Abstract
Cardiovascular (CV) disease is a major factor in mortality rates around the world and contributes to more than one-third of deaths in the US. The underlying cause of CV disease is atherosclerosis, a chronic inflammatory process that is clinically manifested as coronary artery disease, carotid artery disease, or peripheral artery disease. It has been predicted that atherosclerosis will be the primary cause of death in the world by 2020. Consequently, developing a treatment regimen that can slow or even reverse the atherosclerotic process is imperative. Atherogenesis is initiated by endothelial injury due to oxidative stress associated with CV risk factors including diabetes mellitus, hypertension, cigarette smoking, dyslipidemia, obesity, and metabolic syndrome. Since the renin–angiotensin–aldosterone system (RAAS) plays a key role in vascular inflammatory responses, hypertension treatment with RAAS-blocking agents (angiotensin-converting enzyme inhibitors [ACEIs] and angiotensin II receptor blockers [ARBs]) may slow inflammatory processes and disease progression. Reduced nitric oxide (NO) bioavailability has an important role in the process of endothelial dysfunction and hypertension. Therefore, agents that increase NO and decrease oxidative stress, such as ARBs and ACEIs, may interfere with atherosclerosis. Studies show that angiotensin II type 1 receptor antagonism with an ARB improves endothelial function and reduces atherogenesis. In patients with hypertension, the ARB olmesartan medoxomil provides effective blood pressure lowering, with inflammatory marker studies demonstrating significant RAAS suppression. Several prospective, randomized studies show vascular benefits with olmesartan medoxomil: reduced progression of coronary atherosclerosis in patients with stable angina pectoris (OLIVUS); decreased vascular inflammatory markers in patients with hypertension and micro- (pre-clinical) inflammation (EUTOPIA); improved common carotid intima-media thickness and plaque volume in patients with diagnosed atherosclerosis (MORE); and resistance vessel remodeling in patients with stage 1 hypertension (VIOS). Although CV outcomes were not assessed in these studies, the observed benefits in surrogate endpoints of disease suggest that RAAS suppression with olmesartan medoxomil may potentially have beneficial effects on CV outcomes in these patient populations.
Introduction
More than 83 million adults in the US (over one-third) have cardiovascular disease (CVD).Citation1 In 2007, mortality data showed CVD to be the underlying cause of death in 33.6% of all deaths in the US.Citation1 Atherosclerosis, a progressive chronic inflammatory condition occurring in the walls of arteries,Citation2 is the leading cause of CVD in the developed world, and atherosclerosis is predicted to be the primary cause of death worldwide by 2020.Citation3 It is difficult to assess the exact frequency and prevalence of atherosclerosis. Typically, it is an asymptomatic condition that can begin as early as childhood, whereas symptomatic organ-specific clinical manifestations often do not appear until 40 years of age or older when it is most commonly diagnosed.Citation4
Atherosclerosis is characterized by the formation of arterial lesions or plaques as a result of an inflammatory response to endothelial injury.Citation5 The plaque primarily comprises macrophages, lipid-dense macrophages (foam cells), low-density lipoproteins, and neutral lipids, with subsequent calcification and ulceration appearing around the outer base of more mature plaques.Citation6,Citation7 Atherosclerosis eventually leads to artery enlargement, arterial stenosis (resulting in insufficient blood supply to the associated organ), and may ultimately produce an arterial rupture.Citation7
Atherosclerosis can develop in any artery and, depending on the specific artery affected, can potentially develop into three main types of disease – coronary, carotid, or peripheral artery disease (PAD).Citation8 In coronary artery disease (CAD), formation of an atherosclerotic plaque in a coronary artery may result in a myocardial infarction, whereas in carotid artery disease, formation of a plaque may lead to a stroke.Citation8 Plaque formation in the arteries of the legs, arms, and/or pelvis leads to PAD, characterized by numbness, pain, and increased risk of infection and necrosis in the affected limb or region.Citation8
Endothelial dysfunction is observed in the early stages of the atherogenic process, and it is initiated by injury to the arterial endothelium. Such injury has been associated with cardiovascular (CV) risk factors including diabetes mellitus or impaired glucose metabolism, hypertension, cigarette smoking, dyslipidemia, obesity, and/or metabolic syndrome.Citation9
Hypertension is an established risk factor for the development of atherosclerosis.Citation10 Evidence suggests that hypertension both promotes and accelerates the atherosclerotic process via inflammatory mechanisms linked to activation of oxidative stress by angiotensin II (Ang II), which subsequently leads to endothelial dysfunction and development of atherogenic lesions and plaque.Citation5
The purpose of this literature-based review is to present a current understanding of the mechanisms of the atherosclerotic process, including the roles of the renin–angiotensin–aldosterone system (RAAS) and nitric oxide (NO). In addition, a summary of the efficacy of several antihypertensive drug classes in improving CV outcomes, endothelial function, and reducing the progression of atherosclerosis is presented. Finally, recent clinical trial data for the angiotensin II receptor blocker (ARB) olmesartan medoxomil, which focuses on the utility of RAAS suppression in reducing atherosclerosis and/or improving endothelial function, is discussed.
Mechanism of plaque formation in atherosclerosis
The endothelium is responsible for the release and regulation of numerous vasoactive factors. In a disease-free state, the endothelium maintains these factors in homeostasis, resulting in normal control of vascular tone and function, as well as protecting against pro-atherogenic processes such as oxidation, monocyte adhesion, and the accumulation of lipids.Citation11–Citation13 However, initial injury to the endothelium, resulting from any of the abovementioned CV risk factors, alters the endothelial cell surface so that it becomes increasingly permeable and adhesive over time. Consequently, normal endothelial function becomes compromised and a series of cellular events are initiated, which ultimately results in the development of an atherosclerotic plaque.Citation11
The increased permeability of arterial endothelial cells allows migration of low-density lipoprotein cholesterol (LDL-C) into the intima where it undergoes free radical oxidation. The presence of oxidized LDL-C then initiates an inflammatory response that includes the increased expression of circulating adhesion molecules including vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), endothelial-leukocyte adhesion molecule-1 (E-selectin), and P-selectin.Citation14 The presence of adhesion molecules and subsequent release of chemokines by macrophages, vascular smooth muscle cells (VSMCs), and endothelial cells results in the migration of peripheral leukocytes to the vascular wall.
