normal IHC and/or MSI.Citation141
Emerging issues
As the complex puzzle of Lynch syndrome genetics has been pieced together, inherited biallelic mutations in MMR genes have been found to cause constitutional MMR deficiency syndrome, a unique pediatric syndrome.Citation142 Symptoms include extremely early onset cancers, including colon and small bowel cancers. Café-au-lait macules are common, seen among 70% of individuals with inherited biallelic MMR mutations, and the presence of colon polyposis has been reported as well. The average age of onset of colon cancer is around 15–16 years, and the average age of onset of small bowel cancer is 20 years.Citation143,Citation144
When younger family members of childbearing age are identified with Lynch syndrome, the issue of prenatal or pre-implantation diagnosis may be raised. A small study of individuals at risk for Lynch syndrome found that childbearing decisions were impacted by whether or not a mutation was identified.Citation145 Most surveyed believed that it was ethical to offer prenatal testing for Lynch syndrome, despite the fact that few intended to do so themselves. Genetic counselors and other specialty reproductive service providers are available to discuss options with families who indicate an interest in exploring reproductive options.
Studies are beginning to focus on gene–environment interactions, to attempt to explain some of the variability between families with mutations in the same gene, or even within families with the same gene mutation. For example, body mass index seems to be a risk factor for colorectal cancer but not endometrial cancer in Lynch syndrome.Citation146,Citation147 This type of information may be helpful in providing a sense of control to individuals at risk, and additional studies are needed in this complex area of gene–environment interactions.
Summary
The last 20 years have seen tremendous gains in knowledge about the underlying cause of Lynch syndrome, as well as increasing understanding of the spectrum of associated disease. Despite clear evidence that the most substantial cancer risks can be readily mitigated, Lynch syndrome remains underdiagnosed. It is critical to adapt to evolving criteria and universal screening to identify individuals with Lynch syndrome. Effective strategies to identify and appropriately manage the health of people with Lynch syndrome will continue to evolve. Astute clinicians can save lives by identifying patients with Lynch syndrome and managing their health accordingly as knowledge continues to evolve.
Disclosure
The authors have no conflicts of interest in this work.
References
- PalomakiGEMcClainMRMelilloSHampelHLThibodeauSNEGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndromeGenet Med2009111426519125127
- EgoavilCAlendaCCastillejoAPrevalence of Lynch syndrome among patients with newly diagnosed endometrial cancersPLoS One2013811e7973724244552
- WeissmanSBurtRChurchJIdentification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer Joint Practice GuidelineJ Genet Couns201221448449322167527
- TutlewskaKLubinskiJKurzawskiGGermline deletions in the EPCAM gene as a cause of Lynch syndrome: literature reviewHered Cancer Clin Pract2013111923938213
- VasenHMecklinJPKhanPMLynchHTThe International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC)Dis Colon Rectum19913454244252022152
- VasenHFWatsonPMecklinJPLynchHTNew clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCCGastroenterology199911661453145610348829
- LiuTWahlbergSBurekELindblomPRubioCLindblomAMicrosatellite instability as a predictor of a mutation in a DNA mismatch repair gene in familial colorectal cancerGenes Chromosomes Cancer2000271172510564582
- SyngalSFoxEAEngCKolodnerRDGarberJESensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1J Med Genet200037964164510978352
- LindorNMFamilial colorectal cancer type X: the other half of hereditary nonpolyposis colon cancer syndromeSurg Oncol Clin N Am200918463764519793571
- HampelHStephensJAPukkalaECancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onsetGastroenterology2005129241542116083698
- ProvenzaleDJaspersonKAhnenDJNCCN guidlines v1.2014. Colorectal cancer screeningNCCN Guidelines 2014 Available from: http://www.nccn.orgAccessed June 5, 2014
- RamsoekhDWagnerAvan LeerdamMCancer risk in MLH1, MSH2 and MSH6 mutation carriers; different risk profiles may influence clinical managementHered Cancer Clin Pract2009711720028567
