
Abstract
“Angelman syndrome” (AS) is a neurodevelopmental disorder whose main features are intellectual disability, lack of speech, seizures, and a characteristic behavioral profile. The behavioral features of AS include a happy demeanor, easily provoked laughter, short attention span, hypermotoric behavior, mouthing of objects, sleep disturbance, and an affinity for water. Microcephaly and subtle dysmorphic features, as well as ataxia and other movement disturbances, are additional features seen in most affected individuals. AS is due to deficient expression of the ubiquitin protein ligase E3A (UBE3A) gene, which displays paternal imprinting. There are four molecular classes of AS, and some genotype–phenotype correlations have emerged. Much remains to be understood regarding how insufficiency of E6-AP, the protein product of UBE3A, results in the observed neurodevelopmental deficits. Studies of mouse models of AS have implicated UBE3A in experience-dependent synaptic remodeling.
Introduction
Harry Angelman, an English pediatrician, first described this condition in 1965 when he reported three children that he referred to as “Puppet Children” because of their unusual arm position and jerky movements.Citation1 In addition to the characteristic movements, Angelman noted severe intellectual disability, absent speech, and bouts of inappropriate laughter. In the nearly 50 years since that original report, the AS phenotype has been elaborated, and the etiology of the disorder identified as deficiency of UBE3A.Citation2,Citation3 The molecular pathogenesis of how UBE3A deficiency leads to this phenotype is beginning to be clarified. What follows is a description of our current understanding of the clinical and molecular aspects of AS.
Clinical review
“AS” is a neurodevelopmental disorder whose main features are intellectual disability, lack of speech, seizures, and a characteristic behavioral profile.Citation4–Citation9 It has a prevalence of between 1/10,000 and 1/20,000 individuals.Citation10,Citation11 See for consistent, frequent, and occasional features of AS.
Table 1 Clinical features of Angelman syndrome
Performance
Development delays in AS are usually evident within the first year of life, with delayed attainment of gross motor, fine motor, receptive language, expressive language, and social skills. Reportedly, individuals with AS plateau at a developmental level of between 24 and 30 months,Citation12 and cognitive performance is usually in the range of severe functional impairment.Citation13 Language development in those with AS is significantly impaired. Most individuals lack speech entirely, a few individuals have small single-word vocabularies,Citation13 and rare individuals are able to use phrases. The combination of deficits exhibited by individuals with AS make the commonly used developmental assessment tools difficult to apply, and these tests underestimate the abilities of AS children (author’s personal observations). Receptive language is superior to expressive language.Citation14 Nonverbal communication using a variety of systems (picture exchange cards, communication devices, modified sign language) is possible in a substantial proportion of individuals with AS (author’s personal observations). Although all skills are delayed, there is variability in adaptive behavior functioning, with relative strength in socialization and relative weakness in motor skills.Citation12,Citation14 A substantial subset of children with AS qualify for a comorbid diagnosis of autism,Citation15–Citation21 independent of the severity of their cognitive and adaptive behavior functioning.Citation21 AS adults are not able to live independently, but most are able to use feeding utensils and perform some household tasks with supervision.Citation22–Citation25 Dressing skills are dependent on the degree of fine-motor dexterity. Daytime continence is possible with prompted voiding and habit training.
Seizures
Seizures occur in 80%–95% of children with AS and usually start in childhood.Citation26–Citation29 The onset of seizures is before age 3 years in 75% of affected individuals.Citation29 Seizure types include myoclonic, atypical absence, generalized tonic–clonic, and atonic (“drop”) seizures. Many individuals exhibit multiple seizures types.Citation27,Citation28 Seizures usually require broad-spectrum anticonvulsant medication and often combination therapy. Efficacy appears to be highest with valproate and clonazepam and lowest with phenobarbital and carbamazepine;Citation28 vigabatrin and carbamazepine may exacerbate seizures.Citation26,Citation29 Some patients have responded to vagal nerve stimulation or ketogenic dietCitation30 for seizures that were medically refractory.Citation28,Citation29 There can be attenuation of seizure activity in adolescents.Citation27–Citation29 There is a characteristic electroencephalogram (EEG) “signature” in AS,Citation31,Citation32 which can sometimes be useful in pointing toward the diagnosis.Citation31 Various combinations of very high amplitude rhythmic (primarily anterior) delta activity, diffuse high amplitude rhythmic theta activity, and posterior-predominant spike and sharp waves are seen in >90% of individuals with AS.Citation33–Citation38
Behavior
The behavioral features of AS include a happy demeanor, easily provoked laughter, short attention span, hypermotoric behavior, mouthing of objects, sleep disturbance with reduced need for sleep, and an affinity for water.Citation39–Citation41 Though infancy can be difficult due to feeding problems and general irritability, happy disposition and increased smiling characterize most children.Citation39 Rarely, unhappy or irritable affect persists, and gastrointestinal difficulties such as dysmotility and gastroesophageal reflux disease may play a role (author’s personal observations). Mouthing of objects becomes very prominent in the young child, along with drooling and tongue thrusting; these behaviors can be lessened or extinguished with behavioral modification. Individuals with AS have an apparently increased desire for social interaction.Citation39 Children are described as easily excited. Though paroxysms of laughter are said to occur in AS, the laughter is not truly “unprovoked”, since an inciting event can usually be identified; however, the responding laughter is frequently excessive or inappropriate to the triggering stimulus. The majority of AS patients exhibit a short attention span, though this characteristic does not discriminate from other conditions with intellectual disability,Citation42 and most children are hypermotoric/hyperactive, becoming calmer in adolescence and adulthood. Disruptive behaviorsCitation43,Citation44 are displayed by the majority of patients, including biting, pinching, hair-pulling, and grabbing. Rarely are these behaviors intended to cause harm; they usually result from easy excitability, desire for attention, poor control over movements, reduced repertoire of need expression, and occasionally frustration over an inability to communicate effectively. Behavioral noncompliance, tantrums, and repetitive or stereotyped behaviors have also been described.Citation43,Citation44
Sleep
Most children have an apparently reduced need for sleep (sometimes as little as 5–6 hours per night) and abnormalities of the sleep–wake cycle, with long or frequent periods of wakefulness during the night.Citation45–Citation47 Sleep problems can involve the initiation and/or maintenance of sleep and early morning awakening.Citation45–Citation48 In a small study, melatonin levels were found to be low in those with AS,Citation49 corroborating prior observations that melatonin improves the sleep of children with AS.Citation50,Citation51 Despite sleep disruption, most individuals with AS do not exhibit daytime somnolence. With behavior modificationCitation52 and/or pharmacologic treatment, sleep difficulties can be overcome in most patients, and sleep patterns improve with age.Citation48 Epilepsy severity correlates with sleep problems, but whether more severe seizures create sleep disturbance or whether poor sleep patterns exacerbate epilepsy remains unclear.Citation53
Other
Growth in AS varies with molecular diagnosis (see the “Genotype–phenotype correlations” section), and microcephaly is common (80%).Citation54 Individuals with AS are generally non-dysmorphic as infants, but a subtle craniofacial phenotype develops with time, consisting of midface recession, prognathism, and broad mouth (the latter two are possibly consequences of tongue thrusting, mouthing behaviors and increased smiling). A subset of patients have hypopigmentation of the skin, hair, and eyes; this is more common in those with a deletion,Citation55 who lack the maternal copy of OCA2 and presumably have a hypomorphic allele on the paternal chromosome.Citation56 Patients with AS and oculocutaneous albinism type 2 (OCA2) have been reported, mostly commonly due to deletion of the maternal OCA2 and mutation in the paternal OCA2.Citation57 A non-deletion patient with AS and OCA2 presumably has this constellation due to mutation in the paternal OCA2 and isodisomy for paternal chromosome 15 (author’s personal observations). AS patients with UBE3A mutations may have hypopigmentation on the basis of UBE3A’s regulation of the melanocortin-1 receptor, which is downregulated in Ube3a null mice.Citation58
Ocular problems in AS include refractive errors (usually hyperopia and astigmatism), iris and choroidal hypopigmentation, and esotropia or exotropia.Citation59–Citation61 Nystagmus is reported but is not common.Citation61 Ocular hypopigmentation is seen in all molecular classes but is more common in those with deletion.Citation61 Patchy retinochoroidal atrophy was reported in two adults with AS.Citation62 The author knows of one deletion patient with bilateral ocular pterygia requiring corneal transplantation to restore vision.
