 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Artificially synthesized RNA molecules have recently come under study since such molecules have a potential for creating a variety of novel functional molecules. When designing artificial RNA sequences, secondary structure should be taken into account since functions of noncoding RNAs strongly depend on their structure. RNA inverse folding is a methodology for computationally exploring the RNA sequences folding into a user-given target structure. In the present study, we developed a multi-objective genetic algorithm, MODENA (Multi-Objective DEsign of Nucleic Acids), for RNA inverse folding. MODENA explores the approximate set of weak Pareto optimal solutions in the objective function space of 2 objective functions, a structure stability score and structure similarity score. MODENA can simultaneously design multiple different RNA sequences at 1 run, whose lowest free energies range from a very stable value to a higher value near those of natural counterparts. MODENA and previous RNA inverse folding programs were benchmarked with 29 target structures taken from the Rfam database, and we found that MODENA can successfully design 23 RNA sequences folding into the target structures; this result is better than those of the other benchmarked RNA inverse folding programs. The multi-objective genetic algorithm gives a useful framework for a functional biomolecular design. Executable files of MODENA can be obtained at http://rna.eit.hirosaki-u.ac.jp/modena/.
Introduction
To date, a variety of cellular functions of noncoding RNAs (ncRNAs) has been elucidated using experimental and computational methodologies. Since RNA secondary structures play important roles in their functions, various RNA folding algorithms (eg, those based on minimization of free-energy [MFE],Citation1,Citation2 formal grammar,Citation3,Citation4 and maximum expected accuracyCitation5) have been developed to analyze RNA sequences. Recently, RNA secondary structure has become an important notion not only to unveil the cellular functions of natural ncRNAs but also to create artificial RNA molecules which have a desired function (such as artificial ribozymes,Citation6 artificial miRNAs,Citation7 and RNA nanostructuresCitation8,Citation9). In such artificial applications of RNA sequence, we have to design an RNA sequence that folds into a desired structure (target structure) to realize a desired function. This ‘RNA sequence design’ problem can be defined as an inverse problem of the RNA folding. Since no RNA inverse folding algorithm that can find all RNA sequences folding to a desired structure is yet known, heuristic algorithms, RNAinverse,Citation10 RNA-SSDCitation11 and INFO-RNA,Citation12 have been proposed to solve approximately the RNA inverse folding problem. RNA inverse is a pioneering RNA inverse folding algorithm which uses a divide-and-conquer type strategy, where the inverse folding proceeds from sub-sequences to a whole sequence, and an adaptive walk is used to iteratively improve the designed sequence. RNA-SSD also uses a divide-and-conquer type strategy (hierarchical decomposition of the target secondary structure) and a stochastic local searchCitation13 to improve the RNA sequence given by a sophisticated sequence initialization procedure. INFO-RNA is well characterized by its strong sequence initialization procedure; in this initialization, a dynamic programming algorithm is employed to find the RNA sequence which has the lowest free energy in all compatible RNA sequences, where the free energies are calculated by assuming all compatible RNA sequences are folded into the target structure (the RNA sequences that can form a structure strictly the same as that of a given target structure are called ‘compatible sequences’,Citation10 where the target structure does not need to be the optimal structure of each sequence). This initialization is followed by a stochastic local searchCitation13 which iteratively examines neighboring RNA sequences in the RNA sequence space. This local search is done according to the order determined based on the free energy differences between the current and neighboring sequences.
Motivation
Although the previous RNA inverse folding algorithms have shown high performance in various benchmarks, they have the following drawbacks. i) First, these algorithms tend to output the RNA sequences with a narrow range of free energies. This could be due to the search algorithms and objective function (OF) used in the previous algorithms. The previous algorithms adopt local search algorithms to minimize the structure distance between designed and target structures, and do not use a free energy as an OF. As a result, they tend to output the RNA sequences near to the initial RNA sequence. For example, INFO-RNA tends to output an RNA sequence much more stable than the corresponding natural ncRNAs,Citation14 and this is likely to be due to the initial guess of INFO-RNA, which already has a very low free energy. If possible, selecting the stability of the designed RNA sequence from a wider range would be more favorable, since the stability can affect the function of the designed RNA sequence. ii) Second, the direct problem solver (ie, an RNA folding program) of the previous methods is fixed. In RNA inverse folding, a direct problem solver is iteratively used to predict the structure of the designed sequence, which is necessary to compare the structure of the designed sequence with the target structure; eg, RNAinverse, RNA-SSD and INFO-RNA use a fold function of Vienna RNA PackageCitation2 as a direct problem solver. Although it may be theoretically possible to change the direct problem solver in the previous methods to the other one, such an implementation is currently not available. Since more accurate RNA folding algorithms based on new paradigms, such as SfoldCitation15 and CentroidFold,Citation5 have been proposed, an RNA inverse folding algorithm should also has an option to use a new RNA folding program as a direct problem solver.