- WatsonPVasenHFMecklinJPThe risk of extra-colonic, extra-endometrial cancer in the Lynch syndromeInt J Cancer2008123244444918398828
- Lin-HurtubiseKMYheulonCGGaglianoRALynchHTExcess of extracolonic non-endometrial multiple primary cancers in MSH2 germline mutation carriers over MLH1J Surg Oncol2013108743343724122742
- BagliettoLLindorNMDowtyJGRisks of Lynch syndrome cancers for MSH6 mutation carriersJ Natl Cancer Inst2010102319320120028993
- BonadonaVBonaitiBOlschwangSCancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndromeJAMA2011305222304231021642682
- SenterLClendenningMSotamaaKThe clinical phenotype of Lynch syndrome due to germ-line PMS2 mutationsGastroenterology2008135241942818602922
- LigtenbergMLKuiperRGeurts van KesselAHoogerbruggeNEPCAM deletion carriers constitute a unique subgroup of Lynch syndrome patientsFam Cancer201312216917423264089
- JärvinenHJAarnioMMustonenHControlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancerGastroenterology118582983410784581
- de Vos tot Nederveen CappelWHNagengastFMGriffioenGSurveillance for Hereditary Nonpolyposis Colorectal CancerDis Colon Rectum200245121588159412473880
- JohnsonPMGallingerSMcLeodRSSurveillance colonoscopy in individuals at risk for hereditary nonpolyposis colorectal cancer: an evidence-based reviewDis Colon Rectum2006491809516284887
- MesseriniLMoriSZampiGPathologic features of hereditary non-polyposis colorectal cancerTumori19968221141168644372
- ShiaJKlimstraDSNafaKValue of immunohistochemical detection of DNA mismatch repair proteins in predicting germline mutation in hereditary colorectal neoplasmsAm J Surg Pathol20052919610415613860
- WatsonPLinKMRodriguez-BigasMAColorectal carcinoma survival among hereditary nonpolyposis colorectal carcinoma family membersCancer19988322592669669808
- StiglianoVAssisiDCosimelliMSurvival of hereditary non-polyposis colorectal cancer patients compared with sporadic colorectal cancer patientsJ Exp Clin Cancer Res2008273918803843
- DrescherKMSharmaPLynchHTCurrent hypotheses on how microsatellite instability leads to enhanced survival of Lynch syndrome patientsClin Dev Immunol2010201017043220631828
- BarrowEHillJEvansDGCancer risk in Lynch SyndromeFam Cancer201312222924023604856
- ChoiYHCotterchioMMcKeown-EyssenGPenetrance of colorectal cancer among MLH1/MSH2 carriers participating in the colorectal cancer familial registry in OntarioHered Cancer Clin Pract2009711419698169
- KempersMJEKuiperRPOckeloenCWRisk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort studyLancet Oncol2011121495521145788
- BagliettoLLindorNMDowtyJGRisks of Lynch syndrome cancers for MSH6 mutation carriersJ Natl Cancer Inst2010102319320120028993
- Pérez-CaborneroLInfanteMVelascoELastraEMinerCDuránMGenotype–phenotype correlation in MMR mutation-positive families with Lynch syndromeInt J Colorectal Dis20132891195120123588873
- GoeckeTSchulmannKEngelCGenotype-phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch syndrome: a report by the German HNPCC ConsortiumJ Clin Oncol200624264285429216908935
- StoffelEMukherjeeBRaymondVMCalculation of risk of colorectal and endometrial cancer among patients with Lynch syndromeGastroenterology200913751621162719622357
- PlaschkeJEngelCKrügerSLower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer ConsortiumJ Clin Oncol200422224486449415483016
- American Cancer SocietyColorectal Cancer Facts and Figures 2011–2013Atlanta, GAAmerican Cancer Society2011
- HampelHFrankelWLMartinEFeasibility of screening for Lynch syndrome among patients with colorectal cancerJ Clin Oncol200826355783578818809606
- WinAParrySParryBRisk of metachronous colon cancer following surgery for rectal cancer in mismatch repair gene mutation carriersAnn Surg Oncol20132061829183623358792
- EdelsteinDLAxilbundJBaxterMRapid development of colorectal neoplasia in patients with Lynch syndromeClin Gastroenterol Hepatol20119434034321070872
- WinawerSFletcherRRexDColorectal cancer screening and surveillance: clinical guidelines and rationale: update based on new evidenceGastroenterology2003124254456012557158
- LuKHDinhMKohlmannWGynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndromeObstet Gynecol2005105356957415738026
- WalshCSBlumAWaltsALynch syndrome among gynecologic oncology patients meeting Bethesda guidelines for screeningGynecol Oncol2010116351652120034658