Monocytes adhere to the endothelium via the interaction of adhesion molecules and infiltrate into the intima where they differentiate into macrophages, which is facilitated by proteins such as macrophage colony-stimulating factor.Citation11 These macrophages then proceed to ingest the oxidized lipids via their scavenger receptors, thereby accumulating LDL-derived cholesterol esters. This process results in the formation of lipid-dense macrophage foam cells.Citation11 Interactions between foam cells and T-helper 1 and 2 (Th1 and Th2) cells cause the release of various inflammatory molecules and cytokines, including interferon-gamma (IFN-γ), CD40 ligand, and interleukins (IL).Citation15 Tumor necrosis factor (TNF)-α, IFN-γ, and other cytokines then activate vascular endothelial cells, macrophages, and VSMCs.Citation15 VSMCs proliferate and migrate from the media to the intima and accumulate along with the macrophages. Once there, interactions between T-helper cells, macrophages, and VSMCs stimulate secretion of additional inflammatory mediators, resulting in a chronic inflammatory response.
Advanced atherosclerotic lesions have a lipid-rich necrotic core due to the death of macrophages and VSMCs, and the continued release of lipids and other cellular components.Citation16 Subsequent deposition of an interstitial fibrous matrix, composed of fibrin, collagen, and proteoglycans, results in the formation of a fibrous cap enclosing and stabilizing the underlying plaque.Citation17
Outcomes and potential consequences of plaque formation
Following formation of a stable plaque, plaque destabilization can occur in the presence of one or more CV risk factors, particularly dyslipidemia. The vulnerable plaque can then rupture, resulting in platelet aggregation and thrombosis.Citation6,Citation16 The mechanisms of plaque destabilization are complex.Citation18 Initially, macrophages release proinflammatory cytokines that precipitate changes in the plaque surface and create a prothrombotic state.Citation19 Levels of protein-S and tissue plasminogen are reduced, and the release of metalloproteinases from macrophages and T cells, together with a reduction in collagen synthesis by VSMCs, results in degradation of the plaque’s elastin and collagen fibrous cap.Citation20 Simultaneous neovascularization occurs, resulting in further destabilization of the plaque and, ultimately, rupture.Citation6 The main determinants of plaque vulnerability and rupture are related to their composition rather than size, and include lipid content and macrophage-related inflammation, as well as impaired healing and repair mechanisms.Citation16
Following rupture, activation of the coagulation cascade, fibrin deposition, and platelet activation lead to the formation of a localized thrombus, which can cause obstruction of blood flow in the affected artery (arterial occlusion) and tissue ischemia.Citation19 Plaque rupture can be fatal. If the plaque ruptures suddenly and is in a coronary artery, this can result in acute coronary syndrome (unstable angina, acute myocardial infarction, and/or sudden death).Citation21 Alternatively, plaque rupture in a carotid artery leads to symptomatic carotid artery disease and increased risk of ischemic stroke.Citation4
The endothelium possesses an endogenous regenerative capacity mediated primarily by bone marrow-derived endothelial progenitor cells. This repair process is ongoing and takes place after endothelial injury, both during plaque formation and after plaque rupture. However, the ability to undertake such repair and regeneration of damaged endothelium is adversely affected by CV risk factors, as shown by a marked reduction in the level of endothelial progenitor cells in high-risk patients.Citation22
Role of the renin–angiotensin–aldosterone system in inflammation and atherosclerosis
In the nonpathological state, hemodynamic stability is preserved by the RAAS via the regulation of fluid balance and sodium levels, as well as direct and indirect vascular effects.Citation15 However, excessive RAAS activity is associated with atherogenesis via its role in the initiation and maintenance of the vascular inflammatory response, and is implicated in the development of atherosclerosis and vascular events.Citation15 Binding of Ang II, the major effector peptide of the RAAS, to the angiotensin II type 1 (AT1) receptor produces a range of effects including vasoconstriction and both sodium and fluid retention. In addition, Ang II mediates both mitogenic and proliferative effects on vascular endothelial and smooth muscle cells.Citation23
There is substantial evidence supporting the hypothesis that Ang II plays a significant role in the initiation and progression of atherogenesis.Citation24 For example, the recruitment of inflammatory cells to injured arteries by Ang II is one of the initial events in atherogenesis. Inflammatory cells at the site of injury then subsequently produce Ang II, resulting in a positive feedback loop that perpetuates a proinflammatory cycle.Citation15 In addition, Ang II-related mechanisms have been implicated in arterial fibrosis,Citation25 endothelial dysfunction,Citation26 oxidative stress,Citation24 and plaque instability.Citation27
Role of nitric oxide in atherosclerosis
The endothelium is also responsible for the release and regulation of numerous vasoactive substances, including NO. NO mediates the relaxation and vasodilation of VSMCs; prevents leukocyte adhesion and migration into the arterial wall; inhibits platelet activation, adhesion, and aggregation; and inhibits VSMC proliferation.Citation9,Citation29
Impaired NO bioavailability has been shown to play an important role in the process of endothelial dysfunctionCitation9 and subsequent atherogenesis.Citation29 Oxidative stress generated by CV risk factors is a major cause of NO inactivity,Citation30 creating an imbalance that results in increased levels of superoxide anions (O2−). All CV risk factors increase levels of reactive oxygen species (ROS; or oxygen-derived free radicals). For example, in patients with hypertension, the main source of oxygen-derived free radicals is the cyclooxygenase (prostanoid) pathway.Citation31,Citation32 Subsequent increased reactions between NO and superoxide anions create the highly reactive oxidant peroxynitrite (ONOO−).Citation33 Loss of NO bioavailability is associated with enhanced levels of endothelin-1 and Ang II. These factors contribute to reduced vasodilation, increased adhesion of platelets and leukocytes at the site of endothelial injury, VSMC migration and proliferation, and deposition of lipids into the intima.Citation12,Citation13,Citation29,Citation34
