Abstract
Background
Primary hyperoxaluria type 1 (PH1) is characterized by progressive renal insufficiency culminating in end-stage renal disease, and a wide range of clinical features related to systemic oxalosis in different organs. It is caused by autosomal recessive deficiency of alanine:glyoxylate aminotransferase due to a defect in AGXT gene.
Case report
Two brothers (one 6 months old; the other 2 years old) presented with acute renal failure and urinary tract infection respectively. PH1 was confirmed by high urinary oxalate level, demonstration of oxalate crystals in bone biopsy, and pathogenic homozygous known AGXT gene mutation. Despite the same genetic background, same sex, and shared environment, the outcome of the two siblings differs widely. While one of them died earlier with end-stage renal disease and multiorgan failure caused by systemic oxalosis, the older brother is pyridoxine responsive with normal development and renal function.
Conclusion
Clinicians should be aware of extreme intrafamilial variability of PH1 and international registries are needed to characterize the genotype-phenotype correlation in such disorder.
Introduction
Primary hyperoxaluria type 1 (MIM#259900) (PH1) is an autosomal recessive disorder due to deficiency of alanine:glyoxylate aminotransferase (AGT), which catalyzes the conversion of glyoxylate to glycine. This results in an increase of the glyoxylate pool, which is converted to oxalate. This forms insoluble calcium salts that accumulate in kidneys and target organs including bone marrow, retina, oral cavity, peripheral nerves, vascular system, thyroid, heart, and many more. While the age of onset can vary, the classical presentation starts at childhood with nephrolithiasis or nephrocalcinosis, impairment in renal function, and progression to end-stage renal disease.Citation1–Citation4 Diagnosis relies on measurement of 24 hours of urine collection of oxalate, plasma oxalate level, enzymatic assay of AGT catalytic activity from a liver biopsy, and DNA molecular testing of the AGXT gene, which can detect 50%–70% of mutations. Treatment includes: maintenance of high fluid intake; pyridoxine supplements for those who are pyridoxine responsive; and use of potassium or sodium citrate or neutral orthophosphate and magnesium oxide to minimize stone formation.Citation2–Citation5 Little is known about the intrafamilial clinical heterogeneity in PH1. We describe two brothers with different outcomes. Despite having the same AGXT mutation, one brother died in early infancy, while the older brother, who has a milder phenotype with normal development, responded to pyridoxine administration.
Clinical report
Patient 1
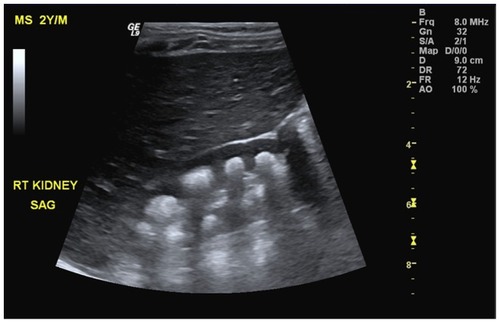
Patient 1 was the product of a full-term normal spontaneous vaginal delivery born to first-cousin Saudi parents with appropriate growth parameters and no significant antenatal history. He was apparently well until 6 months of age, at which point he demonstrated poor feeding and decreased activity for 10 days. After gradual deterioration, he presented to the emergency department with shortness of breath and anuria. Physical examination showed a nondysmorphic infant with appropriate growth parameters including weight: 8.7 kg (10–25th percentile), height: 64.5 cm (5th percentile), and head circumference: 42 cm (5th percentile). He was pale, irritable, hypertensive, and edematous. Lab investigations were consistent with acute renal failure, with urea > 44 mmol/L (1.1–8 mmol/L), creatinine = 1614 mmol/L (18–35 umol/L), K = 5.6 mmol/L (4.1–5.3 mmol/L), and bicarbonate 5 mmol/L (20–28 mmol/L). Therefore, the patient was admitted to the pediatric intensive care unit (PICU) and started on renal replacement therapy. However, the patient did not improve despite continuous dialysis and other supportive therapy. Renal ultrasound and computed tomography scan showed bilateral severe nephrocalcinosis with mild to moderate hydronephrosis (). Serum lactate was high approaching 20 mmol/L (0.5–2.2 mmol/L). Bone biopsy showed deposits of crystalline material with features compatible with oxalate crystals. Urine collection for oxalate was not performed because the patient was anuric. Despite full support in PICU, he did not improve. He developed multiorgan failure due to systemic oxalosis and subsequently died at 7 months of age.