- ObermairAYouldenDRYoungJPRisk of endometrial cancer for women diagnosed with HNPCC-related colorectal carcinomaInt J Cancer2010127112678268420533284
- BroaddusRRLynchHTChenLMPathologic features of endometrial carcinoma associated with HNPCC: a comparison with sporadic endometrial carcinomaCancer20061061879416323174
- GargKSoslowRALynch syndrome (hereditary non-polyposis colorectal cancer) and endometrial carcinomaJ Clin Pathol200962867968419638537
- LeenenCHvan LierMGvan DoornHCProspective evaluation of molecular screening for Lynch syndrome in patients with endometrial cancer ≤70 yearsGynecol Oncol2012125241442022306203
- SolimanPTBroaddusRRSchmelerKMWomen with synchronous primary cancers of the endometrium and ovary: do they have Lynch syndrome?J Clin Oncol200523369344935016361634
- WalshMDCummingsMCBuchananDDMolecular, pathologic, and clinical features of early-onset endometrial cancer: identifying presumptive Lynch syndrome patientsClin Cancer ResMarch 1520081461692170018310315
- KimMKSongSYDoI-GSynchronous gynecologic malignancy and preliminary results of Lynch syndromeJ Gynecol Oncol201122423323822247799
- WatsonPBützowRLynchHTThe clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancerGynecol Oncol200182222322811531271
- RyanPMulliganAMAronsonMComparison of clinical schemas and morphologic features in predicting Lynch syndrome in mutation-positive patients with endometrial cancer encountered in the context of familial gastrointestinal cancer registriesCancer2012118368168821721000
- MasudaK BKYanokuraMCarcinoma of the lower uterine segment (LUS): clinicopathological characteristics and association with Lynch syndromeCurr Genomics2011121252921886452
- WestinSNLacourRAUrbauerDLCarcinoma of the lower uterine segment: a newly described association with Lynch syndromeJ Clin Oncol200826365965597119001318
- MalanderSRambechEKristofferssonUThe contribution of the hereditary nonpolyposis colorectal cancer syndrome to the development of ovarian cancerGynecol Oncol2006101223824316360201
- DomanskaKMalanderSMasbackANilbertMOvarian cancer at young age: the contribution of mismatch-repair defects in a population-based series of epithelial ovarian cancer before age 40Int J Gynecol Cancer200717478979317343610
- EngelCLoefflerMSteinkeVRisks of less common cancers in proven mutation carriers with Lynch syndromeJ Clin Oncol201230354409441523091106
- PalTPermuth-WeyJSellersTAA review of the clinical relevance of mismatch-repair deficiency in ovarian cancerCancer2008113473374218543306
- ChuiMHGilksCBCooperKClarkeBAIdentifying Lynch syndrome in patients with ovarian carcinoma: the significance of tumor subtypeAdv Anat Pathol201320637838624113308
- LynchHTCaseyMJSnyderCLHereditary ovarian carcinoma: heterogeneity, molecular genetics, pathology, and managementMol Oncol2009329713719383374
- KetabiZBartumaKBernsteinIOvarian cancer linked to lynch syndrome typically presents as early-onset, non-serous epithelial tumorsGynecol Oncol2011121346246521388660
- AarnioMSankilaRPukkalaECancer risk in mutation carriers of DNA-mismatch-repair genesInt J Cancer199981221421810188721
- CapelleLGVan GriekenNCLingsmaHFRisk and epidemiological time trends of gastric cancer in Lynch syndrome carriers in The NetherlandsGastroenterology2010138248749219900449
- ChunNFordJMGenetic testing by cancer site: stomachCancer J201218435536322846738
- ParkJGKimDWHongCWGerm line mutations of mismatch repair genes in hereditary nonpolyposis colorectal cancer patients with small bowel cancer: International Society for Gastrointestinal Hereditary Tumours Collaborative StudyClin Cancer Res20061211 Pt 13389339316740762
- GuptaRPanerGPAminMBNeoplasms of the upper urinary tract: a review with focus on urothelial carcinoma of the pelvicalyceal system and aspects related to its diagnosis and reportingAdv Anat Pathol200815312713918434765
- van der PostRSKiemeneyLALigtenbergMJRisk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriersJ Med Genet201047746447020591884
- AarnioMSäilyMJuholaMUroepithelial and kidney carcinoma in Lynch syndromeFam Cancer201211339540122476430
- CrockettDGWagnerDGHolmängSJohanssonSLLynchHTUpper urinary tract carcinoma in Lynch syndrome casesJ Urol201118551627163021419447