Truncal hypotonia and distal extremity hypertonia/hyperreflexia characterize the neurological examination of children with AS.Citation63 Movement disturbances, abnormalities of tone, and impaired balance contribute to the delayed acquisition of motor skills (sitting after 12 months, walking between 2 and 6 years). Movement disorders include jerkiness, ataxic gait, and tremors.Citation63,Citation64 Many walk with arms held up and flexed at the elbows, true to the original description. The incidence of nonambulation is said to be 10%,Citation65 but whether this will remain true in the modern era (of earlier diagnosis and prompt intervention and continued therapy) remains to be seen. The early institution and continuation of physical therapy may change the natural history of scoliosis, previously reported to occur in 10% of children and up to 70% of adults,Citation66 by improving truncal tone.
Life expectancy appears to be normal;Citation22–Citation26 however, early death by accidental drowning has claimed the lives of some children, and the author is aware of premature deaths due to choking, pneumonia, suffocation, and seizures (personal communication).
Genetic basis
AS is caused by a lack of expression of the maternally inherited UBE3A gene in the brain. UBE3A is one of a small subset of human genes that are imprinted – that is, expressed depending on parent of origin, in a tissue-specific manner.Citation65,Citation66 While in most tissues, UBE3A appears to be expressed from both alleles (though perhaps unequally favoring the maternal alleleCitation67), in the brain, the paternally derived UBE3A gene is silenced, and only the maternally inherited copy is active.Citation68–Citation70 UBE3A was initially discarded as a potential candidate for the AS gene because it appeared not to be imprinted when studied in lymphocytes and fibroblasts,Citation71 and its widespread expression ran counter to expectation since the phenotype is exclusively neurological. Establishing brain-only imprinting of UBE3A in mice resurrected UBE3A’s status as a candidate gene for AS.Citation70 Analysis of Ube3a in AS patients with biparental contribution to 15q11-q13 and no imprinting abnormalities showed point mutations in several unrelated patients, identifying it as the gene responsible for the AS phenotype.Citation2,Citation3
AS is caused by deficient expression of the maternal copy of the UBE3A gene due to one of four molecular etiologies: deletion of the AS critical region on maternal chromosome 15q11-q13, paternal uniparental disomy (UPD) for chromosome 15, an imprinting defect causing lack of expression of the maternal copy of UBE3A, and mutations in the maternally inherited copy of UBE3A.Citation55 There is a subgroup of patients with a clinical diagnosis of AS for whom no abnormality of UBE3A can be identified. Two potential explanations for these test-negative patients are 1) novel mechanisms for repression of UBE3A expression yet to be identified and 2) misdiagnoses of phenotypically similar conditions. Patients with AS and negative molecular analyses are now being recognized to have a variety of Angelman-like syndromes.Citation72,Citation73
In a cohort of AS patients participating in a natural history study conducted as part of the Rare Diseases Clinical Research Network, the distribution of molecular diagnoses among 286 patients was as follows: 31.1% class II deletion, 23.8% class I deletion, 10.8% unspecified deletion, 3.8% atypical deletion, 8.7% UPD, 7.7% imprinting defect, and 11.2% UBE3A mutation (unpublished data). Among individuals with a deletion, 40.5% have a common 5.9 megabases (Mb) (class I) deletion; 53% have a smaller 5.0 Mb (class II) deletion, differing only by the location of the proximal (centromeric) breakpoint; and 6.5% have atypical deletions. Deletions are mediated by homologous misalignment and meiotic recombination between low-copy-number repeats (duplicons) that have been identified in proximal and distal 15q11-q13. These duplicons arose with the amplification of an ancestral gene, homologous to the E6-AP carboxy terminus (HECT) and regulator of chromatin condensation 1-like domain (RLD)-containing E3 ubiquitin protein ligase 2 (HERC2). Duplicons having 90%–99% identity to the first 79 exons of HERC2 are found in at least ten copies in the 15q11-q13.Citation74
Paternal UPD is isodisomic in almost all cases. The most likely origin of this event is maternal nondisjunction producing a monosomy 15 conception, with post-zygotic rescue by duplication of the paternal chromosome 15.Citation75 As in the deletion cases, this class of AS represents de novo mutational events and has a very low risk for recurrence.
“Imprinting defect” occurs when a paternal imprint is erroneously assigned to the maternally inherited allele. Two types of imprinting defects are known: those due to a submicroscopic deletion of the imprinting center, and those with no detectable mutation.Citation76–Citation78 Most submicroscopic imprinting center deletions are familial and carry a 50% risk for recurrence. Thus far, all imprinting defects with undetectable mutations have been sporadic events. They are presumed to result from failure to establish or maintain the imprint during oogenesis, due to a stochastic event or perhaps (as yet unknown) environmental factors.Citation78
Intragenic UBE3A mutations include insertion, deletion, nonsense, missense, and splice site mutations.Citation79,Citation80 A substantial portion of UBE3A mutations are inherited from the mother’s paternally acquired allele. In this circumstance, a 50% recurrence risk pertains.Citation81
Mechanisms of imprinting and gene regulation
Genetic imprinting is the process of conferring functional differences onto specific genes such that their expression occurs from only one parent’s allele.Citation65 There is incomplete understanding of the mechanism(s) of imprint establishment in the germ line, imprint maintenance during development and postnatal life, and imprint reversal in the germ line of the next generation. Further complexities, such as tissue-specific imprinting and age-related changes in imprinting are poorly understood.