Figure 1 Sagittal ultrasound of the right kidney revealing presence of multiple crescent-shaped hyperechoic foci involving the renal pyramids consistent with renal medullary nephrocalcinosis.
Patient 2
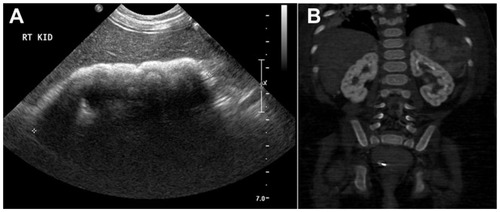
Patient 2 was the product of a full-term normal spontaneous vaginal delivery with appropriate growth parameters and no significant antenatal history. He was apparently well until 2 years of age when he was admitted with symptoms of a urinary tract infection. Physical examination showed a weight of 13 kg (50–75th percentile), height of 85 cm (50th percentile), a macrocephalic child with head circumference of 52 cm (>97th percentile) (of note, his father’s head circumference was 61 cm [>97th percentile]). At that time, his brother was admitted to PICU. Therefore, serial 24 hours of urine collection of oxalate was collected, which confirmed hyperoxaluria = 1.14 mmol/L (0.14–0.42 mmol/L). Renal ultrasound showed bilateral medullary nephrocalcinosis (). Urine aminoacids, calcium/creatinine ratio, renal function, electrolytes, and liver enzymes were all within the normal range. Bone X-ray showed no osteopenia or signs of rickets. Thyroid function test was normal and echocardiogram showed normal cardiac function.
Figure 2 Radiological findings in patient 2.
Patient 2 started on high fluid intake, polycitra, and pyridoxine 6 mg/kg/day and showed more than a 50% reduction in urinary oxalate within 4 weeks, which was 0.58 mmol/L normalized after 2 months to 0.32 mmol/L, and he continued to have normal growth and normal urinary oxalate. Currently, at the age of 3 years he shows appropriate growth parameters and normal urinary oxalate at the same dose of pyridoxine.
After obtaining informed consent from the parents, the AGXT gene was analyzed by polymerase chain reaction and sequencing of both the DNA strand of the entire coding region and the highly conserved exon-intron splice junctions which revealed homozygous missense mutation in exon 2 of AGXT gene, c.187G > C (p.Gly63Arg).Citation6 The parents were tested and found to be carriers for this mutation.
Discussion
The clinical outcome of PH1 is variable, ranging from early death in infancy to the late-onset form with occasional stones diagnosed in adulthood.Citation7 In this report we demonstrate the intrafamilial variability of the clinical outcomes of two brothers with PH1. Although they shared the same environment, sex, and AGXT mutations, they differed in their degrees of oxalosis. The younger brother died at 7 months despite extensive support after multiorgan failure preceded by acute renal failure, while the older brother is pyridoxine-respondent with normal renal function.
