Abstract
A number of epidemiological and clinical studies have demonstrated that plasma high-density lipoprotein (HDL) level is a strong inverse predictor of cardiovascular events. HDL is believed to retard the formation of atherosclerotic lesions by removing excess cholesterol from cells and preventing endothelial dysfunction. Lecithin cholesterol acyltransferase (LCAT) plays a central role in the formation and maturation of HDL, and in the intravascular stage of reverse cholesterol transport: a major mechanism by which HDL modulates the development and progression of atherosclerosis. A defect in LCAT function would be expected to enhance atherosclerosis, by interfering with the reverse cholesterol transport step. As such, one would expect to find more atherosclerosis and cardiovascular events in LCAT-deficient patients. But this relationship is not always evident. In this review, we describe contradictory reports in the literature about cardiovascular risks in this patient population. We discuss the paradoxical finding of severe HDL deficiency and an absence of subclinical atherosclerosis in LCAT-deficient patients, which has been used to reject the hypothesis that HDL level is important in the protection against atherosclerosis. Furthermore, to illustrate this paradoxical finding, we present a case study of one patient, referred for evaluation of global cardiovascular risk in the presence of a low HDL cholesterol level, who was diagnosed with LCAT gene mutations.
Introduction
Several studies have demonstrated an inverse correlation between plasma high-density lipoprotein (HDL) and the risk of coronary artery disease, and have concluded that HDL is a powerful predictor of the disease.Citation1,Citation2
Mutations in lecithin-cholesterol acyltransferase (LCAT), apolipoprotein A-1 (ApoA-1), and ATP binding cassette A1 have been shown to underlie familial hypoalphalipoproteinemia, whereas cholesteryl ester transfer protein (CETP) gene defects underlie familial hyperalphalipoproteinemia. Homozygous mutations in the LCAT gene cause LCAT deficiency – a very rare metabolic disorder with two hypoalphalipoproteinemia syndromes: (1) familial LCAT deficiency (FLD), characterized by a complete lack of enzyme activity, and (2) “fish eye disease” (FED), characterized by a partially defective enzyme. It has been suggested that LCAT exhibits two activities in normal plasma: (1) alpha-LCAT activity, specific for lipoproteins that migrate with alpha mobility upon gel electrophoresis, and (2) beta-LCAT activity, which is specific for pre-beta, and beta-migrating lipoprotein (very-low-density lipoprotein [VLDL] and LDL).
FED is classified as an alpha-LCAT deficiency, whereas FLD has been proposed to result from a lack of both alpha-and beta-LCAT activity. Alpha- and beta-LCAT activity represents two functional aspects of the same protein.
LCAT plays a central role in the formation and maturation of HDL, and in the intravascular stage of reverse cholesterol transport, a major mechanism by which HDL modulates the development and progression of atherosclerosis. Deficiency in LCAT is characterized by a deficit in the esterification of plasma cholesterol. A defect in LCAT function would be expected to enhance atherosclerosis, by interfering with the reverse cholesterol transport step. But there have been contradictory reports regarding this in the literature.Citation3–Citation6
HDL and LCAT biochemistry and physiology
The human LCAT gene, localized at 16q22, is primarily expressed in the liver, but is also produced in smaller amounts in the brain and testes.Citation5 LCAT is a 416-amino acid-long protein that circulates in the blood, either bound to lipoproteins or in lipid-free form. ApoA-1 is the principal physiological activator of LCAT.Citation4
The LCAT reaction occurs in two steps. After binding to a lipoprotein, LCAT cleaves the fatty acid in the sn-2 position of phosphatidylcholine and transfers it onto a serine residue. Next, the fatty acid is transesterified to the 3-β-hydroxyl group on the A-ring of cholesterol, to form cholesterol ester (). Since cholesterol ester is more hydrophobic than free cholesterol, it migrates into the hydrophobic core of lipoprotein particles. Approximately 75% of plasma LCAT activity is associated with HDL. But LCAT is also able to bind and produce cholesteryl ester on LDL and other ApoB-containing lipoproteins.Citation5
Figure 1 High-density lipoprotein metabolism.