Mechanisms of controlling gene expression include DNA insulators (DNA elements which prevent nearby chromatin domains from interacting); histone modifications (such as acetylation, phosphorylation, and methylation), which alter chromatin structure and influence transcriptional accessibility; DNA methylation; and transcriptional enhancer competition (promoters of linked imprinted genes competing for access to enhancers).Citation82–Citation84 Each of these epigenetic mechanisms overlays the information contained within the nucleotide sequence. Imprinted genes are found in clusters in specific areas of the genome, suggesting coordinated regulation by a regional element, which has been designated the “imprinting control region” (ICR).
DNA methylation is the most well understood of the recognized epigenetic mechanisms. The addition of a methyl group to the cytosine base of a CpG dinucleotide is found in most imprinted genes and in all ICRs.Citation82 Loss and reacquisition of DNA methylation occurs during specific phases of germ cell development, and probably represents erasure of the imprint from the previous generation and re-establishment of the parent-of-origin specific epigenotype. DNA methylation may play a role in establishing and/or maintaining the imprint.Citation82–Citation84
Deficits in the imprinted gene cluster on chromosome 15q11-13 cause Prader–Willi syndrome (PWS) and AS.Citation7,Citation66,Citation78,Citation85,Citation86 Loss of expression of maternally derived UBE3A causes AS, while loss of expression of paternally derived gene(s) causes PWS. The main features of PWS are neonatal hypotonia and failure to thrive, childhood onset hyperphagia and obesity, small hands and feet, short stature, hypogonadism, and cognitive impairment.Citation87 Though many features of PWS can be reproduced by absence of a small nucleolar organizing RNA gene, SNORD116, the full PWS phenotype requires the loss of expression of several genes, indicating that PWS is truly a contiguous gene syndrome.Citation87
Differential methylation of chromosome 15q11-q13 provides the basis for diagnostic testing for PWS and AS. The maternal chromosome is highly methylated while the paternal chromosome is mostly unmethylated in the 15q11-q13 region, and this can be demonstrated using methylation-sensitive restriction enzymes and Southern blot analysis or polymerase chain reaction assay of the promoter region of the small nuclear ribonucleoprotein polypeptide N (SNRPN) gene. A maternal-only contribution is diagnostic of PWS, while an exclusively paternal contribution indicates AS. A normal methylation profile having both a maternal and paternal contribution excludes PWS but does not rule out AS, because 10%–20% of cases of AS are due to maternally inherited UBE3A mutations.Citation79,Citation80
The ICR for the 15q11-q13 cluster has been designated the “IC”.Citation76,Citation85 It was defined by small submicroscopic deletions in a subgroup of PWS and AS multiplex families that demonstrated biparental inheritance of the 15q11-13 region yet uniparental imprint.Citation76 The IC regulates in cis the establishment and maintenance of the imprint for the entire cluster. The IC has a bipartite structure, the Prader–Willi syndrome imprinting center (PWS-IC) and the Angelman syndrome imprinting center (AS-IC), separated by 35 kb (). All AS patients with IC deletions are missing the AS-IC (880 bp centromeric of PWS-IC).Citation88 The PWS-IC establishes and maintains paternal gene expression. During oogenesis, the AS-IC negatively regulates the PWS-IC and prevents the paternal imprint from being established. Deletion of the AS-IC leads to a paternal imprint of the entire 15q11-q13 region, and AS occurs when this is transmitted through the maternal germ line. The PWS-IC and AS-IC must be proximal and correctly related in order for the maternal imprint to be established.Citation89
Figure 1 Imprinting map of the human chromosome 15q11-13 region. Paternal and maternal chromosome 15q11-13 regions around the Angelman syndrome imprinting center (AS-IC) and Prader–Willi syndrome imprinting center (PWS-IC) are represented in (A and B), respectively.
Abbreviations: MKRN3, makorin ring finger protein 3; NDN, necdin; MAGEL2, melanoma-associated antigen-like 2; SNURF-SNRPN, small nuclear ribonucleoprotein polypeptide N upstream reading frame; PWS-IC, Prader–Willi syndrome imprinting center; UBE3A, ubiquitin protein ligase E3A; PWRN2, Prader–Willi Region Non-Protein Coding RNA 2; HBII-436, Small Nucleolar RNA, C/D Box 107; PAR5, Prader Willi/Angelman Region RNA 5; IPW, Imprinted In Prader–Willi Syndrome (Non-Protein Coding); HBII-52; Small Nucleolar RNA, C/D Box 115; ATP10A, ATPase, Class V, Type 10A; GABRB3, Gamma-Aminobutyric Acid (GABA) A Receptor, Beta 3; PWARSN, Prader Willi/Angelman region RNA, SNRPN neighbor; C15ORF2, Chromosome 15 Open Reading Frame 2; HBII-85, cluster of C/D box containing small nucleolar RNAs.
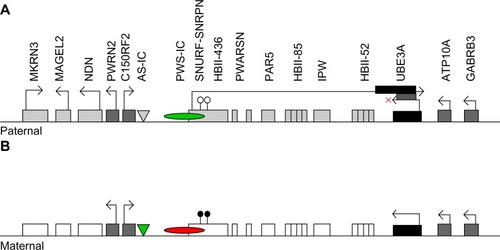
The genes makorin ring finger protein 3 (MKRN3), necdin (NDN), melanoma-associated antigen-like 2 (MAGEL2) and SNRPN upstream reading frame (SNURF-SNRPN) are expressed only from the paternal chromosome where their promoter regions are unmethylated.Citation66,Citation90,Citation91 Between SNURF-SNRPN and UBE3A are located more than 70 small nucleolar RNA (snoRNA) genes. These noncoding genes produce snoRNAs, which modify ribosomal RNA.Citation66 Though not differentially methylated, snoRNAs in this region are indirectly under the control of methylation, because they are processed from the differentially methylated and paternally expressed SNURF-SNRPN sense/UBE3A antisense transcript.
Studies of the orthologous mouse region (chromosome 7) form the basis for much of what is known about methylation status and imprinting of human chromosome 15q11-q13. However, important differences exist.Citation92 The Frat3 gene, for which there is no human homologue, has joined the mouse PWS/AS region, acquiring the paternal pattern of methylation and expression. Transgenic studies, where human elements of the PWS/AS region are inserted in the mouse, have shown that the regulatory elements of the imprinting machinery have diverged between the two species. No mouse equivalent of the human AS-IC has been identified. Further study of the mouse PWS/AS region imprinting will probably yield important insights,Citation93 but some may not apply to human UBE3A imprinting.