To address the drawback i) mentioned above, we have to consider 2 objective functions simultaneously, a stability measure (such as a free energy) and a structure similarity measure (such as the structure distance between the designed and target structures), during the optimization. This indicates that RNA inverse folding can be formulated as a multi-objective problem, whereas the previous algorithms have adopted a structure distance alone as an objective function to be optimized. Multi-objective optimization (MOO)Citation16 is a methodology for solving multi-objective problems. So far, many applications of MOO in bioinformatics have been reported.Citation17,Citation18 On the basis of MOO, we can simultaneously optimize multiple objective functions without assigning a relative weight to each objective function. To solve the MOO problems, multi-objective genetic algorithms (MOGAs),Citation16 eg, nondominated sorting genetic algorithm 2 (NSGA2),Citation16,Citation19 have been developed and widely used. In the present paper, we show that MOGA gives a useful framework for biomolecular sequence design, and our new RNA inverse folding algorithm, called MODENA (Multi-Objective DEsign of Nucleic Acids), can improve the 2 drawbacks pointed out for the previous RNA inverse folding algorithms.
Material and methods
In RNA inverse folding, we explore an RNA sequence space to find the RNA sequences that fold into a user-given target structure, where ‘a sequence folds into a structure’ means that the structure is the optimal structure (eg, the lowest free energy structure or the centroid structure) of the sequence. The sequence length N is determined by the length of the inputted target structure. A point of the RNA sequence space for the sequences with a length of N is denoted by S = s1··si··sN (1 ≤ i ≤ N), where si ∈ {A, C, G, U} for all i; an A, C, G and U indicates an adenine, cytosine, guanine and uracil, respectively. An RNA structure is composed of loop nucleotides and base pairs, where a base pair (i, j) indicates formation of the hydrogen bonds between nucleotide positions i and j (where 1 ≤ i < j ≤ N and usually |i – j| > 3). Moreover, the RNA structure can be represented by using a connection information ci, where if position i forms a base pair with position j, ci = j; when position i is a loop nucleotide, ci = 0.
To explore the RNA sequence which folds into the desired target structure and has the lowest possible free energy, we have to take into account 2 objective functions simultaneously: a measure for structure stability (such as the free energy E computed by a direct problem solver) and a measure for structure similarity (eg, the structure distance d between the structure predicted for the designed RNA sequence and the target structure given by a user). In the present study, we use a structure stability score ɛ and a structure similarity score σ as the objective functions. The ɛ and σ are defined as ɛ = –E and σ = (N – d)/N, respectively, where E and N are the lowest free energy of the designed sequence and the total number of nucleotides, respectively; a structure distance d is the number of the nucleotide positions whose structure in the designed sequence is different from that in the target structure (). A larger value of the ɛ corresponds to more stable structure. The similarity score σ scale ranges from 0.0 to 1.0 and a similarity score of 1.0 indicates a perfect consensus between 2 structures.
Figure 1 Examples of a structure distance d and similarity score σ between a target structure (denoted as ‘target structure’) and 3 structures of designed sequences. Nucleotide positions whose structure in the designed sequence is different from that in the target structure are represented in red. The structure shown at the bottom has a σ = 0.0, ie, the target structure and the structure shown at the bottom are completely different.

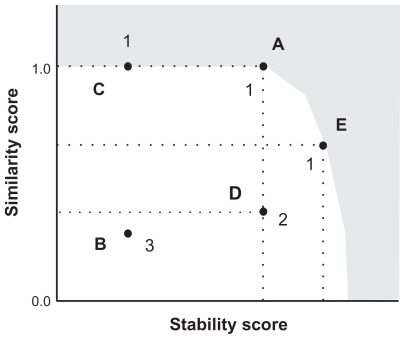
In the present paper, we introduce MOO to take the 2 objective functions into account in the RNA inverse folding. In MOO,Citation16 we evaluate solutions by using a dominance, which is defined between 2 solutions. Let us consider exploring the solutions that maximize all objective functions. Solution A is said to dominate solution B if both of the following 2 conditions are satisfied: i) fiA ≥ fiB (∀i ∈1 … M), where fiA and fiB are the i-th objective functions of solutions A and B, respectively, and M is the number of objective functions; ii) fiA ≠ fiB (∃i ∈1 … M). In addition, solution A is said to strongly dominate solution B when fiA > fiB (∀i ∈1 … M). A Pareto optimal solution is a solution that is not dominated by any other solution, and a solution not strongly dominated by any other solution is called a weak Pareto optimal solution. In accordance with the definitions, 2 solutions with exactly the same values of objective functions are neither dominated nor strongly dominated each other. The definitions of the terminology (eg, dominance) written in italic in this paragraph are taken from Deb.Citation16 A schematic illustration of a dominance relationship in RNA inverse folding is shown in .
