Abstract
Farnesyltransferase inhibitors (FTIs) are a new class of biologically active anticancer drugs. The exact anti-tumorigenic mechanism is currently unknown. FTIs inhibit farnesylation of a wide range of target proteins. In preclinical models, tipifarnib (R115777, Zarnestra®), a non-peptidomimetic competitive FTI, showed great potency against leukemic cells. Although it has recently demonstrated clinical responses in adults with refractory and relapsed acute myeloid leukemia (AML), and in older adults with newly diagnosed poor-risk AML, its activity was far less than anticipated. However, it appears that tipifarnib as a single agent may be important in selected groups of patients. Much remains to be learned to optimize such therapy in patients with AML. To this end, trials that combine tipifarnib with cytotoxics are ongoing.
Introduction
The outcome of therapy for acute myeloid leukemia (AML) has improved over the recent years, mainly in patients of younger age. However, the challenges in this area have remained considerable. AML is primarily a disease of the elderly and this patient population has a very poor prognosis, which is attributed to having disease that is inherently more resistant to current standard cytotoxic agents in relationship with acquired genetic characteristics of the leukemia, and/or relatively poor tolerance of these agents because of comorbidity and reduced tolerance of adverse effects. The unmet therapeutic need is therefore greatest among patients with AML of older age, in whom response rates are comparatively low (50% for those older than 60 years old), relapse rates are exceedingly high (more than 85%), and long-term survival rates are less than 10% (CitationLöwenberg et al 1999; CitationRowe 2000).
The traditional chemotherapeutic approach to the patient with AML has been based on treatment with a combination of an anthracycline (or anthracenedione) with cytarabine. New drugs are currently in early clinical development with the aim of circumventing chemotherapy resistance. Biological insight into the mechanisms of defective molecular pathways in malignant cells has resulted in the identification of novel targets for drug development. Farnesyltransferase inhibitors (FTIs) represent a new class of signaling inhibitors that may inhibit critical growth and survival signals. These agents are competitive inhibitors of intracellular farnesyl protein transferase (FTase), an enzyme that catalyzes the transfer of a farnesyl moiety to the cysteine terminal residue of a substrate protein. A host of intracellular proteins are substrates for prenylation via Ftase. Interrupting the normal prenylation process of these substrate proteins has been shown useful for inhibiting cellular events that are governed by them (CitationEnd 1999).
Four main approaches to blocking FTase have been designed: (i) competition with the farnesyl pyrophosphate (FPP) group using synthetic analogs, (ii) competition with the target protein or its CAAX binding site or both using peptides (peptidomimetics), (iii) competition with FPP and CAAX using analogs that combine features of both the FPP analogs and peptidomimetics, and (iv) competition with protein/CAAX using nonpeptide analogs (Rowansky et al 1999).
At least six FTIs have been tested in clinical trials, including BMS-214662 (Bristol-Myers Squibb, Princeton, NJ, http://www.bms.com), L778123 (Merck and Co., Inc., Whitehouse Station, NJ, http://www.merck.com), lonafarnib (experimental name, SCH66336; Sarasar™; Schering-Plough Corporation, Kenilworth, NJ, http://www.sch-plough.com), FTI-277 (Calbiochem, EMD Bio-Sciences, San Diego, http://www.emdbiosciences.com), L744832 (Biomol International L.P., Plymouth Meeting, PA, http://www.biomol.com), and tipifarnib (experimental name, R115777; Zarnestra®; Ortho Biotech Products, L.P., Bridgewater, NJ, http://www.orthobiotech.com), which is the most advanced FTI in clinical development (CitationBrunner et al 2003).
This review outlines the characteristics of tipifarnib and its potential mechanisms of action, and describes the early results with this FTI in the treatment of AML.
Tipifarnib
Tipifarnib belongs to the nonpeptidomimetic FTIs. It is a 4,6-disubstituted-1-methylquinolin-2-one derivative that was obtained by optimization of a quinolone lead identified from compound library screening (Venet et al 2003) (). Tipifarnib is synthesized by the condensation of the anion of 1-methylimidazole with a 6-(4-chlorobenzoyl) quinolone derivative, followed by dehydration. The quinolone intermediate was prepared in four steps by cyclization of N-phenyl-3-(3-chlorophenyl)-2-propenamide, acylation, oxidation and N-methylation. Tipifarnib was identified from Janssen’s ketoconazole and retinoic acid catabolism programs as a key structural feature into Ras prenylation process. Tipifarnib is a potent inhibitor of FTase in vitro and is orally active in a variety of animal models (CitationEnd et al 2001). Tipifarnib was the first FTI tested in a clinical trial. It is reasonably well tolerated in man and requires twice-daily dosing to obtain effective plasma concentrations (CitationKurzrock et al 2004). Phase I studies showed that myelosuppression and neurotoxicity were dose-limiting toxicities. Gastrointestinal toxicities and fatigue were observed as well (CitationZujewski et al 2000; CitationCrul et al 2002).