Endothelial dysfunction itself has been shown to be directly associated with hypertension, and is thought to contribute to functional abnormalities in resistance vessels (eg, impaired endothelial-dependent vasodilation) observed in patients with hypertension.Citation35,Citation36 Not only is free radical-related reduction in NO bioavailability a major cause of endothelial dysfunction in patients with hypertension, but it is thought that oxidative stress could be a common mechanism of endothelial dysfunction associated with other CV risk factors.Citation31
Restoration of vascular function with antihypertensive agents: clinical data and mechanism of action studies
Antihypertensive agents that increase NO, decrease oxidative stress, and/or reduce RAAS-associated inflammation have been shown to slow the progression of the atherogenic process.Citation37,Citation38
ARBs and angiotensin-converting enzyme inhibitors (ACEIs) are agents that directly affect the RAAS, either by blocking the binding of Ang II to the AT1 receptor or decreasing the production of Ang II, respectively, and therefore may be beneficial in treating patients with atherosclerosis.Citation37 These RAAS antagonists, as well as some dihydropyridine calcium channel blockers (CCBs), possess ancillary and synergistic effects that increase NO bioavailability, reduce oxidative stress, and suppress inflammatory responses, thereby improving both endothelial activity and vascular function.Citation39
ACEIs and CCBs: effect on CV outcomes in high-risk patients
Several large, long-term clinical studies have investigated the effect of ACEIs and CCBs on CV outcomes in high-risk patients.Citation36 The HOPE (Heart Outcomes Prevention Evaluation) study was a 2 × 2 factorial trial undertaken in 9297 patients aged ≥ 55 years at high risk of adverse CV outcomes (existing vascular disease or diabetes plus one other CV risk factor, but no reduction in ejection fraction or heart failure was required). HOPE compared the ACEI ramipril 10 mg once daily vs placebo over 5 years for effect on the primary study outcome – composite of myocardial infarction, stroke, or death from CV causes.Citation40 The HOPE findings demonstrated that ramipril significantly improved the rate of the primary composite endpoint vs placebo (14.0% vs 17.8%; P < 0.001). Ramipril also improved rates of individual endpoints significantly more than placebo (all P ≤ 0.005). These effects were thought to be mainly independent of blood pressure (BP) reduction since the majority of patients did not have hypertension at baseline, and the mean BP reduction was very low.Citation40
The effect of amlodipine on the progression of atherosclerosis and the occurrence of clinical CV events was determined in 825 patients with angiographically documented CAD in the PREVENT (Prospective Randomized Evaluation of the Vascular Effects of Norvasc Trial) study.Citation41 In PREVENT, amlodipine significantly slowed the 36-month progression of atherosclerosis in carotid arteries as assessed by B-mode ultrasonography; intima-media thickness (IMT) decreased by 0.0126 mm in the amlodipine-treated patients compared with an increase of 0.033 mm in the placebo group (P = 0.007 vs placebo).Citation41
The CAMELOT (Comparison of Amlodipine vs Enalapril to Limit Occurrences of Thrombosis) study showed that in patients with normal BP (mean baseline BP = 129/78 mmHg) and documented CAD, amlodipine therapy resulted in a significant reduction in adverse CV events vs placebo (P = 0.003), and a trend toward reduced progression of atherosclerosis vs placebo (P = 0.12). In a subgroup of patients with baseline systolic BP greater than the mean, the rate of atherosclerosis progression was significantly lower with amlodipine compared with placebo (P = 0.02). A trend towards a correlation between BP reduction and progression of atherosclerosis was observed (P = 0.07).Citation42
SECURE (Study to Evaluate Carotid Ultrasound Changes in Patients Treated With Ramipril and Vitamin E) was a randomized, double-blind substudy of the HOPE trial in 732 patients aged ≥ 55 years with vascular disease or diabetes, and at least one other CV risk factor. In addition, patients could not have heart failure or a low left ventricular ejection fraction. SECURE demonstrated that the rate of progression of the mean maximum carotid artery IMT was significantly lower in the ramipril 10 mg once-daily treatment group vs placebo (P = 0.028) over an average follow-up period of 4.5 years. The difference in mean IMT progression between ramipril and placebo remained significant (P < 0.05) after adjustment for BP changes and after multivariate adjustment.Citation43
Mechanisms of action
The mechanisms of action behind the improvement in CV outcomes reported with ACEIs and CCBs are not fully understood. ACEIs are known to interfere with the breakdown of bradykinin, which stimulates NO release.Citation9 ACEIs also inhibit the production of endothelin-1 and Ang II by the endothelium, which reduces the production of superoxide anion, further increasing the levels of available NO.Citation44 Mechanistic studies have shown that in patients with hypertension, vessel wall elasticity is improved and arterial stiffness reduced by calcium channel or RAAS blockade,Citation39 as shown by a significant reduction in the central aortic augmentation index.Citation45,Citation46 Long-acting CCBs, eg, amlodipine, are also known to protect against oxidation of low-density lipoprotein (LDL) and membrane lipids via their antioxidant effects, which are independent of BP lowering.Citation47
Mechanisms and clinical effect of ARBs on endothelial function and atherosclerosis
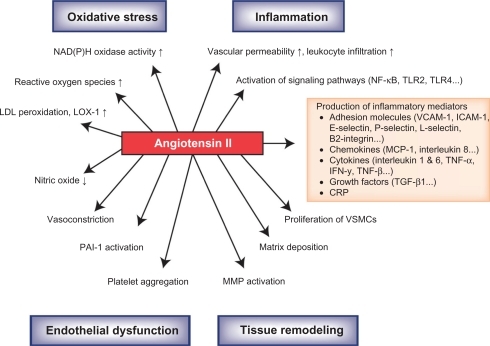
shows some of the known effects on the mechanisms of atherosclerosis resulting from the binding of Ang II to the AT1 receptor.Citation48 Clinical trials have demonstrated that ARBs can improve endothelial function and/or reduce markers of atherosclerosis via AT1 receptor antagonism. Candesartan improves endothelial function as evidenced by significantly increasing flow-mediated dilation in patients with hypertension, stable CAD, and endothelial dysfunction.Citation49 Candesartan significantly improves the percent flow-mediated dilator response to hyperemia (P = 0.019); significantly decreases plasma levels of plasminogen activator inhibitor type-1 (PAI-1) antigen (P < 0.001) as well as monocyte chemoattractant protein-1 (P = 0.004),Citation50 and significantly reduces circulating levels of ICAM-1 and VCAM-1 (P < 0.05).Citation51 Candesartan has also been shown to slow the progression of carotid remodeling in patients with hypertension and type 2 diabetes.Citation52 Experimental in vitro and in vivo studies with candesartan suggest that a possible mechanism of action for the anti-inflammatory effect seen with ARBs may include the suppression of toll-like receptor (TLR) 2 and TLR4 expression.Citation53 TLR2 and TLR4 are thought to participate in the inflammatory process of atherosclerosis.Citation2
Figure 1 Effects of angiotensin II on the mechanisms associated with the arthrosclerotic process.