- SkeldonSCSemotiukKAronsonMPatients with Lynch syndrome mismatch repair gene mutations are at higher risk for not only upper tract urothelial cancer but also bladder cancerEur Urol201363237938522883484
- DasguptaTWilsonLDYuJBA retrospective review of 1349 cases of sebaceous carcinomaCancer2009115115816518988294
- RobertsMRiegert-JohnsonDThomasBScreening for Muir–Torre syndrome using mismatch repair protein immunohistochemistry of sebaceous neoplasmsJ Genet Couns201322339340523212176
- SouthCDHampelHComerasIWestmanJAFrankelWLde la ChapelleAThe frequency of Muir–Torre syndrome among Lynch syndrome familiesJ Natl Cancer Inst2008100427728118270343
- PontiGLosiLPedroniMValue of MLH1 and MSH2 mutations in the appearance of Muir–Torre syndrome phenotype in HNPCC patients presenting sebaceous gland tumors or keratoacanthomasJ Invest Dermatol2006126102302230716826164
- KastrinosFMukherjeeBTayobNRisk of pancreatic cancer in families with Lynch syndromeJAMA2009302161790179519861671
- VasenHFASandersEACMTaalBGThe risk of brain tumours in hereditary non-polyposis colorectal cancer (HNPCC)Int J Cancer19966544224258621220
- HaraldsdottirSHampelHWeiLProstate cancer incidence in males with Lynch syndromeGenet Med Epub1162014
- RyanSJenkinsMAWinAKRisk of prostate cancer in Lynch syndrome: a systematic review and meta-analysisCancer Epidemiol Biomarkers Prev201423343744924425144
- RaymondVMMukherjeeBWangFElevated risk of prostate cancer among men with Lynch syndromeJ Clin Oncol201331141713171823530095
- BuerkiNGautierLKovacMEvidence for breast cancer as an integral part of Lynch syndromeGenes Chromosomes Cancer2012511839122034109
- WinALindorNJenkinsMRisk of breast cancer in Lynch syndrome: a systematic reviewBreast Cancer Res2013152R2723510156
- WenYHBrogiEZengZDNA mismatch repair deficiency in breast carcinoma: a pilot study of triple-negative and non-triple-negative tumorsAm J Surg Pathol201236111700170822992699
- D’ArcyCWenYHStadlerZKBrogiEShiaJSynchronous breast cancers with different morphologic and molecular phenotypes occurring in Lynch syndrome: what does the heterogeneity imply?Am J Surg Pathol201135111743174821997695
- JensenUSundeLTimshelSMismatch repair defective breast cancer in the hereditary nonpolyposis colorectal cancer syndromeBreast Cancer Res Treat2010120377778219575290
- SchmelerKMLynchHTChenLMProphylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndromeN Engl J Med2006354326126916421367
- YurgelunMBMercadoRRosenblattMImpact of genetic testing on endometrial cancer risk-reducing practices in women at risk for Lynch syndromeGynecol Oncol2012127354455122940489
- MecklinJPAarnioMLääräEDevelopment of colorectal tumors in colonoscopic surveillance in Lynch syndromeGastroenterology200713341093109817919485
- VasenHFABlancoIAktan-CollanKRevised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European expertsGut201362681282323408351
- GroverSSyngalSRisk assessment, genetic testing, and management of Lynch syndromeJ Natl Compr Canc Netw2010819810520064292
- LindorNMPetersenGMHadleyDWRecommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic reviewJAMA2006296121507151717003399
- StupartDAGoldbergPAAlgarURamesarRSurveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutationColorectal Dis200911212613019143775
- JarvinenHJRenkonen-SinisaloLAktan-CollanKPeltomakiPAaltonenLAMecklinJPTen years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation-negative family membersJ Clin Oncol200927284793479719720893
- StoffelEMTurgeonDKStockwellDHMissed adenomas during colonoscopic surveillance in individuals with Lynch syndrome (hereditary nonpolyposis colorectal cancer)Cancer Prev Res (Phila)20081647047519138994
- LuKHDanielsMEndometrial and ovarian cancer in women with Lynch syndrome: update in screening and preventionFam Cancer201312227327723765559
- BaucomRBWisePEEndoscopic and surgical management of hereditary nonpolyposis colorectal cancerClin Colon Rectal Surg2012252909623730223
- ParrySWinAKParryBMetachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgeryGut201160795095721193451
- HaanstraJFde Vos Tot Nederveen CappelWHGopieJPQuality of life after surgery for colon cancer in patients with Lynch syndrome: partial versus subtotal colectomyDis Colon Rectum201255665365922595844
- Rodriguez-BigasMMöesleinGSurgical treatment of hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome)Fam Cancer201312229530023508345