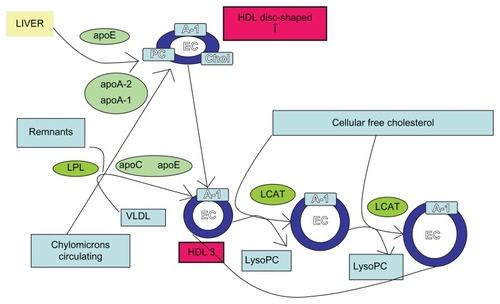
LCAT has been shown to promote the efflux of cholesterol from peripheral cellsCitation3 (). The esterification of cholesterol on HDL increases the concentration gradient from free cholesterol between cell membranes and HDL. The esterification of cholesterol transforms the discoidal-shaped nascent HDL, with a pre-β migration position on agarose gels, into spherical-shaped HDL, termed α-HDL. As cholesteryl esters are much more hydrophobic than cholesterol, another consequence of LCAT is that it prevents the spontaneous back-exchange of cholesterol from HDL to cells, and thus promotes net cellular removal of cholesterol.Citation3 Cholesteryl esters on HDL and LDL are essentially trapped on these lipoproteins, until they can be removed from the circulation by the liver.Citation5
Without ongoing esterification of cholesterol, the capacity of HDL to remove and bind additional cholesterol would eventually be diminished. CETP may further enhance this process by transferring cholesteryl esters formed by LCAT from HDL onto LDL, creating additional capacity for HDL to bind cholesterol.
LCAT deficiency: epidemiology and clinical presentation
LCAT deficiency is a rare disease. Thirty families have been described in the literature; the prevalence is below 1/1,000,000; only sporadic cases have been reported. The disease is transmitted as an autosomal recessive trait. LCAT deficiency results in two clinical entities: (1) FLD – complete LCAT deficiency – in which the lack of activity of the enzyme affects both the esterification on HDL (α-LCAT activity) and LDL (β-LCAT activity), and (2) FED – partial LCAT deficiency – in which only α-LCAT activity is impaired.Citation7,Citation8 In both cases, there is decreased plasma HDL (<10% of normal level) and the presence of corneal opacities, due to cholesterol deposits in the cornea, which begin most often in childhood, affecting the peripheral cornea before reaching the center. These anomalies are the only elements found in FED and, in some cases, may require a corneal transplant. Patients with FLD may also manifest hypertriglyceridemia, normochromic hemolytic anemia, and proteinuria. In this form, the prognosis depends mainly on the evolution of renal function (due to abnormal deposition of lipoprotein, leading to kidney failure).
LCAT deficiency and atherosclerosis
HDL cholesterol level is a strong inverse predictor of cardiovascular events. HDL is believed to retard the formation of atherosclerotic lesions by removing excess cholesterol from cells and preventing endothelial dysfunction.Citation9,Citation10
LCAT deficiency is a metabolic disorder characterized by very low HDL levels. Theoretically, hypoalphalipoproteinemia patients with LCAT deficiency should be at increased cardiovascular risk due to their HDL deficiency and defective reverse cholesterol transport. Thus, it would be expected that more atherosclerosis and cardiovascular events would be detected in LCAT-deficient patients. However, this relationship is not always evident. Previous studies have reported conflicting findings about potential increased cardiovascular risks in this patient population.