Reprinted from Schmieder RE, et al. Renin-angiotensin system and cardiovascular risk. The Lancet. 2007;369:1208–1219. Copyright with permission from Elsevier.Citation48
Clinical studies have shown that in patients with hypertension, irbesartan significantly increases endothelium-dependent and endothelium-independent vasodilation while significantly decreasing plasma endothelin (P = 0.001) and restoring the vasoconstriction capacity of NOS inhibitors.Citation54,Citation55 Irbesartan has also been shown to reduce fibrinogen and thrombomodulin;Citation56 PAI-1;Citation56,Citation57 nitrotyrosine;Citation58 C-reactive protein (CRP), ICAM-1, and IL-6;Citation57,Citation58 and plasma levels of 8-isoprostane (a marker of oxidative stress).Citation57 In addition, clinical data demonstrate that irbesartan significantly increases flow-mediated vasodilation.Citation57,Citation58 Overall, blockade of the RAAS by irbesartan has been shown to improve endothelial function beyond that expected by BP lowering alone.Citation59
In patients with hypertension, losartan has been shown to prevent the production of connective tissue growth factor and transforming growth factor-β (known mediators of Ang II-induced remodeling of resistance arteries) as well as improve the media:lumen ratio in resistance arteries.Citation60 Furthermore, clinical trial data demonstrate that losartan can attenuate the oxidation of LDL (P = 0.001) in patients with type 2 diabetes,Citation61 significantly reduce 8-isoprostane (P = 0.01),Citation62 and significantly decrease common carotid artery IMT (P < 0.05).Citation63
Several valsartan studies have demonstrated RAAS suppression and an associated reduction in inflammation. Valsartan has been shown to significantly suppress the generation of ROS in polymorphonuclear and mononuclear cells of normal subjects (P < 0.01). It has also been shown to suppress the activity of NF-kB binding activity (P < 0.01), which is responsible for regulating the transcription of genes for proinflammatory cytokines, adhesion molecules, chemokines, and protein subunits of ROS-generating enzymes. Valsartan significantly increased the expression of inhibitor kB (P < 0.05), which binds to NF-kB and prevents it from translocating into the nucleus, thus preventing transcription of proinflammatory genes, and has also been shown to significantly decrease plasma CRP concentration (P < 0.01).Citation64 Furthermore, valsartan significantly reduced TNF-α (P = 0.006) and IL-6 (P = 0.005) in patients with essential hypertension.Citation65
The mechanism of the anti-inflammatory effects of telmisartan was investigated in a 12-week, double-blind, placebo-controlled study in patients with hypertension and CAD. A range of inflammatory markers were assessed, ie, high-sensitivity CRP (hsCRP), IL-6, and cell adhesion molecules, soluble ICAM-1, leukocyte adhesion molecule soluble-L-selectin, and the β2 integrin Mac-1; however, only Mac-1 expression was significantly decreased by telmisartan.Citation66 Associated in vitro studies conducted to elucidate the mode of action showed that telmisartan dose-dependently inhibited β2-integrin expression in lymphocytes in either the absence or presence of Ang II, suggesting that the atheroprotective effects associated with telmisartan are independent of the AT1 receptor.Citation66
Studies investigating the anti-atherogenic mechanism of action of olmesartan medoxomil
The ARB olmesartan medoxomil has demonstrated effective BP lowering in numerous clinical trials with a tolerability profile similar to that of placebo.Citation67 Moreover, olmesartan medoxomil has been shown to be efficacious across a wide range of patient subgroups.Citation68,Citation69 In vitro and in vivo animal studies have suggested anti-atherogenic and anti-inflammatory effects of olmesartan medoxomil. In an in vitro study investigating the effect of olmesartan on Ang II-induced migration of VSMCs from the rat aorta, data showed olmesartan to be a potent inhibitor of VSMC migration, with the inhibitory effect being mediated via Src- and mitogen-activated protein kinase pathways.Citation70
These findings suggest that olmesartan medoxomil may potentially prevent vascular remodeling associated with VSMC migration. In addition, animal studies have demonstrated the effects of olmesartan on the suppression or regression of fatty streak plaque,Citation71,Citation72 and reduction of superoxide generation and overload of oxidative stress on the aortic walls.Citation71 In vitro experiments have shown that olmesartan also suppressed levels of IFN-γ, macrophage inflammatory protein-2, and thioredoxin (a marker of oxidative stress) in cultured cells.Citation71
Clinical effect of olmesartan medoxomil on endothelial function and atherosclerosis
Study data from several recent olmesartan medoxomil clinical trials demonstrate the utility of RAAS suppression in reducing atherosclerosis and/or improving endothelial function.Citation73–Citation76 These studies are summarized in .
Table 1 Olmesartan medoxomil (OM) studies investigating the utility of renin–angiotensin–aldosterone system suppression in reducing atherosclerosis and/or improving endothelial function
OLIVUS
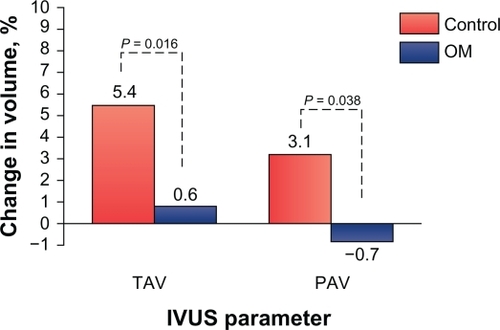
The most recent of these trials was the OLIVUS (Impact of Olmesartan on Progression of Coronary Atherosclerosis: Evaluation by Intravascular Ultrasound) study, which was a prospective, randomized trial designed to assess the effect of olmesartan medoxomil on the progression of coronary atherosclerosis.Citation73 More specifically, this study evaluated the effect of olmesartan medoxomil on coronary atherosclerotic changes assessed by volumetric intravascular ultrasound (IVUS) in 247 patients with stable angina pectoris with native CAD. Patients were randomly assigned to receive olmesartan medoxomil 10, 20, or 40 mg or control (placebo), and also received a combination of β-blockers, CCBs, diuretics, nitrates, glycemic control agents, and/or statins, according to their physician’s guidance (). Serial IVUS examinations were performed at baseline and at 14 months to assess coronary atheroma volume. IVUS was performed in nonculprit vessels (without angiographically documented coronary stenosis; <50%) when patients underwent percutaneous coronary intervention for culprit lesions.Citation73 Volumetric IVUS analyses included lumen, plaque, and vessel volume; percent atheroma volume (PAV); and total atheroma volume (TAV). After 14 months of active therapy, IVUS showed a significant decrease in TAV and a significant percentage improvement in PAV in olmesartan medoxomil recipients: 5.4% vs 0.6% for TAV and 3.1% vs −0.7% for PAV, for placebo vs olmesartan medoxomil, respectively (P < 0.05 for all vs placebo; and ).Citation73 BP control was identical between olmesartan medoxomil and placebo (control) groups. OLIVUS demonstrated that olmesartan medoxomil decreased the rate of coronary atheroma progression in patients with stable angina pectoris, independent of BP lowering.
