
Abstract
Necrotizing autoimmune myopathy is characterized by predominant muscle fiber necrosis and regeneration with little or no inflammation. We describe a 58-year-old woman with previous breast cancer and statin use who complained of rapidly progressive weakness of lower limbs without pain, making walking, running and climbing stairs difficult. The creatine kinase level was 2,843 U/L, and muscle biopsy showed a dystrophic pattern. The genetic test for muscular dystrophies was negative and for anti-3-hydroxy-3-methylglutaryl coenzyme A reductase was positive. Intravenous immunoglobulin was administered, which showed mild improvement. Unfortunately, she took a step and collapsed to the floor, which led to the fracture of right femur delaying her improvement. The diagnosis of necrotizing autoimmune myopathy is sometimes delayed due to the atypical pathologic findings on muscle biopsy. As the disease is a severe condition, prompt recognition can lead to a successful outcome. We advise to consider this entity as a differential diagnosis among muscular dystrophies.
Introduction
Since 2003, an international workshop exposed different points of view to improve the classification criteria of different inflammatory myopathies designed by Anthony Amato, allowing us an accurate diagnosis.
Among the different inflammatory myopathies, a special histologic pattern defined a new subgroup called immune-mediated necrotizing myopathy or necrotizing autoimmune myopathy (NAM), which is characterized by predominant muscle fiber necrosis and regeneration with little or no inflammation.Citation1,Citation2
It has been known that patients with NAM have specific autoantibodies that are associated with specific clinical phenotypes. Thus, they can be grouped into myositis-specific autoantibodies, myositis-associated autoantibodies, signal recognition particle (SRP) and, more recently, antibodies to 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR). Although there are other risk factors recognized for NAM as cancer and, rarely, the HIV infection, prompt recognition of anti-SRP and anti-HMGCR myopathy is very important as the early immuno-suppressive therapy is the treatment of choice often followed by improvement in strength.Citation3,Citation4
Case presentation
A 58-year-old woman with no previous medical history of neuromuscular disorder was referred to our outpatient clinic in August 2014, complaining of weakness of the right foot for 1 year that progressed after 3 months with weakness of the left foot without pain. The worsening was progressive, making walking, running and climbing stairs difficult within 1 year.
She had unrelated parents and a familial history that revealed no abnormalities.
A breast neoplasia (invasive ductal carcinoma) was diagnosed in January 2007. She underwent a mastectomy with lymphadenectomy followed by chemotherapy. Currently, the breast neoplasia has been controlled with no complaints.
Between 2012 and 2014, she had taken 20 mg/day of simvastatin without side effects. She discontinued the medication 3 months after the beginning of symptoms.
In July 2017, she was diagnosed with colon adenoma without surgery.
Her first assessment revealed muscle weakness, which was proximal–distal in the lower limbs, revealing a distal atrophy. The muscle strength was 4-out of 5 (Medical Researcher Council) in the quadriceps and hamstring muscles and 4 in the gastrocnemius and soleus muscles. The tibialis anterior muscle strength was 5. The upper limbs showed a normal strength of proximal and distal muscles. No facial weakness was detected. Her deep tendon reflexes were preserved. No sensitive abnormalities were present. No retractions were found. No cardiac involvement or respiratory dysfunction was observed.
Serologic tests for autoimmune disorders were all normal. Creatine kinase (CK) was 2,843 U/L (26–140) and aldolase was equivalent to 25.9 U/L (<7.6). Other laboratory findings were within normal limits. The cerebrospinal fluid showed no neoplastic cells.
Conduction studies were normal and needle electromyography showed a myogenic pattern with positive waves.
Magnetic resonance imaging of the lower limbs revealed diffuse edema of the right semimembranosus and left semimembranosus and femoral biceps. There was discreet fatty infiltration of the soleus muscle and the medial and lateral heads of both gastrocnemii ().
Figure 1 Axial muscle MRI of thighs and legs and muscle biopsy findings (gastrocnemius).
Abbreviations: MHC, major histocompatibility complex; MRI, magnetic resonance imaging.
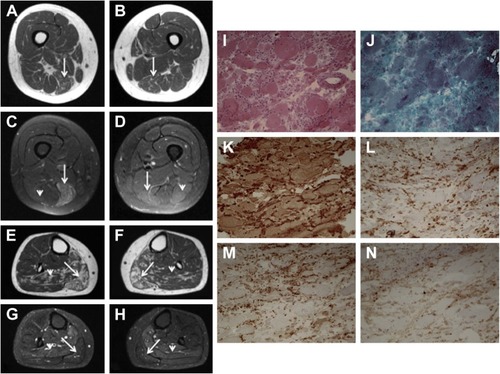
A muscle biopsy of the gastrocnemius was performed, which revealed myopathic changes, including marked variation in fiber size, connective tissue proliferation and necrosis, suggesting a dystrophic pattern. Immunohistochemistry studies, including dysferlin, dystrophin, sarcoglycans, merosin, caveolin-3 and collagen VI, showed normal immunoexpression. Diffuse major histocompatibility complex (MHC) class I overexpression was observed. CD4, CD8 and CD68 were also overexpressed ().