Early cross-sectional studies reported both decreased and increased LCAT activity in patients with angiographically-confirmed coronary artery disease.Citation11,Citation12
Some studies have reported increased atherosclerosis in patients with LCAT deficiency. Hovingh et al demonstrated that patients who are heterozygous for LCAT mutations have increased atherosclerosis, compared with familial controls.Citation13 This was illustrated by the fact that 47 heterozygotes for the LCAT gene mutation presented with a thicker intimal medial thickness (IMT) than 58 family members, as controls (0.623 mm versus 0.591 mm, respectively; P < 0.0015, adjusted for age, sex, and alcohol), and a 2.1-fold increase in high-sensitivity C-reactive protein (hsCRP) levels (4.40 mg/L versus 2.0 mg/L, respectively; P < 0.0001). Compared with family controls, heterozygotes exhibited a mean 36% decrease in HDL levels (0.812 ± 0.238 mmol/L versus 1.276 ± 0.387 mmol/L), and a mean decrease of 22% in ApoA-1 levels (1.21 ± 0.16 g/L versus 1.56 ± 0.28 g/L; P < 0.0001). Given the small number of study subjects, cardiovascular events were not compared.Citation13
In another study, LCAT-deficient patients had not only a reduced HDL level but also a marked increase in both hsCRP and IMT.Citation6 The study investigated 68 carriers of LCAT gene mutations, with an HDL level of 0.70 ± 0.32 mmol/L, and 74 family controls, with an HDL level of 1.59 ± 0.57 mmol/L. Carriers of LCAT gene mutations displayed a significant, twofold increase in median hsCRP levels, compared with controls. IMT progression in heterozygotes was 0.00538 mm/year, versus 0.00301 mm/year in controls (P < 0.0001).Citation6
It is worth noting that the reported increase of atherosclerosis in LCAT-deficient patients was based on hsCRP levels – a controversial cardiovascular marker – and IMT, with values in LCAT carriers within a “normal” range. Given these data, even if LCAT deficient patients have an increase cardiovascular risk, this increased risk appears to be minimal.
In a larger study, Calabresi et al did not find evidence for subclinical atherosclerosis in patients with LCAT deficiency. Citation8 They investigated a total of 120 patients: 40 carriers of LCAT gene mutations and 80 matched blood donor controls. Age, body mass index (BMI), blood glucose, and smoking status were comparable between the two groups. HDL levels were low in carriers (0.772 ± 0.446 mmol/L) versus controls (1.497 ± 0.307 mmol/L) (P < 0.0001). Maximum and average IMT values were significantly lower in carriers than in controls, even after adjustment for age, sex, BMI, smoking status, and blood pressure. Low HDL levels in the carriers of LCAT gene mutations did not result in subclinical atherosclerosis. Indeed, these authors found that the inheritance of a mutated LCAT genotype had a remarkable gene- and dose-dependent effect on reducing carotid IMT.Citation8
In a Canadian study, two homozygote and seven heterozygote patients with FLD, aged up to 70 years, were followed for 25 years, during which time none of the patients suffered any vascular events. The IMT in homozygote patients did not progress over the last four years of the study, and they were shown to have normal endothelial function (as measured by flow-mediated dilation of the brachial artery).Citation14
These seemingly contradictory results may potentially be explained by the fact that most of these studies were relatively small, and did not examine other important proteins and enzymes of the reverse cholesterol transport pathway, which can potentially alter the effect of LCAT on atherosclerosis. Furthermore, individuals with LCAT mutations have very few clinical events, for a small number of patients.
Clinical case
To further illustrate the paradoxical relationship between LCAT deficiency, very low HDL levels, and cardiovascular risk factors, we report one patient, referred for evaluation of global cardiovascular risk in the presence of a low HDL level.
A 52 year-old women presented with no significant familial or past medical history, other than an ophthalmologic anomaly with corneal opacity but normal vision. The patient had no cardiovascular symptoms, a normal clinical examination, and a normal ECG. Risk factors and atherosclerosis burden are reported in . Standard anthropometric measures, blood analyses, and atherosclerosis markers were obtained, as previously reported.Citation15,Citation16 The patient’s HDL level was significantly lower than the level typically reported for carriers (0.15 mmol/L) Cardiovascular evaluation found no trace of subclinical atherosclerosis (IMT, carotid plaques, ankle brachial index, and stress test), or associated risk factors such as hsCRP. Genetic analysis identified two mutations, heterozygous on the LCAT gene: (1) Exon 3, c.321C > A, Tyr83Stp, and (2) Exon 3 c.367C > T, Arg99cys. These two mutations have been previously described in one other patient (with similar findings: very low HDL and no cardiovascular disease). It was not possible to complete a family history of this patient, as she did not know her family.
Table 1 Patient characteristics
The paradoxical finding of severe HDL deficiency and the reported absence of subclinical atherosclerosis in LCAT-deficient patients has been used to reject the hypothesis that HDL is important in protection against atherosclerosis. Potential mechanisms to explain these findings have been postulated, such as preferential clearance of HDL fractions that have less atherogenic potential.Citation17 Decreased LDL and ApoB levels have also been suggested as potential explanations for the lack of marked cardiovascular disease. However, there is no clear evidence that LDL levels in these patients are decreased.Citation8 A recent report demonstrated that the capacity of HDL to accept cholesterol from macrophages (cholesterol efflux capacity – a metric of HDL function) has a strong inverse association with both carotid IMT and the likelihood of angiographic coronary artery disease, independent of HDL level.Citation18
These current insights into LCAT disorders further add to our understanding of the variable relationship between HDL metabolism and atherosclerosis risk.