- HampelHFrankelWPanescuJScreening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patientsCancer Res200666157810781716885385
- DownesMRAlloGMcCluggageWGReview of findings in prophylactic gynaecological specimens in Lynch syndrome with literature review and recommendations for grossingHistopathology Epub242014
- KaramurzinYSoslowRAGargKHistologic evaluation of prophylactic hysterectomy and oophorectomy in Lynch syndromeAm J Surg Pathol201337457958523426126
- BurnJGerdesAMMacraeFLong-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trialLancet201137898092081208722036019
- LuKHLooseDSYatesMSProspective multicenter randomized intermediate biomarker study of oral contraceptive versus depo-provera for prevention of endometrial cancer in women with Lynch syndromeCancer Prev Res (Phila)20136877478123639481
- RubinsteinWSWeissmanSMManaging hereditary gastrointestinal cancer syndromes: the partnership between genetic counselors and gastroenterologistsNat Clin Pract Gastroenterol Hepatol200851056958218797444
- ClaesE DLEvers-KieboomsPredictive testing for hereditary non-polyposis colorectal cancer: motivation, illness representations and short-term psychological impactPatient Educ Couns200465226527415530764
- MeiserBCollinsVWarrenRPsychological impact of genetic testing for hereditary non-polyposis colorectal cancerClin Genet200466650251115521977
- EsplenMJStucklessNGallingerSDevelopment and validation of an instrument to measure the impact of genetic testing on self-concept in Lynch syndromeClin Genet201180541542321883167
- Aktan-CollanKKääriäinenHJärvinenHPsychosocial consequences of predictive genetic testing for lynch syndrome and associations to surveillance behaviour in a 7-year follow-up studyFam Cancer201312463964623512527
- StoffelEMMercadoRCKohlmannWPrevalence and predictors of appropriate colorectal cancer surveillance in Lynch syndromeAm J Gastroenterol201010581851186020354509
- KetabiZMosgaardBJGerdesA-MLadelundSBernsteinITAwareness of endometrial cancer risk and compliance with screening in hereditary nonpolyposis colorectal cancerObstet Gynecol201212051005101223090516
- HampelHFrankelWLMartinEScreening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer)N Engl J Med Overseas Ed20053521818511860
- EGAPPRecommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relativesGenet Med2009111354119125126
- ClarkeBACooperKIdentifying Lynch syndrome in patients with endometrial carcinoma: shortcomings of morphologic and clinical schemasAdv Anat Pathol201219423123822692286
- Rodriguez-BigasMABolandCRHamiltonSRA National Cancer Institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and bethesda guidelinesJ Natl Cancer Inst19978923175817629392616
- UmarABolandCRTerdimanJPRevised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instabilityJ Natl Cancer Inst200496426126814970275
- RamsoekhDWagnerAvan LeerdamMEA high incidence of MSH6 mutations in Amsterdam criteria II-negative families tested in a diagnostic settingGut200857111539154418625694
- DinhTARosnerBIAtwoodJCHealth benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general populationCancer Prev Res (Phila)20114192221088223
- GudgeonJWilliamsJBurtRWSamowitzWSnowGWilliamsMLynch Syndrome screening implementation: business analysis by a healthcare systemAm J Manag Care2011178e288e30021851136
- MvunduraMGrosseSDHampelHPalomakiGEThe cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancerGenet Med20101229310420084010
- LadabaumUWangGTerdimanJStrategies to identify the Lynch syndrome among patients with colorectal cancerAnn Intern Med20111552697921768580
- WilliamsJLWilliamsMSInformed consent and immunohistochemistry screening for Lynch syndromeGenet Med201113984884921885923
- ChubakBHealdBSharpRRInformed consent to microsatellite instability and immunohistochemistry screening for Lynch syndromeGenet Med201113435636021407081
- CohenSCurrent Lynch syndrome tumor screening practices: a survey of genetic counselorsJ Genet Couns2013231110
- BellcrossCABedrosianSRDanielsEImplementing screening for Lynch syndrome among patients with newly diagnosed colorectal cancer: summary of a public health/clinical collaborative meetingGenet Med201214115216222237445
- JaspersonKColorectal cancer: cascade genetic testing in Lynch syndrome: room for improvementNat Rev Gastroenterol Hepatol201310950650823835491