Figure 2 An example of a dominance relationship in the RNA inverse folding problem. Stability and similarity scores are used as objective functions. Each solid circle indicates a solution in the objective function space. In this figure, solutions A and E dominate solutions B and D; solution A also dominates solution C. Since solution C has a similarity score the same as that of solution A, solution A does not strongly dominate solution C. In this figure, a gray region schematically indicates the region where no solution exists; hence solutions A and E are Pareto optimal solutions. Since solution A is the Pareto optimal solution with a similarity score of 1.0, solution A has the highest stability score in all RNA sequences folding into a target structure. Solution C is not a Pareto optimal solution, but is included in weak Pareto optimal solutions. The integers assigned to each solution are the dominance ranks calculated for the 5 solutions, where the dominance ranks are calculated based on strong dominance.
Multi-objective genetic algorithm
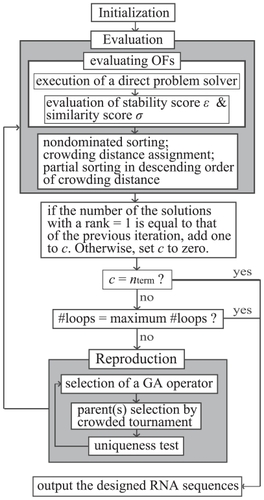
To obtain an approximate set of weak Pareto optimal solutions for RNA inverse folding, we developed an RNA inverse folding algorithm, MODENA, on the basis of NSGA2,Citation16,Citation19 which is one of the standard MOGAs. A flowchart of MODENA is shown in , where initialization, evaluation and reproduction procedures are used in accordance with the standard procedures of genetic algorithm (GA); GA is a population-based metaheuristics for a search and combinatorial optimization. Citation20 In the initialization step, initial Np solutions are generated in a random manner, where Np is a GA population size. In MODENA, an RNA nucleotide sequence is used as a solution, and duplication of identical RNA sequences in one population is prohibited throughout a MODENA run. In the evaluation step, a direct problem solver is invoked to assign stability and similarity scores to each solution. After the evaluation step, reproduction procedures generate child solutions by applying mutations and a crossover to a parent population. The iteration between the evaluation and reproduction procedures stops when one of the following conditions is satisfied: i) a GA iteration number reaches a given maximum number of iterations; ii) the number of non-strongly dominated solutions in a GA population does not change during continuous nterm iterations. In the present study, nterm = 30 was used. Details of the initialization, evaluation and reproduction procedures are described below.
Figure 3 Flowchart of MODENA algorithm.
Initialization
In this step, we generate an initial population of Np RNA sequences in a random manner. To generate the i-th individual of the initial population (1 ≤ i ≤ Np), first, we define a probability pAG = (i – 1.0)/(Np – 1.0). Then we scan the nucleotide positions of i-th solution to assign a nucleotide as follows: if the position is a loop position in the target structure, we assign an adenine to the position with a probability of pAG + (1.0 – pAG) × 0.25, otherwise a cytosine, guanine, or uracil is assigned to the position with an equal probability; if the position has a base pair, a guanine is assigned to the position with a probability of pAG + (1.0 – pAG) × 0.25, otherwise an adenine, cytosine, or uracil is assigned to the position with an equal probability.
In addition to the procedure mentioned above, we perform 2 procedures for the initialization of the i-th individual. The first one is a ‘random change from a guanine to cytosine’. In this procedure, a guanine assigned to each nucleotide position is randomly changed to a cytosine with a probability of 0.5. This procedure is introduced to shuffle a guanine and cytosine in the base paired positions without changing a base pair type (a G:C and C:G are the same base pair type). The second one is a ‘propagation procedure’ where the generated RNA sequence is scanned from the 5′ end to the 3′ end, and if we find a position i which forms a base pair with a position j (where i < j), a nucleotide compensatory to the nucleotide at the position i is assigned to the position j, where only canonical A:U and G:C pairs are used (eg, if G is found at position i, C is assigned to position j; a G:U pair is not used). This procedure guarantees that the randomly generated RNA sequence is a compatible RNA sequence for the target structure.
By using the procedures mentioned above with a high pAG, we can obtain an RNA sequence that has GC-rich base paired positions and A-rich loop positions, and we can expect that such an RNA sequence has a high stability score. In contrast, when we use a low pAG, we can obtain an RNA sequence with a lower stability score. By using the formula pAG = (i – 1.0)/(Np – 1.0) for the i-th solution, we can fill the initial population with RNA sequences that have diverse values of stability scores, and such a set of RNA sequences can give a good initial guess for obtaining an approximate set of weak Pareto optimal solutions.
Evaluation
In the evaluation step, the stability score ɛ and similarity score σ for each solution are evaluated. To calculate the 2 objective functions, we have to run a direct problem solver for each solution since both the lowest free energy (which is needed to obtain the stability score) and a structure (which is needed to calculate the structure similarity with the target structure) are computed by the direct problem solver. As a direct problem solver, we can utilize RNAfoldCitation2 and CentroidFoldCitation5 in MODENA. A default direct problem solver is RNAfold. These direct problem solvers can be specified through an option of MODENA.