Acknowledgments
We would like to thank Prof Moulin (CHU de Lyon) for support and helpful discussion.
Disclosure
The authors report no conflicts of interest in this work.
References
- GordonDJRifkindBMHigh-density lipoprotein – the clinical implications of recent studiesN Engl J Med198932119131113162677733
- BarterPGottoAMLaRosaJCHdl cholesterol, very low levels of ldl cholesterol, and cardiovascular eventsN Engl J Med2007357131301131017898099
- CzarneckaHYokoyamaSRegulation of cellular cholesterol efflux by lecithin: Cholesterol acyltransferase reaction through nonspecific lipid exchangeJ Biol Chem19962714202320288567654
- KuivenhovenJAPritchardHHillJFrohlichJAssmannGKasteleinJThe molecular pathology of lecithin: Cholesterol acyltransferase (lcat) deficiency syndromesJ Lipid Res19973821912059162740
- CalabresiLPisciottaLCostantinAThe molecular basis of lecithin: Cholesterol acyltransferase deficiency syndromes: A comprehensive study of molecular and biochemical findings in 13 unrelated italian familiesArterioscler Thromb Vasc Biol20052591972197815994445
- HovinghGKde GrootEvan der SteegWInherited disorders of hdl metabolism and atherosclerosisCurr Opin Lipidol200516213914515767853
- KleinHGSantamarina-FojoSDuvergerNFish eye syndrome: A molecular defect in the lecithin-cholesterol acyltransferase (lcat) gene associated with normal alpha-lcat-specific activity. Implications for classification and prognosisJ Clin Invest19939214794858326012
- CalabresiLBaldassarreDCastelnuovoSFunctional lecithin: Cholesterol acyltransferase is not required for efficient atheroprotection in humansCirculation2009120762863519687369
- CalabresiLGomaraschiMFranceschiniGEndothelial protection by high-density lipoproteins: From bench to bedsideArterioscler Thromb Vasc Biol200323101724173112969988
- CuchelMRaderDJMacrophage reverse cholesterol transport: Key to the regression of atherosclerosis?Circulation2006113212548255516735689
- WellsICPeitzmeierGVincentJKLecithin: Cholesterol acyltransferase and lysolecithin in coronary atherosclerosisExp Mol Pathol19864533033103466803
- Solajic-BozicevicNStavljenic-RukavinaASestoMLecithin-cholesterol acryltransferase activity in patients with coronary artery disease examined by coronary angiographyClin Investig19947212951956
- HovinghGKHuttenBAHolleboomAGCompromised lcat function is associated with increased atherosclerosisCirculation2005112687988416061733
- AyyobiAFMcGladderySHChanSJohn ManciniGBHillJSFrohlichJJLecithin: Cholesterol acyltransferase (lcat) deficiency and risk of vascular disease: 25 year follow-upAtherosclerosis2004177236136615530911
- LafitteMPradeauVLerouxLEfficacy over time of a short overall atherosclerosis management programme on the reduction of cardiovascular risk in patients after an acute coronary syndromeArch Cardiovasc Dis20091021515819233109
- RiedelMLafitteMPucheuYLatryKCouffinhalTPrognostic value of high-sensitivity c-reactive protein in a population of post-acute coronary syndrome patients receiving optimal medical treatmentEur J Cardiovasc Prev Rehabil201197 [Epub ahead of print]
- RaderDJIkewakiKDuvergerNMarkedly accelerated catabolism of apolipoprotein a-ii (apoa-ii) and high density lipoproteins containing apoa-ii in classic lecithin: Cholesterol acyltransferase deficiency and fish-eye diseaseJ Clin Invest19949313213308282802
- KheraAVCuchelMde la Llera-MoyaMCholesterol efflux capacity, high-density lipoprotein function, and atherosclerosisN Engl J Med2011364212713521226578