UBE3A itself is not differentially methylated;Citation55 its imprinted expression is indirectly regulated by a long noncoding antisense RNA transcript (UBE3A-ATS) which is part of a larger SNURF-SNRPN transcript.Citation66 UBE3A-ATS is active on the paternal chromosome and blocks UBE3A transcription in cis.Citation94–Citation97 Changes in DNA methylation and histone acetylation of the PWS-IC control production of the UBE3A-ATS from the paternal allele.Citation98 The mechanism by which UBE3A-ATS blocks UBE3A transcription is unknown, but may involve histone-mediated repression, transcriptional interference, or repressive three-dimensional chromatin structure.Citation66
Molecular pathogenesis
UBE3A spans 120 kb of genomic DNA.Citation9 Mutations have been detected throughout all regions of the gene. Sixteen exons have been identified; since this region displays alternative splicing, additional exons at the 5′ end of the gene are possible. UBE3A is transcribed in the direction from telomere to centromere, producing RNA transcripts of 5–6 kb that include 2 kb of 3′ untranslated region sequence. There are three main transcripts producing three isoforms of UBE3A, and eight to ten additional transcripts of uncertain function. The function of the three different isoforms of UBE3A is unknown and the significance of tissue-specific variations in RNA splicing and isoform predominance is unclear.
UBE3A encodes E6-associated protein (E6-AP), an E3 ubiquitin ligase.Citation9 E6-AP derives its name from its initial characterization, in which it was found to be associated with the E6 protein of papillomaviruses to promote degradation of p53. However, E6-AP does not maintain a stable association with p53, and its main function is believed to be participation in protein degradation in proteasomes via the ubiquitin pathway. The ubiquitin-proteasome system targets cellular proteins for destruction by covalently attaching ubiquitin to one or more lysine residues of proteins destined for degradation. Ubiquitination involves a three-step process: 1) activation of ubiquitin by an E1 enzyme, 2) transfer to an E2 conjugating enzyme, and 3) covalent ligation of ubiquitin to the protein substrate by an E3 ligase; E6-AP is one of many E3 ligases. E6-AP also interacts with proteins involved in such cellular functions as cell-cycle regulation and synaptic function and plasticity,Citation99,Citation100 and acts as a transcriptional co-activator of steroid hormone receptors.Citation101,Citation102 The C-terminus of E6-AP is a functionally important and highly conserved domain that is shared by a family of proteins (HECT domain), of which E6-AP is the founding member. The last six amino acids of the E6-AP C-terminus are essential for activity in vitro. It is unknown how substrate specificity is determined. E6-AP is found in all tissues that have been studied.
Presumably, defective UBE3A activity results in failure to degrade its substrates because of impaired ubiquitination. Several E6-AP targets have been identified, including activity-regulated cytoskeleton-associated protein (Arc) and Ephexin5.Citation9 E6-AP regulates Arc levels either by regulating estradiol-induced transcription of ArcCitation103 or by direct ubiquitination of Arc.Citation104 Arc regulates surface expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionate receptors (AMPARs). When E6-AP is deficient, the excitatory postsynaptic AMPARs are internalized, which impairs synaptic transmission.Citation104 Ephexin5 has a role in controlling synapse number.Citation105 Recently proposed as a substrate for E6-AP is sacsin,Citation104 defects of which cause a form of spastic ataxia;Citation106 given the ataxic gait in AS, sacsin is an attractive target. There is also mounting evidence that UBE3A and methyl CpG binding protein 2 interact to regulate the expression of target genes.Citation107 Recently, a role for E6-AP in Golgi acidification and protein sialylation was proposed.Citation108
Mouse models have facilitated the progress in understanding the molecular pathogenesis of AS.Citation109 The Ube3a knockout mouse, generated with target disruption of Ube3a on the maternal chromosome 7, nicely recapitulates the human disorder.Citation110,Citation111 Ube3am-p+ mice demonstrate reduced brain size, ataxia, motor impairment, abnormal EEG, sleep disturbance,Citation112 learning and memory impairment, and deficits in hippocampal long-term potentiation (LTP).Citation110,Citation111 In the hippocampus of Ube3am-p+ mice, the calcium/calmodulin-dependent protein kinase II (CaMKII), which plays a role in induction of LTP that is critical for memory, has an increased level of inhibitory phosphorylation and reduced activity.Citation113 Ube3am-p+ mice with a concomitant mutation in CaMKII that blocks inhibitory phosphorylation are indistinguishable from wild-type mice,Citation114 indicating the defect of E6-AP can be overcome with excess CaMKII activity. E6-AP appears to play a role in activity-dependent synaptic plasticity; that is, the remodeling of synapses depending on experience.Citation99,Citation100 The observations of abnormal dendritic spine morphologyCitation115 and increase in synapses lacking AMPARsCitation104 in Ube3am-p+ mice fit with this hypothesis. Ube3a acts as a transcriptional co-activator of many steroid hormone receptors, including the glucocorticoid receptor, and glucocorticoid receptor-mediated signaling is dysregulated in the brains of Ube3am-p+ mice, which show increased serum levels of corticosterone and increased anxiety-like behavior.Citation116 Ube3am-p+ mice also have enhanced neuregulin-ErbB4 signalingCitation117 that correlates with abnormal synaptic plasticity and memory impairment, not mediated through differences in AMPARs and N-methyl-D-aspartate receptors and not mediated through direct interaction of neuregulin or ErbB4 with UBE3A. The deficits in LTP can be rescued by ErbB inhibitors infused directly into the hippocampus of Ube3am-p+ mice.Citation117
Diagnostic algorithm
The most sensitive test for AS is a methylation analysis of the chromosome 15q11-13 region, using either methylation-specific polymerase chain reaction or methylation-sensitive multiplex ligation-dependent probe amplification. Further testing is needed to parse those with abnormal methylation testing into deletion, UPD, and imprinting defect categories.Citation118 depicts the diagnostic algorithm for testing for AS.
Figure 2 Molecular diagnostic algorithm for AS.