Figure 1 Structure of tipifarnib: R115777(R)-6-amino[(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolone. Tipifarnib is a farnesyltransferase inhibitor developed by Johnson & Johnson Pharmaceutical Research and Development LLC. It is a nonpeptidomimetic oral quinolone analog of imidazole-containing heterocyclic compounds. It competitively inhibits the CAAX binding site of farnesyl transferase. The imidazole group is the central pharmacophore and the imidazole may interact with the coordination structure of the zinc catalytic site.
![Figure 1 Structure of tipifarnib: R115777(R)-6-amino[(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolone. Tipifarnib is a farnesyltransferase inhibitor developed by Johnson & Johnson Pharmaceutical Research and Development LLC. It is a nonpeptidomimetic oral quinolone analog of imidazole-containing heterocyclic compounds. It competitively inhibits the CAAX binding site of farnesyl transferase. The imidazole group is the central pharmacophore and the imidazole may interact with the coordination structure of the zinc catalytic site.](/cms/asset/0462d266-460a-48ff-af6f-aa94d0cd5c9f/dbtt_a_12160303_f0001_b.jpg)
Possible mechanisms of tipifarnib biological activity
FTIs were initially developed to specifically inhibit the activity of oncogenic ras in tumor cells by inhibiting the farnesylation of Ras. Evidence pointing to the importance of ras in myeloid leukemogenesis emanates from an in vivo model, in which irradiated mice, reconstituted with bone marrow transfected with activated N-ras, developed AML and myelodysplastic syndrome (CitationMackenzie et al 1999). At present, the mechanisms behind the anti-tumorigenic effects of FTIs appear complex. The role of ras inhibition in the antitumor activity of tipifarnib is a topic of debate, and other farnesylated targets have been identified.
The Ras protein
The family of ras genes consists of three functional genes, H-ras, K-ras, and N-ras. These genes are highly homologous and encode for four 21-kDa proteins: H-Ras, the splice variants Ki4A-Ras and Ki4B-Ras, and N-Ras, respectively. The N-ras gene is predominantly mutated in AML (30%) (CitationBos 1989).
Ras proteins are GTPases that play a central role in growth signal transduction pathways. Following isoprenylation in the cytosol, the Ras protein migrates to the cell membrane, where it is capable of activating downstream signaling events (CitationEnd et al 1999; CitationKhosravi-Far et al 1992; CitationCox et al 2001). Ras proteins contain 188 or 189 amino acids and exhibit high sequence homology, with the first 86 amino acids being identical, the next 78 having 79% homology, and the following 25 amino acids being highly variable. The final four amino acids play an important role in specifying subcellular localization of the Ras protein. All Ras proteins have a specific amino acid sequence motif at the COOH-terminal region, commonly referred to as the CAAX box, in which C represents a cysteine residue; AA represent aliphatic amino acids, usually valine, leucine, or isoleucine; and X is either methionine or serine. The membrane-targeting domain contains cysteine palmitoylation sites in H-Ras, N-Ras, and K-Ras4A or a polylysine domain in K-Ras4B. Palmitoylation or the presence of a polybasic domain is essential for efficient transportation to the plasma membrane. The transfer of a farnesyl group is mediated by the enzyme FTase, whereas transfer of a geranyl group is mediated by geranylgeranyl transferase (). Ras protein isoforms differ in their affinity for specific isoprenyl groups. K-Ras is a higher affinity substrate for Ftase than H-Ras (CitationZhang et al 1997). Both N-Ras and K-Ras, but not H-Ras, are weak substrates for GGTase-1. In cell culture, FTIs prevent H-Ras farnesylation. In contrast K- and N-Ras are alternatively prenylated by GGTase-1 in FTI-treated cells (CitationWhyte et al 1997). While membrane bound and upon binding an active deoxyguanosine triphosphate (GTP), Ras transduces the signal to various effector proteins (). Subsequently, it becomes inactivated through conversion of GTP to an inactive guanosine diphosphate (GDP) by an intrinsic GTPase (CitationBoguski and McCormick 1993). A point mutation in codon 12, 13, or 61 of the ras gene leads to insensitivity of Ras to the GTPase-activating protein (GAP) and a significantly lower GTPase activity, resulting in deranged or aberrant signal transduction (CitationLowy and Willumsen 1993).
Figure 2 Ftase catalyses the farnesylation step by recognizing the CAAX motif of the Ras C-terminus and transferring a 15-carbon farnesyl isoprenoid from farnesyl diphosphate to form a thioether bond with the Ras cysteine. In another principal prenylation reaction, protein geranylgeranyl transferases transfer either one or two 20-carbon geranylgeranyl isoprenoids from geranylgeranyl diphosphate to proteins (Rowinski et al 1999).