- SouthCDYearsleyMMartinEArnoldMFrankelWHampelHImmunohistochemistry staining for the mismatch repair proteins in the clinical care of patients with colorectal cancerGenet Med2009111181281719752738
- WardRLHicksSHawkinsNJPopulation-based molecular screening for Lynch syndrome: implications for personalized medicineJ Clin Oncol201331202554256223733757
- CragunDMaloTLPalTShibataDVadaparampilSTColorectal cancer survivors’ interest in genetic testing for hereditary cancer: implications for universal tumor screeningGenet Test Mol Biomarkers201216649349922224634
- HallMJHerdaMMHandorfEADirect-to-patient disclosure of results of mismatch repair screening for Lynch syndrome via electronic personal health record: a feasibility studyGenet Med Epub512014
- HealdBPlesecTLiuXImplementation of universal microsatellite instability and immunohistochemistry screening for diagnosing Lynch syndrome in a large academic medical centerJ Clin Oncol201331101336134023401454
- CragunDDebateRVadaparampilSTBaldwinJHampelHPalTComparing universal Lynch syndrome tumor-screening programs to evaluate associations between implementation strategies and patient follow-throughGenet Med Epub3202014
- SharafRNMyerPStaveCDDiamondLCLadabaumUUptake of genetic testing by relatives of lynch syndrome probands: a systematic reviewClin Gastroenterol Hepatol20131191093110023669308
- TomiakESamsonASpectorNReflex testing for Lynch syndrome: if we build it, will they come? Lessons learned from the uptake of clinical genetics services by individuals with newly diagnosed colorectal cancer (CRC)Fam Cancer2014131758224002367
- StoffelEMFordBMercadoRCSharing genetic test results in Lynch syndrome: communication with close and distant relativesClin Gastroenterol Hepatol20086333333818258490
- MensenkampARVogelaarIPvan Zelst-StamsWASomatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumorsGastroenterology20141463643646. e64824333619
- CoppolaDNicosiaSDotyAUncertainty in the utility of immunohistochemistry in mismatch repair protein expression in epithelial ovarian cancerAnticancer Res201232114963496923155266
- LeeJHCragunDThompsonZAssociation between IHC and MSI testing to identify mismatch repair-deficient patients with ovarian cancerGenet Test Mol Biomarkers201418422923524592941
- AbbasOMahalingamMCutaneous sebaceous neoplasms as markers of Muir–Torre syndrome: a diagnostic algorithmJ Cutan Pathol200936661361919515040
- OrtaLKlimstraDSQinJTowards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristicsAm J Surg Pathol200933693494419342947
- PlocharczykEFFrankelWLHampelHPetersSBMismatch repair protein deficiency is common in sebaceous neoplasms and suggests the importance of screening for Lynch syndromeAm J Dermatopathol201335219119522722469
- RobertsMERiegert-JohnsonDLThomasBCA clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir–Torre variant of Lynch syndromeGenet Med Epub362014
- WilliamsASHuangW-YThe analysis of microsatellite instability in extracolonic gastrointestinal malignancyPathology201345654055224018804
- KaramurzinYZengZStadlerZKUnusual DNA mismatch repair–deficient tumors in Lynch syndrome: a report of new cases and review of the literatureHum Pathol201243101677168722516243
- WimmerKKratzCPVasenHFADiagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium “Care for CMMRD” (C4CMMRD)J Med Genet201451635536524737826
- PoleyJWWagnerAHoogmansMMBiallelic germline mutations of mismatch-repair genes: a possible cause for multiple pediatric malignanciesCancer2007109112349235617440981
- DurnoCAHolterSShermanPMGallingerSThe gastrointestinal phenotype of germline biallelic mismatch repair gene mutationsAm J Gastroenterol2010105112449245620531397
- DewanwalaAChittendenARosenblattMAttitudes toward childbearing and prenatal testing in individuals undergoing genetic testing for Lynch SyndromeFam Cancer201110354955621567236
- WinAKDowtyJGEnglishDRBody mass index in early adulthood and colorectal cancer risk for carriers and non-carriers of germline mutations in DNA mismatch repair genesBr J Cancer2011105116216921559014
- WinAKDowtyJGAntillYCBody mass index in early adulthood and endometrial cancer risk for mismatch repair gene mutation carriersObstet Gynecol2011117489990521422863
- Surveillance, Epidemiology, and End Results Program (SEER)Cancer Stat Fact SheetsBethesda, MDNational Institutes of Health Available from: http://seer.cancer.gov/statfacts/html/breast.htmlAccessed June 12, 2014