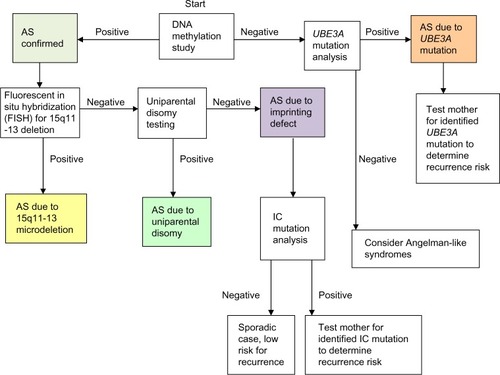
If DNA methylation analysis is negative, then UBE3A sequencing is appropriate for those with a convincing AS phenotype. When both DNA methylation analysis and UBE3A mutation testing are negative, the likelihood of AS is small.Citation18 Though once thought to represent a substantial portion of cases, test-negative (“clinical diagnosis”) patients with AS are probably rare. Patients with an AS phenotype in whom testing returns normal should be considered for an alternative diagnosis, such as Pitt–Hopkins syndrome, Mowat–Wilson syndrome, Kleefstra syndrome, Phelan–McDermid syndrome, Koolen–de Vries syndrome, Christianson syndrome, and MBD5 haploinsufficiency.Citation72,Citation73
Recurrence risk for AS due to microdeletion or UPD is negligible, whereas UBE3A mutations and imprinting defects can have a 50% risk for recurrence if the mother is found to carry the mutation on her paternally inherited chromosome 15.Citation81
Genotype–phenotype correlations
Clinical differences between the molecular classes of AS have been recognized.Citation55,Citation119–Citation123 Those with deletion tend to be shorter and lighter than the general population, while those with UPD or imprinting defects tend to be taller and heavier; growth in those with UBE3A mutations is variable.Citation54 There is a higher incidence of microcephaly in the deletion group compared with non-deletion subtypes.Citation54,Citation119 As a group, patients with deletions tend to have a more severe phenotype (later onset of independent walking, earlier onset and increased severity of seizures, complete absence of speech) than patients with UPD, imprinting defect, or UBE3A mutation (non-deletion patients).Citation14,Citation55 In younger patients with AS, those with UBE3A mutations scored higher in tests of cognition, gross motor and fine motor skills, and receptive language than deletion patients.Citation14 Though speech is usually absent in AS patients with deletions, use of up to 20 words has been reported in AS patients of other molecular classes; however, Gentile et al found no differences in expressive language skills in patients of less than 5 years of age with AS with respect to molecular subtype.Citation14 Compared with patients with a smaller class II deletion (~5 Mb), patients with a larger class I deletion (~6.0 Mb) are more likely to meet criteria for a comorbid diagnosis of autism, have lower cognitive scores, and require more seizure medications.Citation122–Citation124 It has been proposed that other deleted genes in the 15q11-13 region account for the increased phenotypic severity, but the precise contribution and mechanisms remain to be clarified. Three genes for gamma-Aminobutyric acid (GABA)-receptor subunits (GABRB3, GABRA5, GABRG3) are located telomeric of UBE3A and contained within the deleted region. Though these genes show biallelic expression, a role for them in the genesis of epilepsy in AS has been suggested.Citation125 The GABA receptor subunit genes are deleted in both of the common deletion classes (I, II) so these genes do not account for differences seen among deletion-class groups but may contribute to the differences observed between deletion and non-deletion patients. No correlation between genotype and electroencephalographic pattern has emerged.Citation31
Hypopigmentation occurs more frequently in AS deletion patients.Citation55 The (un-imprinted) OCA2 gene located telomeric within the commonly deleted region is responsible for autosomal recessive OCA2. Individuals with AS and OCA2 have been reported, the mechanism being a maternal chromosome deletion and paternal OCA2 mutation.Citation57,Citation126 Semidominant behavior of the OCA2 product (expression of a hypomorphic allele) has been offered to explain the hypopigmentation seen in AS deletion patients. However, hypopigmentation has been reported in other classes of AS patients, including siblings with a maternally inherited intragenic deletion of exons 8–16 of UBE3A, suggesting that Ube3a can alter pigmentation.Citation127 The melanocortin-1 receptor, which is downregulated in Ube3a-null mice, is a potential effector.Citation58
Status of clinical research
Clinical trials conducted thus far have produced negative results. Attempts to increase transcription from the paternal allele through the use of pro-methylation vitamin supplements did not result in any noticeable improvement.Citation128,Citation129 There is an ongoing randomized, placebo-controlled trial using levodopa/carbidopa to treat AS.Citation130 The rationale for this trial was based on the observations that levodopa was able to influence phosphorylation of CaMKII threonine residues in a rat model of Parkinson’s disease;Citation131,Citation132 the finding of dopaminergic neuronal loss in AS mouse models;Citation133 and a report of two adults with AS and Parkinsonian symptoms who responded to levodopa.Citation134 There was a short open-label trial of minocycline treatment,Citation135 results of which have not yet been subjected to peer review. The rationale for minocycline was made based on the observation of elevated matrix metalloproteinase-9 activity in the hippocampi of AS mice (personal communication) and minocycline’s ability to reduce matrix metalloproteinase-9 activity.
Status of basic research
Interrupting the AS pathophysiology may be undertaken at the level of gene transcription or at the point of protein interactions.Citation136 Mouse models for AS (the history of which was summarized recentlyCitation137), have been invaluable in exploring both of these avenues. The Ube3am-p+ mouse has reduced brain size, ataxia, motor deficits, abnormal EEG, inducible seizures, and behavioral alterations.Citation111,Citation133 The AS mouse demonstrates impairments of context-dependent learning and memory and hippocampal LTP.Citation110 Abnormalities of function in cerebellar Purkinje cellsCitation110,Citation137,Citation138 and nigrostriatal pathwaysCitation133,Citation139 have been reported. The behavioral phenotype of the Ube3am-p+ mouse was recently characterized in a comprehensive manner,Citation140 which will assist greatly in the evaluation of future potential treatments.
Transcriptional upregulation has been approached by both gene therapyCitation141 and pharmacologic intervention.Citation142 Daily et al showed that direct injection of a recombinant adeno- associated viral vector carrying Ube3a into the hippocampi of adult AS mice could significantly improve associative learning (contextual fear conditioning).Citation141 Huang and colleagues have demonstrated that the topoisomerase topotecan can un-silence the paternal Ube3a allele in cultured mouse primary cortical neurons, apparently by reducing transcription of Ube3a-ats.Citation142 Recently the mechanism of inhibition of Ube3a-ats was shown to be mediated through RNA:DNA hybrid loop stabilization with the paternal snoRNA cluster.Citation143
Identifying the pathways in which Ube3a participates may lead to targeted intervention.Citation134 Studies have shown a role for CaMKII,Citation113,Citation114 neuregulin-ErbB4 signaling,Citation117 EphB/Ephexin5 signaling,Citation105 and Arc (possibly mediated through brain-derived neurotrophic factor-induced TrkB-PSD-95Citation144) signaling.Citation103,Citation104 These observations point to a critical role for Ube3a in experience-dependent synaptic remodeling, possibly through more than one molecular pathway.
Conclusion
AS is a syndromic form of intellectual disability with a distinctive clinical presentation that can be best recognized by behavioral and performance characteristics. Molecular testing can diagnose most, if not all, cases. The causative protein, UBE3A, is critical for the processes of learning and memory through activity-dependent synaptic plasticity. Strides are being made in understanding the molecular pathogenesis, aided by mouse models that faithfully recapitulate the clinical syndrome.
Disclosure
The author declares no conflicts of interest in this work.