After evaluating the objective functions, in accordance with NSGA2,Citation16,Citation19 MODENA assigns a dominance rank to each solution and determines the Np parent solutions for the next GA generation as follows:
Step 1 We set R = (Np parent solutions) + (Np child solutions). It is noted that, at the first GA iteration, the child population is a null; ie, R = (Np parent solutions) at the first GA iteration.
Step 2 A dominance rank is assigned to each solution in R. We use the O(MNp2) nondominated sorting of the original NAGA2,Citation16 where M is the number of objective functions.
Step 3 The solutions in R are sorted in ascending order of dominance rank.
Step 4 If both the Np-th solution and the (Np + 1)-th solution in R share a dominance rank of i, sorting in descending order of crowding distanceCitation16 is performed only for the solutions with a dominance rank of i; this procedure is called a ‘niching’. We use the top Np solutions in R as the parent solutions at the next GA generation. Step 4 is not used at the first GA iteration since (Np + 1)-th solution does not exist.
Reproduction
In the reproduction step of MODENA, in accordance with NSGA2,Citation16,Citation19 we generate Np child solutions from the parent Np solutions. The child solutions are generated with randomly invoked GA operators. After selection of the GA operator, a parent solution is selected using crowded tournament selection,Citation16 where a mutation and crossover operator needs 1 and 2 parent solutions, respectively. In the MODENA algorithm, we use 1 crossover operator (structural n-point crossover) and 2 mutation operators (point accepted mutation and error diagnosis mutation). The 3 GA operators are invoked with an equal probability, ie, 1/3.
Structural n-point crossover is performed with a crossover parameter nc and a randomly determined x0 (= 0 or 1) as follows:
Step 1 Set l = 0 and set xi = x0 for all i (1 ≤ i ≤ N; N is a sequence length). Randomly select a base pair (i, j) (1 ≤ i < j ≤ N).
Step 2 If xk (i ≤ k ≤ j) is 0, change xk to 1, otherwise change xk to 0 for all k; then increment l by 1.
Step 3 If l < nc and ‘the number of the base pairs whose upstream nucleotide position m satisfies i < m (where m < N)’ is larger than or equal to 1, randomly select a base pair (inew, jnew), where i < inew < jnew ≤ N; then we set i = inew and j = jnew, and move to Step 2; otherwise we go to Step 4.
Step 4 Generate a child solution according to xi for all i (1 ≤ i ≤ N); if xi = 0, copy the value of a nucleotide siA in parent A to the corresponding nucleotide sichild of the child solution; if xi = 1, the value of a nucleotide siB in parent B is copied to sichild.
It is noted that we consider only the target structure and do not use a predicted structure in structural n-point crossover. The algorithm of structural n-point crossover is illustrated in . In this study, we use nc = 1 (a user can change this value with a command line option). As can be seen from the figure, structural n-point crossover splices the subsequences of 2 parent RNA sequences in such a way that the 2 nucleotides of each base pair are copied from 1 parent RNA sequence. By using the structural n-point crossover, we can crossover the parent RNA sequences without destroying the compensatory relationships in each parent RNA sequence.
Figure 4 Examples of the genetic algorithm operators used in the MODENA algorithm. a) Structural n-point crossover: parent A and parent B are randomly divided into subsequences (denoted in red and blue) in accordance with the target structure, where base pairs in each parent solution are preserved in each set of the subsequences. A child solution is generated by concatenating the subsequences selected from parent A (denoted in red) and those selected from parent B (denoted in blue). b) Point accepted mutation: randomly selected positions of the child solution are mutated. In this example, mutated positions are indicated in red. c) Error diagnosis mutation: examples corresponding to the rules (i), (ii) and (iii) are denoted in red, blue and green, respectively.

Point accepted mutation copies the parent sequence to the child sequence, and then scans the child sequence from the 5′ end in order to randomly change each nucleotide with a probability of pM. If the position of the changed nucleotide forms a base pair with the other position k (1 ≤ k ≤ N) in the target structure, a nucleotide compensatory to the changed nucleotide is assigned to the position k, where an A:U or G:C base pair is used ().