Figure 2 OLIVUS study: change in intravascular ultrasound (IVUS) parameters from baseline to 14-month follow-up. An olmesartan medoxomil (OM)-based treatment regimen significantly decreased total atheroma volume (TAV) and percent change in percent atheroma volume (PAV) as demonstrated by IVUS in patients with stable angina pectoris and native coronary artery disease.
EUTOPIA
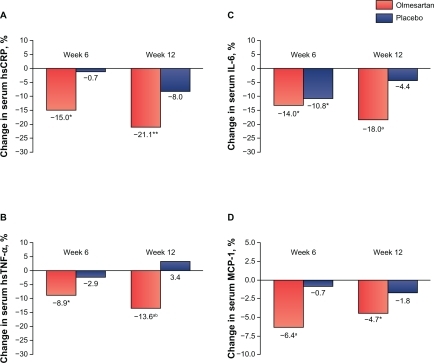
The EUTOPIA (European Trial on Olmesartan and Pravastatin in Inflammation and Atherosclerosis) study was a prospective, double-blind, multicenter trial evaluating the anti-inflammatory effects of olmesartan medoxomil, with or without pravastatin, in 211 patients with hypertension and microinflammation.Citation76 Patients were randomized to olmesartan medoxomil or placebo for 12 weeks, with pravastatin added to both treatment arms at week 6, and hydrochlorothiazide (HCTZ) added as necessary to control BP (). A panel of vascular inflammation markers was assessed at baseline, 6 weeks (primary endpoint), and 12 weeks. At 6 weeks, olmesartan medoxomil significantly reduced the levels of hsCRP, high-sensitivity TNF-α (hsTNF-α), IL-6, and monocyte chemotactic protein, whereas placebo had no effect ( and ). After 12 weeks, hsCRP, hsTNF-α, and IL-6 were further decreased in the olmesartan medoxomil plus pravastatin arm, but no significant reduction in anti-inflammatory markers was observed with pravastatin alone from weeks 6 to 12. Pravastatin monotherapy significantly reduced LDL-C in both arms.Citation76 In the EUTOPIA study, Ang II receptor blockade with olmesartan medoxomil significantly reduced vascular inflammation/inflammatory markers in patients with hypertension.
Figure 3 EUTOPIA study: changes in serum concentrations of hsCRP, hsTNF-α, IL-6, and MCP-1 in patients with essential hypertension after 6 weeks of olmesartan medoxomil therapy or placebo; *P < 0.05, **P < 0.02, aP < 0.01 vs baseline; bP < 0.05 olmesartan vs placebo. Olmesartan medoxomil significantly decreased serum levels of high-sensitivity C-reactive protein (hsCRP), high-sensitivity tumor necrosis factor- alpha (hsTNF-α), interleukin-6 (IL-6), and monocyte chemotactic protein-1 (MCP-1) from baseline to week 6. Reductions for placebo were only significant for IL-6 at week 6.
Reprinted with permission from Fliser D, et al. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation. 2004;110(9):1103–1107.Citation76
MORE
The MORE (Multicenter Olmesartan Atherosclerosis Regression Evaluation) study compared the effect of olmesartan medoxomil vs atenolol on changes in common carotid IMT and plaque volume after 2 years of therapy in 165 patients with atherosclerosis. Patients received olmesartan medoxomil 20–40 mg/day or atenolol 50–100 mg/day for 2 years. Two- and three-dimensional ultrasound was used to determine the change from baseline in common carotid IMT and plaque volume, respectively, after 28, 52, and 104 weeks of therapy ().Citation74
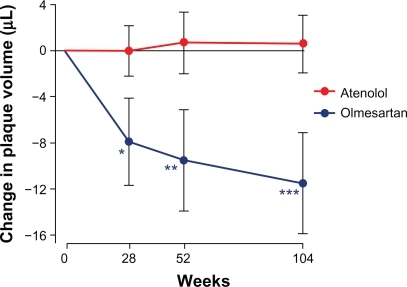
Olmesartan medoxomil and atenolol both resulted in similar and significant reductions in carotid IMT. There were no between-treatment differences in plaque volume. In a post hoc analysis in patients with a baseline plaque volume ≥ median (33.7 μL), plaque volume significantly regressed with olmesartan medoxomil but not with atenolol, even though BP reductions were comparable ( and ).Citation74 Common carotid IMT and BP decreased similarly with olmesartan medoxomil and atenolol. In addition, there was a preferential reduction in the volume of larger atherosclerotic plaques with olmesartan medoxomil compared with atenolol.
Figure 4 MORE study: post hoc analysis of mean changes in plaque volume at weeks 28, 52, and 104 of follow-up in atenolol- (n = 41) and olmesartan- (n = 36) treated patients with baseline plaque volume ≥ median (33.7 μL).