References
- AngelmanH‘Puppet’ children. A report on three casesDev Med Child Neurol196576681688
- KishinoTLalandeMWagstaffJUBE3A/E6-AP mutations cause Angelman syndromeNat Genet19971517073 Erratum in: Nat Genet. 1997;15(4):4118988171
- MatsuuraTSutcliffeJSFangPDe novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndromeNat Genet199715174778988172
- Clayton-SmithJLaanLAngelman syndrome: a review of the clinical and genetic aspectsJ Med Genet2003402879512566516
- WilliamsCAAngelmanHClayton-SmithJAngelman syndrome: consensus for diagnostic criteria. Angelman Syndrome FoundationAm J Med Genet19955622372387625452
- WilliamsCABeaudetALClayton-SmithJAngelman syndrome 2005: updated consensus for diagnostic criteriaAm J Med Genet2006140541341816470747
- ChamberlainSJLalandeMAngelman syndrome, a genomic imprinting disorder of the brainJ Neurosci201030309958996320668179
- WilliamsCADriscollDJDagliAIClinical and genetic aspects of Angelman syndromeGenet Med201012738539520445456
- DagliABuitingKWilliamsCAMolecular and clinical aspects of Angelman syndromeMol Syndromol201223–510011222670133
- PetersenMBBrøndum-NielsenKHansenLKWulffKClinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish countyAm J Med Genet19956032612627573182
- KyllermanMOn the prevalence of Angelman syndromeAm J Med Genet19955934058599374
- PetersSUGoddard-FinegoldJBeaudetALMadduriNTurcichMBacinoCACognitive and adaptive behavior profiles of children with Angelman syndromeAm J Med Genet A2004128A211011315213998
- AndersenWHRasmussenRKStrømmePLevels of cognitive and linguistic development in Angelman syndrome: a study of 20 childrenLogoped Phoniatr Vocol20012612911432411
- GentileJKTanWHHorowitzLTA neurodevelopmental survey of Angelman syndrome with genotype-phenotype correlationsJ Dev Behav Pediatr2011317592601 Erratum in: J Dev Behav Pediatr. 2011;32(3):26720729760
- TrillingsgaardAØStergaardJRAutism in Angelman syndrome: an exploration of comorbidityAutism20048216317415165432
- PetersSUBeaudetALMadduriNBacinoCAAutism in Angelman syndrome: implications for autism researchClin Genet200466653053615521981
- DykensEMSutcliffeJSLevittPAutism and 15q11-q13 disorders: behavioral, genetic, and pathophysiological issuesMent Retard Dev Disabil Res Rev200410428429115666333
- WalzNCParent report of stereotyped behaviors, social interaction, and developmental disturbances in individuals with Angelman syndromeJ Autism Dev Disord200737594094717019625
- BonatiMTRussoSFinelliPEvaluation of autism traits in Angelman syndrome: a resource to unfold autism genesNeurogenetics20078316917817415598
- HogartAWuDLaSalleJMSchanenNCThe comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13Neurobiol Dis201038218119118840528
- PetersSUHorowitzLBarbieri-WelgeRTaylorJLHundleyRJLongitudinal follow-up of autism spectrum features and sensory behaviors in Angelman syndrome by deletion classJ Child Psychol Psychiatry201253215215921831244
- Clayton-SmithJPembreyMEAngelman syndromeJ Med Genet19922964124151619637
- LaanLAden BoerATHennekamRCRenierWOBrouwerOFAngelman syndrome in adulthoodAm J Med Genet19966633563609072912
- Clayton-SmithJClinical research on Angelman syndrome in the United Kingdom: observations on 82 affected individualsAm J Med Genet199346112157684188
- BuntinxIMHennekamRCBrouwerOFClinical profile of Angelman syndrome at different agesAm J Med Genet19955621761837625442
- ValenteKDKoiffmannCPFridmanCEpilepsy in patients with angelman syndrome caused by deletion of the chromosome 15q11-13Arch Neurol200663112212816401744
- PelcKBoydSGCheronGDanBEpilepsy in Angelman syndromeSeizure200817321121717904873
- ThibertRLConantKDBraunEKEpilepsy in Angelman syndrome: a questionnaire-based assessment of the natural history and current treatment optionsEpilepsia200950112369237619453717
- ThibertRLLarsonAMHsiehDTRabyARThieleEANeurologic manifestations of Angelman syndromePediatr Neurol201348427127923498559
- ThibertRLPfeiferHHLarsonAMLow glycemic index treatment for seizures in Angelman syndromeEpilepsia20125391498150222779920
- BoydSGHardenAPattonMAThe EEG in early diagnosis of the Angelman (happy puppet) syndromeEur J Pediatr198814755085133409926
- DanBBoydSGAngelman syndrome reviewed from a neurophysiological perspective. The UBE3A-GABRB3 hypothesisNeuropediatrics200334416917612973656
- LaanLARenierWOArtsWFEvolution of epilepsy and EEG findings in Angelman syndromeEpilepsia19973821951999048672
- MinassianBADeLoreyTMOlsenRWAngelman syndrome: correlations between epilepsy phenotypes and genotypesAnn Neurol19984344854939546330
- ValenteKDAndradeJQGrossmannRMAngelman syndrome: difficulties in EEG pattern recognition and possible misinterpretationsEpilepsia20034481051106312887436
- KorffCMKelleyKRNordliDRJrNotched delta, phenotype, and Angelman syndromeJ Clin Neurophysiol200522423824316093895
- LaanLAVeinAAAngelman syndrome: is there a characteristic EEG? Brain Dev2005272808715668045
- VendrameMLoddenkemperTZarowskiMAnalysis of EEG patterns and genotypes in patients with Angelman syndromeEpilepsy Behav201223326126522341959
- WilliamsCAThe behavioral phenotype of the Angelman syndromeAm J Med Genet C Semin Med Genet2010154C443243720981772
- PelcKCheronGDanBBehavior and neuropsychiatric manifestations in Angelman syndromeNeuropsychiatr Dis Treat20084357758418830393
- HorslerKOliverCThe behavioural phenotype of Angelman syndromeJ Intellect Disabil Res200650Pt 1335316316429
- BarryRJLeitnerRPClarkeAREinfeldSLBehavioral aspects of Angelman syndrome: a case control studyAm J Med Genet A2005132A1812 Erratum in: Am J Med Genet A. 2005;136(1):11115578589
- SummersJAAllisonDBLynchPSSandlerLBehaviour problems in Angelman syndromeJ Intellect Disabil Res199539Pt 2971067787388
- ClarkeDJMarstonGProblem behaviors associated with 15q-Angelman syndromeAm J Ment Retard20001051253110683706
- BruniOFerriRD’AgostinoGMianoSRoccellaMEliaMSleep disturbances in Angelman syndrome: a questionnaire studyBrain Dev200426423324015130689
- DiddenRKorziliusHSmitsMGCurfsLMSleep problems in individuals with Angelman syndromeAm J Ment Retard2004109427528415176919
- WalzNCBeebeDByarsKSleep in individuals with Angelman syndrome: parent perceptions of patterns and problemsAm J Ment Retard2005110424325215941362
- PelcKCheronGBoydSGDanBAre there distinctive sleep problems in Angelman syndrome? Sleep Med20089443444117765640
- TakaesuYKomadaYInoueYMelatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patientsSleep Med20121391164117022841843
- ZhandovaIVWurtmanRJWagstaffJEffects of a low dose of melatonin on sleep in children with Angelman syndromeJ Pediatr Endocrinol Metab1999121576710392349