There are few reports in the literature describing different outcomes in siblings with PH1. Frishberg et al described a family who demonstrated the extreme of intrafamilial phenotypic variability.Citation8 One baby presented in utero with massive nephrocalcinosis and died at the age of 3 days. Four siblings diagnosed between 2 and 7 months of age developed end-stage renal disease associated with severe oxalosis. All died before the age of 3 years, while one older brother was asymptomatic at the age of 20 years. In another report, two adult males were affected but their two younger adult sisters, who had the same mutation, remained free of symptoms.Citation9 Additionally, 12 adults with the same mutation (p.Ile244Thr) showed different outcomes and clinical phenotypes.Citation10
Suggested reasons for this heterogeneity include differences in the activity level of other enzymes essential in oxalate synthesis, modifier genes, the amount of oxalate precursors in the diet, renal oxalate handling, absorption of dietary oxalate, hydration status, infections, and urinary crystallization factors.Citation11 The causes for a poorer outcome in an infant with the same mutation as an older sibling have not been established. However, it may be related to a low glomerular filtration rate in infants, which predisposes them to earlier oxalate deposition. Another reason could be related to normal variation in the number of nephrons present at birth.Citation12
Interestingly, the surviving child in this report is pyridoxine- respondent. This is confirmed by the reduction of the oxalate level by more than 50% within 4 weeks after starting the treatment with pyridoxine. Pyridoxine is an important precursor of the AGT cofactor, which is pyridoxal 5′-phosphate. Although the exact mechanism is not clear, it has been postulated that pyridoxine may increase the proportion of AGT that has a bound co-factor.Citation11 There are several reports that have shown pyridoxine administration reduces oxalate excretion and oxalate level in 25%–50% of cases.Citation13,Citation14 Many studies suggest that the most common AGXT mutation, c.508G > A (p.Gly170Arg), is associated with pyridoxine responsiveness.Citation4,Citation11
Another missense mutation reported to be associated with pyridoxine responsiveness is c.454T > A (p.Phe152Ile).Citation15 The pyridoxine responsive child in this report has a c.187G > C (p.Gly63Arg) mutation, which was not reported previously as pyridoxine-responsive and it is extremely difficult from our report to conclude the association of such a mutation with pyridoxine responsiveness. This demonstrates the need for more studies with larger sample sizes focusing on genotype–phenotype correlation in PH1.
Conclusion
In summary, our report highlights the variability of the natural history of the PH1 even within the same family and with the same sex and genotype. International registries to accommodate more cases of PH1 will lead to a better understanding of the natural history and will facilitate well-designed treatment and outcome studies.
Disclosure
The authors report no conflicts of interest in this work.
References
- XuYXiaoYJZhuKUnfolding the pathophysiological role of bioactive lysophospholipidsCurr Drug Targets Immune Endocr Metabol Disord200331233212570723
- DanpureCJPrimary hyperoxaluriaScriverCRBeaudetALSlyWSValleDThe Metabolic and Molecular Bases of Inherited DiseaseNew YorkMcGraw-Hill2001
- DanpureCJJenningsPRFryerPPurduePEAllsopJPrimary hyperoxaluria type 1: genotypic and phenotypic heterogeneityJ Inherit Metab Dis19941744874997967498
- Coulter-MackieMBWhiteCTHurleyRMChewBHLangeDPrimary Hyperoxaluria Type 1PagonRABirdTDDolanCRStephensKAdamMPGeneReviews™SeattleUniversity of Washington1993
- Coulter-MackieMBRumsbyGGenetic heterogeneity in primary hyperoxaluria type 1: impact on diagnosisMol Genet Metab2004831–2384615464418
- WilliamsELAcquavivaCAmorosoAPrimary hyperoxaluria type 1: update and additional mutation analysis of the AGXT geneHum Mutat200930691091719479957
- CochatPFargueSHarambatJPrimary hyperoxaluria type 1: strategy for organ transplantationCurr Opin Organ Transplant201015559059320733487
- FrishbergYRinatCShalataAIntra-familial clinical heterogeneity: absence of genotype-phenotype correlation in primary hyperoxaluria type 1 in IsraelAm J Nephrol200525326927515961946
- MandrileGRobbianoAGiachinoDFPrimary hyperoxaluria: report of an Italian family with clear sex conditioned penetranceUrol Res200836630931218985333
- LorenzoVAlvarezATorresATorregrosaVHernándezDSalidoEPresentation and role of transplantation in adult patients with type 1 primary hyperoxaluria and the I244T AGXT mutation: single-center experienceKidney Int20067061115111916912707
- DanpureCPrimary hyperoxaluriaScriverCRBeaudetALSlyWSValleDThe Metabolic and Molecular Bases of Inherited Disease28th edNew YorkMcGraw-Hill2001
- LuyckxVABrennerBMLow birth weight, nephron number, and kidney diseaseKidney Int Suppl200597S68S7716014104
- ToussaintCPyridoxine-responsive PH1: treatmentJ Nephrol199811Suppl 149509604811
- MarangellaMTransplantation strategies in type 1 primary hyperoxaluria: the issue of pyridoxine responsivenessNephrol Dial Transplant199914230130310069180
- MonicoCGOlsonJBMillinerDSImplications of genotype and enzyme phenotype in pyridoxine response of patients with type I primary hyperoxaluriaAm J Nephrol200525218318815849466