Reprinted by permission of SAGE from Stumpe KO, et al. Ther Adv Cardiovasc Dis. 2007;1(2):97–106. Copyright © by SAGE Publications.Citation74
VIOS
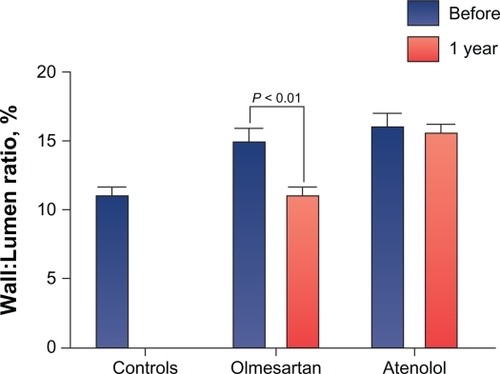
The VIOS (Vascular Improvement With Olmesartan Medoxomil Study) study investigated the impact of RAAS suppression by AT1 receptor blockade on small resistance vessel remodeling – an effect thought to provide more complete end-organ protection than BP lowering alone.Citation75 Patients with stage 1 hypertension were randomized to olmesartan medoxomil 20–40 mg/day or atenolol 50–100 mg/day plus HCTZ, amlodipine, or hydralazine, as needed, to achieve a BP goal of <140/90 mmHg (). Subcutaneous gluteal resistance arteries were examined on a pressurized myograph to evaluate remodeling at baseline and after 1 year of active treatment.Citation75
Data from VIOS showed that the wall:lumen ratio in arteries obtained from olmesartan medoxomil recipients was significantly reduced (P < 0.01) after 1 year of active therapy, whereas no significant change was seen in patients receiving atenolol ( and ).Citation77 BP was reduced to a similar level by both agents. VIOS showed that AT1 receptor blockade with olmesartan medoxomil reduced the wall:lumen ratio in resistance arteries in patients with hypertension, independently of BP reduction, and resulted in ratios similar to normotensive controls after 1 year of treatment.Citation77 In addition, the augmentation index, a surrogate measure for vascular compliance, was significantly (P < 0.05) reduced from baseline with the olmesartan medoxomil-based regimen but not with the atenolol-based regimen. This result provided noninvasive evidence that olmesartan medoxomil improved vascular compliance in association with the aforementioned improvement in resistance vessel morphology.
Figure 5 VIOS study: an olmesartan medoxomil regimen significantly reduced the wall:lumen ratio in arteries to values similar to those observed in normotensive controls at 1 year of therapy. Conversely, no significant change in wall:lumen ratio was observed in arteries from atenolol-treated patients.
Reprinted from Smith RD, et al. Reversal of vascular hypertrophy in hypertensive patients through blockage of angiotensin II receptors. J Am Soc Hypertens. 2008;2(3):165–172. Copyright with permission from Elsevier.Citation77
Summary
Atherosclerosis is a complex disease that is associated with CV events including myocardial infarction, unstable angina, and sudden cardiac death, as well as cerebrovascular events and peripheral thromboses. Endothelial dysfunction is a hallmark early event in atherogenesis. Endothelial cells produce a number of vasoactive substances responsible for maintaining vascular tone. Local impairment of the endothelium associated with CV risk factors creates an imbalance between the normal concentrations of vasodilating and vasoconstricting factors, in particular, an increase in Ang II and a decrease in NO. Impaired NO bioavailability as a consequence of oxidative stress and increased levels of Ang II results in increased vasoconstriction, and both of these processes play key roles in endothelial dysfunction and atherosclerosis. The RAAS, and its primary mediator Ang II, also have a direct influence on the progression of the atherosclerotic process via effects on endothelial function, inflammation, fibrinolytic balance, and plaque stability.
Agents in the ARB, ACEI, and CCB drug classes have demonstrated beneficial effects on the atherosclerotic process. The clinical trial data presented in this review for the ARB olmesartan medoxomil demonstrate anti-atherogenic effects in a range of patient types, as well as provide evidence of slowing atheroma progression, reducing vascular inflammation, decreasing carotid IMT and atherosclerotic plaque volume, and improving the arterial wall:lumen ratio. These data complement the results from several animal model and in vitro studies, which serve to elucidate the mechanisms of the anti-atherogenic effect. The aforementioned clinical changes observed in the olmesartan medoxomil studies may have a beneficial effect on improving endothelial dysfunction and, consequently, slowing atherogenesis. Although long-term CV outcomes were not assessed in these studies, the observed changes provide a rationale for prospective trials to ascertain the role of olmesartan medoxomil in reducing the risk or severity of CVD.
Conclusion
Certain antihypertensive agents have been shown to effectively slow the process of atherogenesis in clinical evaluations. Both ARBs and ACEIs affect the RAAS directly, either by blocking the binding of Ang II to the AT1 receptor or by decreasing production of Ang II. These RAAS antagonists, as well as certain dihydropyridine CCBs, possess both ancillary and synergistic effects that increase NO bioavailability, reduce oxidative stress, and/or suppress RAAS-associated inflammatory response, as shown by the effect of such agents on a range of inflammatory markers. These changes bring about an improvement in endothelial activity and vascular function, and may additionally be associated with benefits beyond BP lowering in the treatment of patients with atherosclerosis and hypertension.
Data from the olmesartan medoxomil clinical trials OLIVUS, EUTOPIA, VIOS, and MORE have demonstrated the specific utility of RAAS suppression in reducing atherosclerotic plaque volume, improving plaque composition and stability, and in improving endothelial dysfunction. These studies have shown that olmesartan medoxomil treatment may slow the progression of atherosclerosis, thereby potentially improving CV outcomes.
Acknowledgements
Medical writing assistance was provided by Alan J Klopp, PhD, and Mary Hines of inScience Communications, a Wolters Kluwer Business, and funded by Daiichi Sankyo, Inc.
Disclosures
Dr Mason did not receive any compensation for the preparation of this manuscript.