In error diagnosis mutation operator, first we copy the nucleotide siparent and connection information ciparent of a parent solution to the sichild and cichild of a child solution for all i (1 ≤ i ≤ N), respectively; here we copy the ciparent, which is assigned to the parent solution by a direct problem solver, to the child solution. Then, we scan the cichild (1 ≤ i ≤ N) of the child solution from the 5′ end. If we find a nucleotide position i which has a structure different from the corresponding one of the target structure (ie, when citarget ≠ cichild), a sichild is modified (and s jchild, where 1 ≤ j ≤ N, is also modified if position i forms a base pair with position j in the target structure) in accordance with the following 3 rules:
when position i forms a base pair with position j in the predicted structure (ie, cichild = j, 1 ≤ i ≤ N, 1 ≤ j ≤ N) whereas position i has a loop nucleotide in the target structure (citarget = 0), we change a nucleotide sichild in accordance with the value of s jchild; when s jchild is an adenine, we set a cytosine or guanine to sichild ; if s jchild is a cytosine, we set an adenine or uracil; if s jchild is a guanine or uracil, we set an adenine or cytosine, respectively; these operations prevent a nondesired base pair formation when the child RNA sequence is folded by a direct problem solver.
when position i does not form a base pair in the predicted structure (cichild = 0) while the corresponding position in the target structure forms a base pair with position j (citarget = j), we set a C:G or G:C pair to a sichild:sjchild pair.
if position i forms a base pair with position k in the predicted structure (citarget = k, 1 ≤ k ≤ N) whereas position i forms a base pair with position j (≠ k) in the target structure (citarget = j, 1 ≤ j ≤ N), we change the nucleotide sichild in accordance with the value of skchild; when skchild is an adenine, we set a cytosine or guanine to sichild; if skchild is a cytosine, we set an adenine or uracil; if skchild is a guanine or uracil, we set an adenine or cytosine, respectively; then a nucleotide compensatory to that of position i is assigned to s jchild, where A:U or G:C base pairs are used.
An example of error diagnosis mutation is shown in . It is noted that the operations by the rules (i)–(iii) are not independent of each other; during the scan from the 5′-end, an operation by a rule may overwrite the previous change made by another rule.
Datasets for the performance evaluation
We evaluated the performance of MODENA and other RNA inverse folding programs with the dataset taken from the seed alignments of Rfam 9.0.Citation21 To evaluate the RNA design performance for RNA secondary structures, we extracted the annotated structures from 29 Rfam entries (from RF00001 to RF00030 except for RF00023), where each Rfam entry corresponds to an RNA family. RF00023 (Rfam ID:tmRNA) was not used since it has too many pseudoknots. In each Rfam entry, we selected the longest RNA sequence and extracted its annotated structure, and we used the extracted structure as the target structure of the Rfam entry. If pseudoknot base pairs are included in the extracted structure, pseudoknot base pairs were deleted from the extracted structure before the performance evaluation. As a result, we obtained 29 target secondary structures. Hereafter, we refer to this dataset as the Rfam dataset.
Results and discussion
Since MODENA is a ‘population-based’ algorithm based on GA, multiple solutions can be obtained at 1 run. In the present study, we use a GA population size Np = 50 to evaluate the performance of MODENA. MODENA with this setting outputs at most 50 successfully designed RNA sequences at one run; we refer to the designed RNA sequence which has a similarity score of 1.0 as a ‘successfully designed RNA sequence’, and we refer to a run which outputs the successfully designed RNA sequence as a ‘successful run’.Citation12 A GA population size = 100 was also used in order to compare the results obtained with population sizes of 50 and 100. Throughout the present paper, we use a maximum GA iteration number equal to the population size (eg, we use a maximum iteration number = 50 when using a population size = 50). Performance comparison between MODENA and previous methods is not straightforward, since the previous methods are ‘nonpopulation-based’ algorithms, which output one solution at 1 run. To compare the performance of MODENA with those of nonpopulation-based algorithms (INFO-RNA 2.1 and RNAinverse of Vienna RNA Package 1.8.3), we performed 50 independent runs for each target structure when evaluating the nonpopulation-based methods. Independent multiple runs of a nonpopulation-based method can give multiple solutions different from each other, since the nonpopulation-based methods used in our performance comparison are stochastic algorithms.
Comparison of computational times of MODENA and the previous nonpopulation-based algorithms is also not straightforward. When we performed 50 independent runs of each nonpopulation-based algorithm for a target structure, we calculated ‘the expected time Et required for finding a solution’, which was used in the previous studies.Citation11,Citation12 The Et is defined as follows:
where Es is the average time of successful runs, Eu is the average time of unsuccessful runs, and fs is a rate of successful runs (fs = the number of successful runs divided by the total number of independent runs). The Et gives an expected computational time for obtaining a successfully designed RNA sequence. When there is no successful run (ie, fs = 0), the Et and the computational time of MODENA are denoted as ‘−’ in our results. We discuss the computational speed of MODENA and other methods by comparing the computational time of 1 MODENA run and the Et of other methods.