- BraamWDiddenRSmitsMGCurfsLMMelatonin for chronic insomnia in Angelman syndrome: a randomized placebo-controlled trialJ Child Neurol200823664965418539989
- AllenKDKuhnBRDeHaaiKAWallaceDPEvaluation of a behavioral treatment package to reduce sleep problems in children with Angelman SyndromeRes Dev Disabil201334167668623123881
- ConantKDThibertRLThieleEAEpilepsy and the sleep-wake patterns found in Angelman syndromeEpilepsia200950112497250019453716
- TanWHBacinoCASkinnerSAAngelman syndrome: Mutations influence features in early childhoodAm J Med Genet A2011155A1819021204213
- LossieACWhitneyMMAmidonDDistinct phenotypes distinguish the molecular classes of Angelman syndromeJ Med Genet2001381283484511748306
- BrilliantMHKingRFranckeUThe mouse pink-eyed dilution gene: association with hypopigmentation in Prader-Willi and Angelman syndromes and with human OCA2Pigment Cell Res1994763984027761348
- FridmanCHosomiNVarelaMCSouzaAHFukaiKKoiffmannCPAngelman syndrome associated with oculocutaneous albinism due to an intragenic deletion of the OCA2Am J Med Genet A2003119A218018312749060
- LowDChenKSUBE3A regulates MC1R expression: a link to hypopigmentation in Angelman syndromePigment Cell Melanoma Res201124594495221733131
- MahMLWallaceDKPowellCMOphthalmic manifestations of Angelman syndromeJ AAPOS20004424824910951304
- SchneiderBBMainoDMAngelman syndromeJ Am Optom Assoc19936475025068376719
- MichielettoPBonanniPPensieroSOphthalmic findings in Angelman syndromeJ AAPOS201115215816121596294
- RufaADottiMTOrricoABattistiCCarlettoFFedericoARetinochoroidal atrophy in two adult patients with Angelman syndromeAm J Med Genet A2003122A215515812955768
- WilliamsCANeurological aspects of the Angelman syndromeBrain Dev2005272889415668046
- LaanLAv HaeringenABrouwerOFAngelman syndrome: a review of clinical and genetic aspectsClin Neurol Neurosurg1999101316117010536901
- ReikWWalterJGenomic imprinting: parental influence on the genomeNat Rev Genet200121213211253064
- ChamberlainSJRNAs of the human chromosome 15q11-q13 imprinted regionWiley Interdiscip Rev RNA20134215516623208756
- GustinRMBichellTJBubserMTissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndromeNeurobiol Dis201039328329120423730
- RougeulleCGlattHLalandeMThe Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brainNat Genet199717114159288088
- VuTHHoffmanARImprinting of the Angelman syndrome gene, UBE3A, is restricted to brainNat Genet199717112139288087
- AlbrechtUSutcliffeJSCattanachBMImprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neuronsNat Genet199717175789288101
- NakaoMSutcliffeJSDurtschiBMutiranguraALedbetterDHBeaudetALImprinting analysis of three genes in the Prader-Willi/Angelman region: SNRPN, E6-associated protein, and PAR-2 (D15S225E)Hum Mol Genet1994323093158004100
- WilliamsCALossieADriscollDRC Phillips UnitAngelman syndrome: mimicking conditions and phenotypesAm J Med Genet2001;1011596411343340
- TanWHBirdLMThibertRLWilliamsCAIf not Angelman, what is it? A review of Angelman-like syndromesAm J Med Genet1292014 Epub
- JiYRebertNAJoslinJMHigginsMJSchultzRANichollsRDStructure of the highly conserved HERC2 gene and of multiple partially duplicated paralogs in humanGenome Res200010331932910720573
- MutiranguraAGreenbergFButlerMGMultiplex PCR of three dinucleotide repeats in the Prader-Willi/Angelman critical region (15q11-q13): molecular diagnosis and mechanism of uniparental disomyHum Mol Genet1993221431518499903
- BuitingKSaitohSGrossSInherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15Nat Genet1995943954007795645
- BuitingKGrossSLichCGillessen-KaesbachGel-MaarriOHorsthemkeBEpimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defectAm J Hum Genet200372357157712545427
- HorsthemkeBBuitingKImprinting defects on human chromosome 15Cytogenet Genome Res20061131–429229916575192
- MalzacPWebberHMonclaAMutation analysis of UBE3A in Angelman syndrome patientsAm J Hum Genet1998626135313609585605
- FangPLev-LehmanETsaiTFThe spectrum of mutations in UBE3A causing Angelman syndromeHum Mol Genet1999811291359887341
- Van BuggenhoutGFrynsJPAngelman syndrome (AS, MIM 105830)Eur J Hum Genet200917111367137319455185
- AbramowitzLKBartolomeiMSGenomic imprinting: recognition and marking of imprinted lociCurr Opin Genet Dev2012222727822195775
- KelseyGFeilRNew insights into establishment and maintenance of DNA methylation imprints in mammalsPhilos Trans R Soc Lond B Biol Sci201336816092011033623166397
- IshidaMMooreGEThe role of imprinted genes in humansMol Aspects Med201334482684022771538
- HorsthemkeBStructure and function of the human chromosome 15 imprinting centerJ Cell Physiol199717322372419365529
- LalandeMCalcianoMAMolecular epigenetics of Angelman syndromeCell Mol Life Sci2007647–894796017347796
- CassidySBSchwartzSMillerJLDriscollDJPrader-Willi syndromeGenet Med2012141102622237428
- BuitingKLichCCottrellSBarnicoatAHorsthemkeBA 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bpHum Genet199910566566610647904
- BuitingKBarnicoatALichCPembreyMMalcolmSHorsthemkeBDisruption of the bipartite imprinting center in a family with Angelman syndromeAm J Hum Genet20016851290129411283796
- NichollsRDKnepperJLGenome organization, function, and imprinting in Prader-Willi and Angelman syndromesAnnu Rev Genomics Hum Genet2001215317511701647
- HorsthemkeBWagstaffJMechanisms of imprinting of the Prader-Willi/Angelman regionAm J Med Genet A2008146A162041205218627066
- JohnstoneKADuBoseAJFuttnerCRElmoreMDBrannanCIResnickJLA human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for Angelman syndrome imprinting defectsHum Mol Genet200615339340416368707
- WuMYJiangMZhaiXBeaudetALWuRCAn unexpected function of the Prader-Willi syndrome imprinting center in maternal imprinting in micePLoS One201274e3434822496793
- RougeulleCCardosoCFontésMColleauxLLalandeMAn imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcriptNat Genet199819115169590281
- RunteMHüttenhoferAGrossSKiefmannMHorsthemkeBBuitingKThe IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3AHum Mol Genet200110232687270011726556
- LandersMBancescuDLLe MeurERegulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/SnrpnNucleic Acids Res200432113480349215226413
- MengLPersonREBeaudetALUbe3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3aHum Mol Genet201221133001301222493002
- ChamberlainSJBrannanCIThe Prader-Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3aGenomics200173331632211350123
- YashiroKRidayTTCondonKHUbe3a is required for experience-dependent maturation of the neocortexNat Neurosci200912677778319430469
- SatoMStrykerMPGenomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3aProc Natl Acad Sci USA2010107125611561620212164