References
- RogerVLGoASLloyd-JonesDMHeart disease and stroke statistics – 2011 update: a report from the American Heart AssociationCirculation20111234e18e20921160056
- GalkinaELeyKImmune and inflammatory mechanisms of atherosclerosis (*)Annu Rev Immunol20092716519719302038
- ScottJThe pathogenesis of atherosclerosis and new opportunities for treatment and preventionJ Neural Transm Suppl20026311712597606
- WassermanBAClinical carotid atherosclerosisNeuroimaging Clin N Am200212340341912486829
- LiJJChenJLInflammation may be a bridge connecting hypertension and atherosclerosisMed Hypotheses200564592592915780486
- RosenfeldMEAn overview of the evolution of the atherosclerotic plaque: from fatty streak to plaque rupture and thrombosisZ Kardiol200089Suppl 72611098552
- MatsushitaMNishikimiNSakuraiTNimuraYRelationship between aortic calcification and atherosclerotic disease in patients with abdominal aortic aneurysmInt Angiol200019327627911201598
- KramerCMAndersonJDMRI of atherosclerosis: diagnosis and monitoring therapyExpert Rev Cardiovasc Ther200751698017187458
- BrunnerHCockcroftJRDeanfieldJEndothelial function and dysfunction. Part II: association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of HypertensionJ Hypertens200523223324615662207
- StandridgeJBHypertension and atherosclerosis: clinical implications from the ALLHAT trialCurr Atheroscler Rep20057213213915727729
- RossRAtherosclerosis – an inflammatory diseaseN Engl J Med199934021151269887164
- DzauVBraunwaldEResolved and unresolved issues in the prevention and treatment of coronary artery disease: a workshop consensus statementAm Heart J19911214 Pt 1124412632008853
- DeanfieldJDonaldAFerriCEndothelial function and dysfunction. Part I: methodological issues for assessment in the different vascular beds: a statement by the Working Group on Endothelin and Endothelial Factors of the European Society of HypertensionJ Hypertens200523171715643116
- HwangSJBallantyneCMSharrettARCirculating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk In Communities (ARIC) studyCirculation19979612421942259416885
- FerrarioCMStrawnWBRole of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular diseaseAm J Cardiol200698112112816784934
- GronholdtMLDalager-PedersenSFalkECoronary atherosclerosis: determinants of plaque ruptureEur Heart J199819Suppl CC24C299597422
- KoenigWKhuseyinovaNBiomarkers of atherosclerotic plaque instability and ruptureArterioscler Thromb Vasc Biol2007271152617082488
- LibbyPChanging concepts of atherogenesisJ Intern Med2000247334935810762452
- SakariassenKSBarstadRMMechanisms of thromboembolism at arterial plaquesBlood Coagul Fibrinolysis1993446156258218859
- KatsudaSKajiTAtherosclerosis and extracellular matrixJ Atheroscler Thromb200310526727414718743
- TherouxPFusterVAcute coronary syndromes: unstable angina and non-Q-wave myocardial infarctionCirculation19989712119512069537346
- BriasoulisATousoulisDAntoniadesCStefanadisCPapageorgiouNThe role of endothelial progenitor cells in vascular repair after arterial injury and atherosclerotic plaque developmentCardiovasc Ther472010 [Epub ahead of print].
- VaragicJTraskAJJessupJAChappellMCFerrarioCMNew angiotensinsJ Mol Med200886666367118437333
- WeissDSorescuDTaylorWRAngiotensin II and atherosclerosisAm J Cardiol2001878 A25C32C
- Ruiz-OrtegaMRuperezMEstebanVAngiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseasesNephrol Dial Transplant2006211162016280370
- MatsumotoKMorishitaRTomitaNImprovement of endothelial dysfunction by angiotensin II blockade accompanied by induction of vascular hepatocyte growth factor system in diabetic spontaneously hypertensive ratsHeart Vessels2003181182512644877
- da CunhaVMartin-McNultyBVinceletteJAngiotensin II induces histomorphologic features of unstable plaque in a murine model of accelerated atherosclerosisJ Vasc Surg200644236437116890870
- DavidSKumpersPLukaszAKielsteinJTHallerHFliserDCirculating angiopoietin-2 in essential hypertension: relation to atherosclerosis, vascular inflammation, and treatment with olmesartan/pravastatinJ Hypertens20092781641164719390459
- MasonRPNitric oxide mechanisms in the pathogenesis of global riskJ Clin Hypertens (Greenwich)200688 Suppl 23138 quiz 40.16894246
- SinghUJialalIOxidative stress and atherosclerosisPathophysiology200613312914216757157
- TaddeiSVirdisAGhiadoniLMagagnaASalvettiAVitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertensionCirculation19989722222222299631871
- BautistaLEInflammation, endothelial dysfunction, and the risk of high blood pressure: epidemiologic and biological evidenceJ Hum Hypertens200317422323012692566
- TouyzRMSchiffrinELReactive oxygen species in vascular biology: implications in hypertensionHistochem Cell Biol2004122433935215338229
- GibbonsGHEndothelial function as a determinant of vascular function and structure: a new therapeutic targetAm J Cardiol1997795A389127614
- PanzaJAQuyyumiAABrushJEJrEpsteinSEAbnormal endothelium-dependent vascular relaxation in patients with essential hypertensionN Engl J Med1990323122272355955
- MizunoYJacobRFMasonRPAdvances in pharmacologic modulation of nitric oxide in hypertensionCurr Cardiol Rep201012647248020809234
- FerrarioCMStrawnWBTargeting the RAAS for the treatment of atherosclerosisDrug Discov Today Ther Strateg200523221229
- HammoudRAVaccariCSNagamiaSHKhanBVRegulation of the renin-angiotensin system in coronary atherosclerosis: a review of the literatureVasc Health Risk Manag20073693794518200812
- MizunoYJacobRFMasonRPEffects of calcium channel and renin-angiotensin system blockade on intravascular and neurohormonal mechanisms of hypertensive vascular diseaseAm J Hypertens200821101076108518756260
- YusufSSleightPPogueJBoschJDaviesRDagenaisGEffects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study InvestigatorsN Engl J Med2000342314515310639539
- PittBByingtonRPFurbergCDEffect of amlodipine on the progression of atherosclerosis and the occurrence of clinical events. PREVENT InvestigatorsCirculation2000102131503151011004140
- NissenSETuzcuEMLibbyPEffect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure: the CAMELOT study: a randomized controlled trialJAMA2004292182217222515536108
- LonnEYusufSDzavikVEffects of ramipril and vitamin E on atherosclerosis: the study to evaluate carotid ultrasound changes in patients treated with ramipril and vitamin E (SECURE)Circulation2001103791992511181464
- GriendlingKKSorescuDUshio-FukaiMNAD(P)H oxidase: role in cardiovascular biology and diseaseCirc Res200086549450110720409
- ChenCHTingCTLinSJDifferent effects of fosinopril and atenolol on wave reflections in hypertensive patientsHypertension1995255103410417737712