An approximate set of weak Pareto optimal solutions
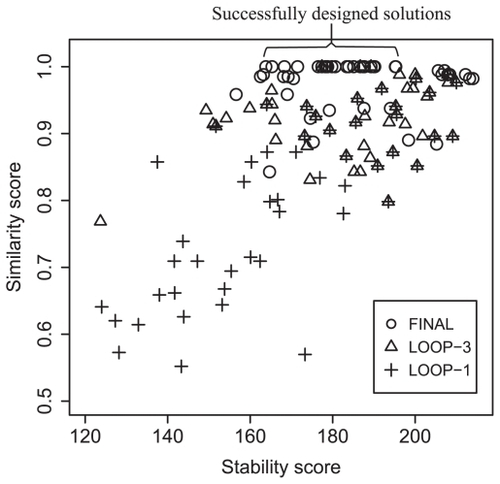
shows an example of how the solutions computed by MODENA evolve to the successfully designed RNA sequences. In the figure, we plot the solution distributions of the initial, third and final populations obtained in the GA iterations. In the initial distribution of this example, there are no successfully designed RNA sequences, ie, no solution has a similarity score of 1.0. These rather randomly distributed initial solutions successfully evolved to an approximate set of weak Pareto optimal solutions after 50 GA iterations, where 23 solutions with a wide range of stability scores were successfully designed. Moreover, we observed a fast convergence in this example; we obtained a solution distribution almost similar to that of the final population even at the tenth GA iteration (data not shown).
Figure 5 Example plots in an objective function space for the solutions obtained by MODENA. An RNA inverse folding result for a target structure (a structure of a rRNA predicted by RNAfold; GenBank accession number of the rRNA:AF107506) is shown. In this figure, the solution distribution of the initial genetic algoritham iteration (denoted by LOOP-1), that of the third iteration (LOOP-3) and the final one (FINAL) are shown. Successfully designed RNA sequences (ie, they have a similarity score of 1.0) are indicated as ‘successfully designed solutions’.
The results for the Rfam dataset
The performance evaluation results for the Rfam dataset are summarized in , where RNAfold was used as a direct problem solver of MODENA. In this table, the results for INFO-RNA and RNAinverse are also shown for the performance comparison. In the target structures taken from the annotations of the 29 RNA families in Rfam, MODENA successfully designed RNA sequences for 23 RNA families. This result is better than those obtained by INFO-RNA and RNAinverse, by which the RNA sequences of 17 and 13 RNA families were successfully designed, respectively. The free energy distributions for the successfully designed RNA sequences are shown in , and , where the results for the RNA families successfully designed by all methods (MODENA, INFO-RNA and RNAinverse) are represented. As can be seen from the figures, the RNA sequences designed by MODENA have much wider free energy ranges compared with those of INFO-RNA and RNAinverse. There are 2 possible reasons for this result. The first is the local search nature of INFO-RNA and RNAinverse. Since INFO-RNA and RNAinverse explore the region near to the initial RNA sequences, the initial guess can strongly affect the final RNA sequence. Interestingly, RNAinverse and INFO-RNA consistently outputs high free energy solutions and low free energy solutions, respectively, in our results. These results clearly correspond to the sequence initialization algorithms of the methods; RNAinverse uses a purely random compatible sequence as an initial sequence, while IFNO-RNA generates an initial sequence with a very low free energy by a dynamic programming. The second is the MOGA used in MODENA. MODENA is developed based on NSGA2; NAGA2 takes niching into account in the optimization procedure, which encourages the solutions in a population to be diverse in the objective function space. As a result, the approximate set of weak Pareto optimal solutions computed by MODENA tends to expand not only to a high stability score side but to a low stability score side. This leads to a wider free energy range of the RNA sequences designed by MODENA.
Figure 6 Free energy distributions of the successfully designed RNA sequences for a part of the Rfam dataset. The RNA family name and the accession number in Rfam are indicated at the top of the figure. The distributions for RNAinverse, INFO-RNA, and MODENA are shown by a boxplot. In the figure, the free energy of the natural RNA sequence corresponding to the target structure is also plotted as a ‘natural’, where the free energy was obtained by folding the RNA sequence with RNAfold.
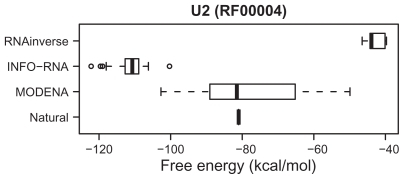
Figure 7 Free energy distributions of the successfully designed RNA sequences for a part of the Rfam dataset (continued from ).
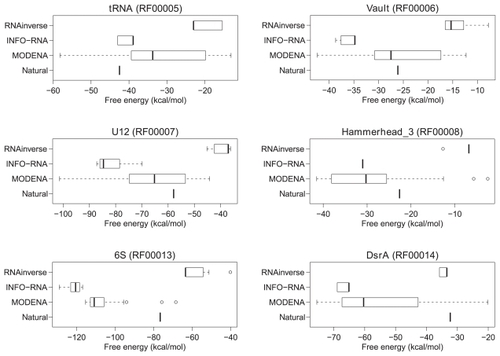
Figure 8 Free energy distributions of the successfully designed RNA sequences for a part of the Rfam dataset (continued from and ).