Based on these findings, several molecular analyses were performed, including a genetic panel of NGS containing ANO5, DYSF, GAA, SGCB, SGCG, SGCA, SGCD, CAPN3, FKRP and TCAP genes. No mutations were found.
As the progression continued, it was decided to treat the patient with prednisolone 1 g/day for 5 days, which showed clinical improvement.
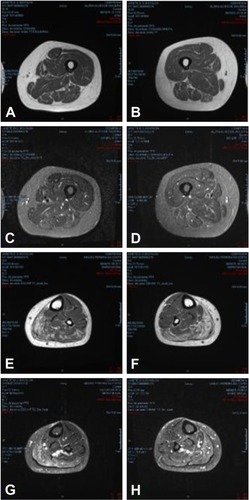
After 1 year, a new muscle magnetic resonance imaging was done to assess the progression of the weakness, which revealed progressive worsening characterized by muscle atrophy and fatty infiltration of the bilateral semimembranosus and fatty infiltration and edema of the gastrocnemius and soleus muscles ().
Figure 2 Axial muscle MRI of thighs and legs.
Abbreviation: MRI, magnetic resonance imaging.
As she did not improve, in March 2016, we started treatment with intravenous immunoglobulin (IV Ig). As a result, the patient noticed a slight improvement in the strength of the right foot.
As all molecular results were negative, anti-SRP and anti-HMGCR antibodies were determined by enzyme-linked immune-sorbent assay (CUSABIO® kit). It was found to be negative for anti-SRP and positive for anti-HMGCR (value=7.516632, cutoff 2.283). A diagnosis of NAM was finally confirmed, but immunoglobulin therapy was stopped due to the fracture.
Unfortunately, in October 2016, she collapsed on the floor. An X-ray revealed a fracture of her right femur. She also suffered from brachial plexus palsy. She underwent surgery and was kept under complete rest for 3 months interrupting IV Ig therapy. Currently, she has started using IV Ig once a month again and is getting better every day despite the limitations resulting from the fall. Currently, she cannot lift her right arm, but is able to walk with assistance.
Ethical approval
Written informed consent has been provided by the patient to have the case details and any accompanying images published.
Discussion
We report here an adult case of anti-HMGCR antibodies-linked NAM for whom diagnosis was delayed mainly due to pathologic findings, suggesting muscular dystrophy (MD).
There are a few reports in the literature about cases of anti-HMGCR+ myopathy previously diagnosed as MD.Citation5–Citation7 shows the demographic and clinical characteristics of these patients.
Table 1 Previous cases reported with anti-HMGCR myopathy simulating dystrophic-like pattern
Some points should be highlighted in this case:
Although there were no other family members with muscle disease, the muscle biopsy findings of our patient were reminiscent of hereditary MDs, while the disease progression was faster than it usually occurs in MD.
Careful history and examination of the patient complemented by review of the muscle biopsy showing overexpression of MHC class I associated with absence of facial weakness and extraocular muscle involvement led us to consider the possibility of an autoimmune myopathy, even in the presence of a dystrophic-like pattern on muscle biopsy.
Although she was a healthy patient in the point of neurologic view, she had a history of significant comorbidities:
The patient had dyslipidemia which had been treated for 2 years with simvastatin 20 mg/day; she discontinued the medication after the onset of muscle symptoms without improvement of muscle strength.
In 2007, 6 years before the myopathy was diagnosed, she was diagnosed with breast cancer for which she underwent surgery and chemotherapy. They gave satisfactory results that are currently controlled.
We must remember that adults with proximal muscle weakness, elevated CK levels and features of myopathy on electromyography have a broad differential diagnosis that includes autoimmune myopathies, toxic myopathies (statin self-limited side effects, anti-retrovirus), paraneoplastic myopathies and MDs. Distinguishing between immune-mediated myopathies and other etiologies is crucial, because only autoimmune muscle diseases routinely respond to immuno-suppressive therapy.Citation8
According to a previous report, weakness in NAM anti-HMGCR in some cases presents an acute onset (within ≤4 weeks), and in a few cases, weakness could evolve slowly (from months to several years), occasionally mimicking MD, the most common and representative myopathy, being part of hereditary or sporadic progressive diseases and characterized by several phenotypic and genetic features.Citation2,Citation6,Citation9
In relation to autoimmune myopathies, anti-SRP-positive patients have a low incidence of pulmonary fibrosis, skin rash and arthritis as well as muscular involvement. In contrast, anti-HMGCR-positive patients usually present only muscle weakness which difficult to differentiate them from MD patients.Citation9,Citation10
Regardless of the upregulation of MHC class I and the deposition of complement on capillaries and non-necrotic muscle fibers, which may suggest the probability of an immune-mediated process, the possibility of nonimmune-mediated myopathic processes should always be considered in patients who have a necrotizing muscle biopsy.Citation10
Although anti-HMGCR myopathy is associated with statin exposure in patients aged 50 years, approximately one-fourth of anti-HMGCR-positive patients develop a similar myopathic process in the absence of statins, including those with self-limited statin intolerance.Citation5,Citation11 Our patient developed the symptoms during statin exposure and showed no improvement after statin withdrawal, maintaining high CK levels.