- NawazZLonardDMSmithCLThe Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamilyMol Cell Biol1999192118211899891052
- El HokayemJNawazZE6AP in the Brain: One Protein, Dual Function, Multiple DiseasesMol Neurobiol1052013 Epub
- KühnleSMothesBMatentzogluKScheffnerMRole of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein ArcProc Natl Acad Sci USA2013110228888889323671107
- GreerPLHanayamaRBloodgoodBLThe Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arcCell2010140570471620211139
- MargolisSSSalogiannisJLiptonDMEphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formationCell2010143344245521029865
- GregianinEVazzaGScaramelEA novel SACS mutation results in non-ataxic spastic paraplegia and peripheral neuropathyEur J Neurol201320111486149123800155
- KimSChahrourMBen-ShacharSLimJUbe3a/E6AP is involved in a subset of MeCP2 functionsBiochem Biophys Res Commun20134371677323791832
- CondonKHHoJRobinsonCGHanusCEhlersMDThe Angelman syndrome protein Ube3a/E6AP is required for Golgi acidification and surface protein sialylationJ Neurosci20133393799381423447592
- MabbAMJudsonMCZylkaMJPhilpotBDAngelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypesTrends Neurosci201134629330321592595
- JiangYHArmstrongDAlbrechtUMutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiationNeuron19982147998119808466
- MiuraKKishinoTLiENeurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient miceNeurobiol Dis20029214915911895368
- ColasDWagstaffJFortPSalvertDSardaNSleep disturbances in Ube3a maternal-deficient mice modeling Angelman syndromeNeurobiol Dis200520247147815921919
- WeeberEJJiangYHElgersmaYDerangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndromeJ Neurosci20032372634264412684449
- van WoerdenGMHarrisKDHojjatiMRRescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alpha-CaMKII inhibitory phosphorylationNat Neurosci200710328028217259980
- DindotSVAntalffyBABhattacharjeeMBBeaudetALThe Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphologyHum Mol Genet200817111111817940072
- GodavarthiSKDeyPMaheshwariMJanaNRDefective glucocorticoid hormone receptor signaling leads to increased stress and anxiety in a mouse model of Angelman syndromeHum Mol Genet20122181824183422215440
- KaphzanHHernandezPJungJIReversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitorsBiol Psychiatry201272318219022381732
- RamsdenSCClayton-SmithJBirchRBuitingKPractice guidelines for the molecular analysis of Prader-Willi and Angelman syndromesBMC Med Genet2010117020459762
- BürgerJKunzeJSperlingKReisAPhenotypic differences in Angelman syndrome patients: imprinting mutations show less frequently microcephaly and hypopigmentation than deletionsAm J Med Genet19966622212268958335
- MonclaAMalzacPVoelckelMAPhenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patientsEur J Hum Genet19997213113910196695
- VarelaMCKokFOttoPAKoiffmannCPPhenotypic variability in Angelman syndrome: comparison among different deletion classes and between deletion and UPD subjectsEur J Hum Genet2004121298799215470370
- SahooTPetersSUMadduriNSMicroarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype-phenotype correlationsJ Med Genet200643651251616183798
- SahooTBacinoCAGermanJRIdentification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlationsEur J Hum Genet200715994394917522620
- ValenteKDVarelaMCKoiffmannCPAngelman syndrome caused by deletion: a genotype-phenotype correlation determined by breakpointEpilepsy Res20131051–223423923352739
- DanBBoydSGAngelman syndrome reviewed from a neurophysiological perspective. The UBE3A-GABRB3 hypothesisNeuropediatrics200334416917612973656
- SaitohSOisoNWadaTNarazakiOFukaiKOculocutaneous albinism type 2 with a OCA2 missense mutation in a patient with Angelman syndromeJ Med Genet200037539239410905897
- BoyesLWallaceAJKrajewska-WalasekMChrzanowskaKHClayton-SmithJRamsdenSDetection of a deletion of exons 8–16 of the UBE3A gene in familial Angelman syndrome using a semi-quantitative dosage PCR based assayEur J Med Genet200649647248016740422
- PetersSUBirdLMKimonisVDouble-blind therapeutic trial in Angelman syndrome using betaine and folic acidAm J Med Genet A2010152A81994200120635355
- BirdLMTanWHBacinoCAA therapeutic trial of pro-methylation dietary supplements in Angelman syndromeAm J Med Genet A2011155A122956296322002941
- Wen-HannTanA trial of levodopa in Angelman syndromeClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2011 [updated November 18, 2013] Available from: http://clinicaltrials.gov/show/NCT01281475. NLM identifier: NCT01281475Accessed February 23, 2014
- PicconiBCentonzeDRossiSBernardiGCalabresiPTherapeutic doses of L-dopa reverse hypersensitivity of corticostriatal D2- dopamine receptors and glutamatergic overactivity in experimental parkinsonismBrain2004127Pt 71661166915155524
- BrownAMDeutchAYColbranRJDopamine depletion alters phosphorylation of striatal proteins in a model of ParkinsonismEur J Neurosci200522124725616029214
- MulherkarSAJanaNRLoss of dopaminergic neurons and resulting behavioural deficits in mouse model of Angelman syndromeNeurobiol Dis201040358659220696245
- HarbordMLevodopa responsive Parkinsonism in adults with Angelman SyndromeJ Clin Neuroscience200185421422
- University of South FloridaMinocycline in the treatment of Angelman syndromeClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2012 [updated June 27, 2013] Available from: http://clinicaltrials.gov/show/NCT01531582. NLM identifier: NCT01531582Accessed February 23, 2014
- DanBAngelman syndrome: current understanding and research prospectsEpilepsia200950112331233919874386
- JanaNRUnderstanding the pathogenesis of Angelman syndrome through animal modelsNeural Plast2012201271094322830052
- CheronGServaisLWagstaffJDanBFast cerebellar oscillation associated with ataxia in a mouse model of Angelman syndromeNeuroscience2005130363163715590147
- RidayTTDankoskiECKrouseMCPathway-specific dopaminergic deficits in a mouse model of Angelman syndromeJ Clin Invest2012122124544455423143301
- HuangHSBurnsAJNonnemanRJBehavioral deficits in an Angelman syndrome model: effects of genetic background and ageBehav Brain Res2013243799023295389
- DailyJLNashKJinwalUAdeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndromePLoS One2011612e2722122174738
- HuangHSAllenJAMabbAMTopoisomerase inhibitors unsilence the dormant allele of Ube3a in neuronsNature2011481738018518922190039
- PowellWTCoulsonRLGonzalesMLR-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensationProc Natl Acad Sci USA201311034139381394323918391
- CaoCRioult-PedottiMSMiganiPImpairment of TrkB-PSD-95 signaling in Angelman syndromePLoS Biol2013112e100147823424281