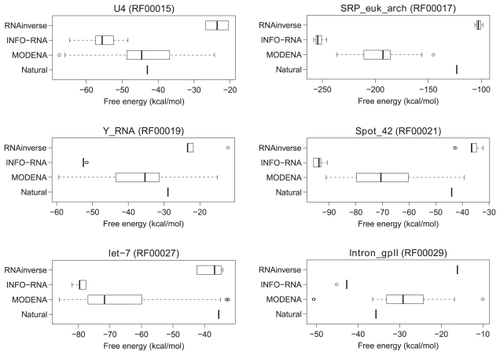
Table 1 The results for the Rfam dataset
In almost all RNA families included in , and , the free energies of the natural RNA sequences corresponding to the target structures are located within the free energy range of MODENA, whereas the energy ranges of RNAinverse and INFO-RNA do not include the energies of the natural ones in almost all RNA families of , and .
To test the initial random number dependence of MODENA, we performed an additional 3 independent runs for the Rfam dataset with different initial random numbers. As a result, we found that the random number dependence of MODENA is very small, where only 1 run for RF00003 in an additional runs failed and the other RNA families, which were successfully designed in , were successfully designed in the additional 3 runs. It is noteworthy that we obtained 1 successful design of RF00028 in an additional run whereas design of this RNA family failed in .
As can be seen from , INFO-RNA is much faster than MODENA in many cases. Computational times of MODENA and RNAinverse are comparable.
To test the performance of the MODENA invoked with an option of a non-MFE direct problem solver (CentroidFold),Citation5 we performed the RNA inverse folding with the Rfam dataset, where a population size of 50 was used. As a result, we successfully designed RNA sequences for 20 RNA families out of the 29 RNA families of the Rfam dataset. Since this result is slightly worse than that obtained with the MFE direct problem solver (RNAfold), we performed larger calculations with the CentroidFold direct problem solver on the failed 9 target structures of the Rfam dataset, where we set both a GA population size and maximum iteration number to 100. These larger calculations improved the results, where two out of the nine target structures were successfully designed. These results imply that RNA inverse folding with a non-MFE direct problem solver is more difficult than that with a MFE direct problem solver.
Why we explore weak Pareto optimal solutions
In the Pareto optimal solutions for the RNA inverse folding problem, where the stability score and similarity score are used as objective functions, there exists only 1 solution (eg, solution A in ) with a similarity score of 1.0 which folds into the target structure; this is because the other solutions (eg, solution C in ) with a similarity score of 1.0 (and a lower stability score) are dominated by the single Pareto optimal solution with a similarity score of 1.0. This indicates that the stability score of the Pareto optimal solution with a similarity score = 1.0 is highest in the stability scores of all RNA sequences folding into the target structure. The other solutions (eg, solution E in ) in the Pareto optimal solutions have a similarity score σ < 1.0.
It is noted that even when we successfully find the single solution which has a similarity score of 1.0 and the best stability score in all RNA sequences folding into the target structure, our search does not finish. This is because the other solutions (eg, solution C in ) with a lower stability score and a similarity score of 1.0 usually exist and they are also acceptable as solutions of RNA inverse folding. Since not only very stable RNA structures but also those with a lower stability can be candidates for artificial functional RNA sequences, it is important to develop a methodology that can design the RNA sequences with a wide range of free energies. To obtain a set of the RNA sequences that fold into the target structure and have a wide range of stability score, we can utilize weak Pareto optimal solutions. In weak Pareto optimal solutions of the RNA inverse folding problem, multiple solutions are allowed to have a similarity score of 1.0 in contrast to the case of the Pareto optimal solutions, where only 1 solution is allowed to have a similarity score of 1.0 (in , solutions A and C are weak Pareto optimal solutions). Thus, weak Pareto optimal solutions can give more a comprehensive solution set for RNA inverse folding. Since it is difficult to obtain the complete set of weak Pareto optimal solutions, we explored the approximate set of weak Pareto optimal solutions in the RNA inverse problem by using a framework of MOGA.
Conclusions
We have developed a new RNA inverse folding algorithm, MODENA, on the basis of MOGA. We have evaluated MODENA with the dataset taken from Rfam, and found that our program can successfully design 23 RNA sequences out of the 29 target secondary structures. This result is better than those of the previous RNA inverse folding algorithms, INFO-RNA and RNAinverse, which successfully designed 17 and 13 RNA sequences, respectively. MODENA can not only design the RNA sequences successfully but also output multiple solutions with a wide range of free energies at 1 run. In addition, we showed that MODENA can design RNA sequences on the basis of a non-MFE-based state-of-the-art RNA folding program.
The advantage of MODENA is the ability to produce successfully designed multiple RNA sequences with a wide range of free energies. Currently, this function has not been realized by the other RNA inverse folding algorithms. This function enables us to select not only a very stable RNA secondary structure but also an RNA sequence whose secondary structure has a free energy similar to that of natural counterparts.