- HirataKVlachopoulosCAdjiAO’RourkeMFBenefits from angiotensin-converting enzyme inhibitor ‘beyond blood pressure lowering’: beyond blood pressure or beyond the brachial artery?J Hypertens200523355155615716696
- MasonRPWalterMFTrumboreMWOlmsteadEGJrMasonPEMembrane antioxidant effects of the charged dihydropyridine calcium antagonist amlodipineJ Mol Cell Cardiol199931127528110072734
- SchmiederREHilgersKFSchlaichMPSchmidtBMRenin-angiotensin system and cardiovascular riskLancet200736995681208121917416265
- Perrone-FilardiPCorradoLBrevettiGEffects of AT1 receptor antagonism with candesartan on endothelial function in patients with hypertension and coronary artery diseaseJ Clin Hypertens (Greenwich)200911526026519534023
- KohKKHanSHChungWJComparison of effects of losartan, irbesartan, and candesartan on flow-mediated brachial artery dilation and on inflammatory and thrombolytic markers in patients with systemic hypertensionAm J Cardiol2004931114321435A141015165934
- RoseiEARizzoniDMuiesanMLEffects of candesartan cilexetil and enalapril on inflammatory markers of atherosclerosis in hypertensive patients with non-insulin-dependent diabetes mellitusJ Hypertens200523243544415662233
- BaguetJPAsmarRValensiPNisse-DurgeatSMallionJMEffects of candesartan cilexetil on carotid remodeling in hypertensive diabetic patients: the MITEC studyVasc Health Risk Manag20095117518319436670
- DasuMRRiosvelascoACJialalICandesartan inhibits toll-like receptor expression and activity both in vitro and in vivoAtherosclerosis20092021768318495130
- BragulatELarrousseMCocaAde la SierraAEffect of long-term irbesartan treatment on endothelium-dependent vasodilation in essential hypertensive patientsBr J Biomed Sci200360419119614725334
- von zur MuhlenBKahanTHaggAMillgardJLindLTreatment with irbesartan or atenolol improves endothelial function in essential hypertensionJ Hypertens200119101813181811593101
- MakrisTKStavroulakisGAKrespiPGFibrinolytic/hemostatic variables in arterial hypertension: response to treatment with irbesartan or atenololAm J Hypertens200013778378810933570
- SolaSMirMQCheemaFAIrbesartan and lipoic acid improve endothelial function and reduce markers of inflammation in the metabolic syndrome: results of the Irbesartan and Lipoic Acid in Endothelial Dysfunction (ISLAND) studyCirculation2005111334334815655130
- CerielloAAssaloniRDa RosREffect of atorvastatin and irbesartan, alone and in combination, on postprandial endothelial dysfunction, oxidative stress, and inflammation in type 2 diabetic patientsCirculation2005111192518252415867169
- NegroREndothelial effects of antihypertensive treatment: focus on irbesartanVasc Health Risk Manag2008418910118629353
- Gomez-GarreDMartin-VenturaJLGranadosRLosartan improves resistance artery lesions and prevents CTGF and TGF-beta production in mild hypertensive patientsKidney Int20066971237124416482098
- RachmaniRLeviZZadokBSRavidMLosartan and lercanidipine attenuate low-density lipoprotein oxidation in patients with hypertension and type 2 diabetes mellitus: a randomized, prospective crossover studyClin Pharmacol Ther200272330230712235451
- FlammerAJHermannFWiesliPEffect of losartan, compared with atenolol, on endothelial function and oxidative stress in patients with type 2 diabetes and hypertensionJ Hypertens200725478579117351370
- SonodaMAoyagiTTakenakaKUnoKNagaiRA one-year study of the antiatherosclerotic effect of the angiotensin-II receptor blocker losartan in hypertensive patients. A comparison with angiotension-converting enzyme inhibitorsInt Heart J20084919510318360068
- DandonaPKumarVAljadaAAngiotensin II receptor blocker valsartan suppresses reactive oxygen species generation in leukocytes, nuclear factor-kappa B, in mononuclear cells of normal subjects: evidence of an antiinflammatory actionJ Clin Endocrinol Metab20038894496450112970329
- ManabeSOkuraTWatanabeSFukuokaTHigakiJEffects of angiotensin II receptor blockade with valsartan on pro-inflammatory cytokines in patients with essential hypertensionJ Cardiovasc Pharmacol200546673573916306795
- LinkALenzMLegnerDBohmMNickenigGTelmisartan inhibits beta2-integrin MAC-1 expression in human T-lymphocytesJ Hypertens20062491891189816915040
- BrunnerHRClinical efficacy and tolerability of olmesartanClin Ther200426Suppl AA28A3215291377
- NeutelJMKereiakesDJAn olmesartan medoxomil-based treatment algorithm is effective in achieving 24-hour BP control in patients with type 2 diabetes mellitus, regardless of age, race, sex, or severity of hypertension: subgroup analysis of the BENIFICIARY studyAm J Cardiovasc Drugs201010528930320712386
- OparilSPimentaEEfficacy of an olmesartan medoxomil-based treatment algorithm in patients stratified by age, race, or sexJ Clin Hypertens (Greenwich)201012131320047622
- KyotaniYZhaoJTomitaSOlmesartan inhibits angiotensin II-induced migration of vascular smooth muscle cells through Src and mitogen-activated protein kinase pathwaysJ Pharmacol Sci2010113216116820508392
- ShimadaKMurayamaTYokodeMKitaTFujitaMKishimotoCOlmesartan, a novel angiotensin II type 1 receptor antagonist, reduces severity of atherosclerosis in apolipoprotein E deficient mice associated with reducing superoxide productionNutr Metab Cardiovasc Dis4142010 [Epub ahead of print].
- TakaiSJinDSakaguchiMMuramatsuMMiyazakiMThe regressive effect of an angiotensin II receptor blocker on formed fatty streaks in monkeys fed a high-cholesterol dietJ Hypertens200523101879188616148612
- HirohataAYamamotoKMiyoshiTImpact of olmesartan on progression of coronary atherosclerosis: a serial volumetric intravascular ultrasound analysis from the OLIVUS (impact of olmesarten on progression of coronary atherosclerosis: evaluation by intravascular ultrasound) trialJ Am Coll Cardiol2010551097698220202514
- StumpeKOAgabiti-RoseiEZielinskiTCarotid intima-media thickness and plaque volume changes following 2-year angiotensin II-receptor blockade. The Multicentre Olmesartan atherosclerosis Regression Evaluation (MORE) studyTher Adv Cardiovasc Dis2007129710619124398
- SmithRDYokoyamaHAverillDBThe protective effects of angiotensin II blockade with olmesartan medoxomil on resistance vessel remodeling (the VIOS study): rationale and baseline characteristicsAm J Cardiovasc Drugs20066533534217083268
- FliserDBuchholzKHallerHAntiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammationCirculation200411091103110715313950
- SmithRDYokoyamaHAverillDBSchiffrinELFerrarioCMReversal of vascular hypertrophy in hypertensive patients through blockade of angiotensin II receptorsJ Am Soc Hypertens20082316517220409899