Clinical and epidemiologic data have confirmed the temporal relationship between the onset of myositis and malignancy, and cancer-associated myositis has been defined as a cancer occurring within 3 years of diagnosing myositis.Citation12 In addition, other studies report an increased risk of cancer up to 5 years.Citation13
In 2016, Allenbach et al examined a large cohort of patients and showed for the first time that patients with NAM and anti-HMGCR antibodies were associated with an increased risk of cancer. Malignancy occurred in 17.3% (9/52) of anti-HMGCR+ patients. The mean duration between the diagnosis of cancer and myopathy was 4.2±4.9 years. Two-thirds of the malignancies occurred within 3 years of or before the diagnosis anti-HMGCR+ myopathy. No specific type of cancer was observed.Citation14
In the same year, Kadoya et al found that the prevalence of cancer within 3 years of myopathy diagnosis (cancer association) among patients with anti-HMGCR myopathy was 36% (12/33). This occurred in most patients (92%) within 1 year of myopathy diagnosis.Citation15
On the other hand, previous large-scale studies showed prevalence rates of cancers in patients with anti-HMGCR myopathy ranging from 4% to 26% (irrespective of the temporal association between cancer detection and myopathy diagnosis).Citation15 Our patient had breast cancer 6 years before the beginning of symptoms.
Based on the findings, we considered the possibility of an autoimmune myopathy, which led us to initiate IV Ig that caused a mild improvement in muscle weakness. However, the IV Ig was interrupted because the patient had a fall that caused fracture.
The poor response to IV Ig may have different explanations:
Would the HMGCR myopathy be a paraneoplastic manifestation and, therefore, without indication of the immunoglobulin? There are some differences between cancer association and history of cancer, the former highlighting the importance of the successful treatment of malignancy improving the myopathy.
Could the poor response to the immunoglobulin treatment be caused by the fracture, making it impossible for the patient to walk?
Could the patient have a self-limited statin myopathy to NAM anti-HMGCR that typically improves by immuno-suppressive therapy?
However, many questions remain unanswered. The new case studies may clarify these.
In summary, the diagnosis of NAM is sometimes delayed due to the atypical pathologic findings on muscle biopsy. As the disease is a severe condition, prompt recognition can lead to a successful outcome.
We advise to consider this entity as a differential diagnosis among MDs.
Acknowledgments
The authors gratefully acknowledge the contributions from the staff of Clinical Analysis Laboratory, Faculty of Medicine of ABC. The authors also thank Dr L Avaizoglou for the comments regarding MRI.
Disclosure
The authors report no conflicts of interest in this work.
References
- HoogendijkJEAmatoAALeckyBR119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis. 10–12 October 2003, Naarden, The NetherlandsNeuromuscul Disord200414533734515099594
- AllenbachYDrouotLRigoletAFrench Myositis NetworkAnti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statinMedicine (Baltimore)201493315015724797170
- MammenALNecrotizing myopathiesCurr Opin Rheumatol201426667968325203117
- MussetLMiyaraMBenvenisteOAnalysis of autoantibodies to 3-hydroxy-3-methylglutaryl-coenzyme A reductase using different technologiesJ Immunol Res2014201440595624741598
- MohasselPFoleyARDonkervoortSAnti-3-hydroxy-3-methylglutaryl-coenzyme a reductase necrotizing myopathy masquerading as a muscular dystrophy in a childMuscle Nerve20175661177118128066895
- TardCTiffreauVJailletteEAnti-HMGCR antibody–related necrotizing autoimmune myopathy mimicking muscular dystrophyNeuropediatrics201748647347628778101
- LiangWCUruhaASuzukiSPediatric necrotizing myopathy associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodiesRheumatology (Oxford)201756228729327818386
- MammenAStatin-associated autoimmune myopathyN Engl J Med2016374766466926886523
- SuzukiSHayashiYKuwanaMTsuburayaRSuzukiNNishinoIMyopathy associated with antibodies to signal recognition particleArch Neurol201269672873222332183
- AllenbachYBenvenisteOAcquired necrotizing myopathiesCurr Opin Neurol201326555456023995277
- MammenALChungTChristopher-StineLAutoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathyArthritis Rheum201163371372121360500
- Landon-CardinalOAllenbachYBenvenisteOAnti-HMGCR necrotizing autoimmune myopathy leading to identification of cancer relapseClin Med Rev Case Rep201744164
- TroyanovYTargoffINTremblayJLGouletJRRaymondYSenécalJLNovel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patientsMedicine (Baltimore)200584423124916010208
- AllenbachYKeraenJBouvierAHigh risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibodyBrain2016139Pt 82131213527086869
- KadoyaMHidaAMaedaMHCancer association as a risk factor for anti-HMGCR antibody-positive myopathyNeurol Neuroimmunol Neuroinflamm201636e29027761483