It is noted that the RNA sequence designed by an RNA inverse folding program is a ‘predicted candidate’; ie, there is no guarantee that the designed RNA sequence folds to the structure exactly same as that of the specified structure in vivo. For this reason, an experimenter will have to assess a number of computationally designed RNA sequences before obtaining an RNA sequence that has a desired function.
For example, riboswitchesCitation6 are interesting targets of the RNA sequence design. Riboswitches show a conformational change when they work; if the designed riboswitch sequence has too stable a structure, the riboswitch may not work correctly since a too stable structure can disturb the conformational change necessary to function as a riboswitch. In such a case, selecting a designed RNA sequence whose free energy is not too low but near that of natural counterparts will become important. Moreover, in other ncRNAs, if the cleavage of the RNA strands of the ncRNA is necessary to work as a functional RNA, a similar situation can occur.
A slow computational speed is the drawback of MODENA. Since MODENA executes a direct problem solver many times (if we use a population size of 50 and a maximum GA iteration number of 50, the direct problem solver is called at most 2500 times), the computational time of MODENA strongly depends on the speed of the direct problem solver. MODENA is based on GA, and GA can be accelerated by using a parallel computation; a parallel computation is a promising way to hasten MODENA and such an implementation is currently in progress.
The objective functions of MODENA still have a degree of freedom. In the current version of MODENA, the probability of the lowest energy structure of the designed RNA sequence is not used in the objective functions.Citation10 By using the probability as the stability score instead of –E, we can explore the RNA sequence whose lowest energy structure has the largest probability. Implementation of this direction is not difficult, and we will include such a function also in the next versions.
It is noteworthy that the framework of the MOO allows us to optimize not only a system with 2 objective functions but also that with more objective functions. This means that the MODENA algorithm can potentially be extended to various different purposes. We believe that the MODENA algorithm gives a useful framework for the inverse folding of a great variety of biomolecules.
Acknowledgments
This work was partially supported by KAKENHI (22700304).
Disclosure
The author reports no conflicts of interest.
References
- MarkhamNRZukerMUNAFold: software for nucleic acid folding and hybridizationMethods Mol Biol200845333118712296
- HofackerIVienna RNA secondary structure serverNucleic Acids Res2003313429343112824340
- SakakibaraYBrownMHugheyRStochastic context-free grammars for tRNA modelingNucleic Acids Res199422511251207800507
- KnudsenBHeinJPfold: RNA secondary structure prediction using stochastic context-free grammarsNucleic Acids Res2003313423342812824339
- HamadaMKiryuHSatoKMituyamaTAsaiKPrediction of RNA secondary structure using generalized centroid estimatorsBioinformatics20092546547319095700
- BauerGSuessBEngineered riboswitches as novel tools in molecular biologyJ Biotechnol200612441116442180
- SchwabROssowskiSRiesterMWarthmannNWeigelDHighly specific gene silencing by artificial microRNAs in ArabidopsisPlant Cell2006181121113316531494
- YinglingYGShapiroBAComputational design of an RNA hexagonal nanoring and an RNA nanotubeNano Lett200772328233417616164
- SevercanIGearyCVerzemnieksEChworosAJaegerLSquare-shaped RNA particles from different RNA foldsNano Lett200991270127719239258
- HofackerIFontanaWStadlerPBonhoefferLTackerMSchusterPFast folding and comparison of RNA Secondary structuresMonatsh Chem1994125167188
- AndronescuMFejesAPHutterFHoosHHCondonAA new algorithm for RNA secondary structure designJ Mol Biol200433660762415095976
- BuschABackofenRINFO-RNA–a fast approach to inverse RNA foldingBioinformatics2006221823183116709587
- HoosHHStutzleTStochastic Local Search: Foundations and ApplicationsSan Francisco, CAElsevier/Morgan Kaufmann2004
- BuschABackofenRINFO-RNA–a server for fast inverse RNA folding satisfying sequence constraintsNucleic Acids Res200735W31031317452349
- DingYChanCYLawrenceCERNA secondary structure prediction by centroids in a Boltzmann weighted ensembleRNA2005111157116616043502
- DebKMulti-Objective Optimization using Evolutionary AlgorithmsChichester, UKJohn Wiley and Sons2001
- HandlJKellDBKnowlesJMultiobjective optimization in Bioinformatics and computational biologyIEEE/ACM Trans Comput Biol Bioinform2007427929217473320
- TanedaAMulti-objective pairwise RNA sequence alignmentBioinformatics2010262383239020679330
- DebKPratapAAgarwalSMeyarivanTA Fast Elitist Multi-Objective Genetic Algorithm: NSGA-IIIEEE Transactions on Evolutionary Computation20006182197
- GoldbergDEGenetic Algorithms in Search, Optimization and Machine learningNew York, NYAddison-Wesley1987
- Griffiths-JonesSBatemanAMarshallMKhannaAEddySRfam: an RNA family databaseNucleic Acids Res20033143944112520045