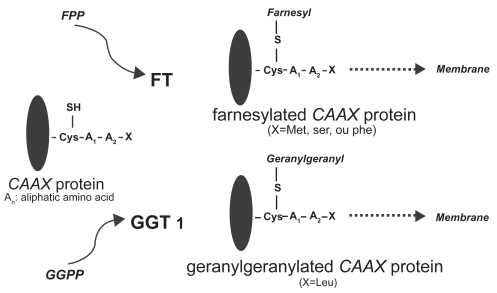
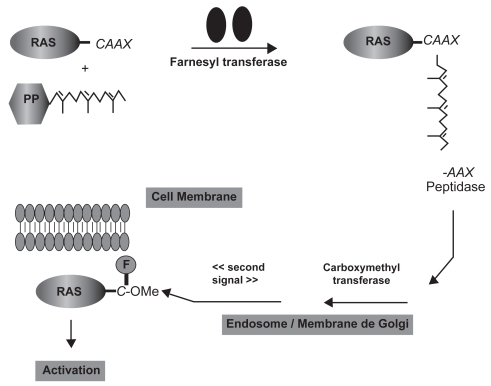
Figure 3 Ras processing and targeting to the plasma membrane. The cytosolic FTase catalyzes the covalent addition of farnesol from farnesylpyrophosphate (FPP) to the cysteine residue of the carboxyl terminal CAAX tetrapeptide sequence (where C is a cysteine residue, A an aliphatic amino acid, and X either methionine or serine). In the endosome/Golgi membranes, transferase enzymes catalyze the removal of the AAX residues and the methylation of the resulting farnesyl-cysteine residue. A “second signal” is required to complete the translocation of Ras from endosomal membranes to the plasma membrane (CitationCox et al 2001).
The pathways that are controlled by activated Ras are designed to prolong cell survival and promote cell proliferation ().
Figure 4 Simplified scheme of Ras activation. Ras proteins are activated by tyrosine kinase receptors as well as cytokine receptors. Once activated, GTP-bound Ras binds to effector molecules such as Raf kinase, Ral-GEF and PI3K. Ras signaling through Raf leads to sequential activation of MEK and ERK, resulting in cellular proliferation, differentiation and cell cycle progression. Ras activation of Ral-GEF causes activation of Rho, which induces stress fiber formation and actin polymerization/depolymerization. Activation of PI3K recruits PDK1/2 and AKT to the plasma membrane, resulting in activation of transcription factors, activation of glycogen synthetase, increased cell survival, and entry into the cell cycle by activating cyclin D1 (CitationMorgan et al 2003).
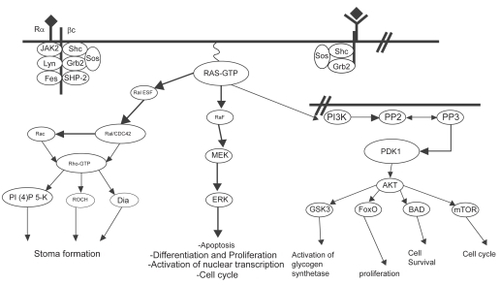
One key pathway is the Raf-MEK-MAPK cascade. The Raf protein is a serine-threonine protein kinase that binds Ras to become active. Raf is able to initiate a downstream cascade of phosphorylation, ultimately resulting in the phosphorylation of mitogen-activated protein kinase (MAPK) also known as extracellular signal-regulated kinase (ERK), localized in the nucleus where it activates transcription factors involved in the control of cellular proliferation and apoptosis (CitationDavies 1995; CitationBonni et al 1999). Transcription-independent mechanisms may also account for the pro-survival effect of this pathway via phosphorylation and subsequent inactivation of the proapoptotic BAD (Bcl-2-associated death promotor) protein. MAPK may be overexpressed or constitutively activated in hematopoietic malignancies, and may be a viable indirect target for FTIs.
The PI3K/AKT pathway occupies a critical position in the transduction of signals beginning with groth-stimulating cytokines and ending with cell proliferation and survival (CitationBurgering and Coffer 1995; CitationKlippel et al 1998). One trigger for this pathway is activated Ras, which interacts with PI3K in the phosphorylation and activation of the serine-threonine kinase AKT, which phosphorylates substrates involved in cell proliferation and survival following DNA damage or other cellular stresses. Vascular endothelial growth factor (VEGF), a pivotal growth and survival factor for leukemias, is also produced by the Ras-dependent AKT pathway (CitationMazure et al 1997). Other substrates, including those involved in cell cycle regulation and DNA repair, are inhibited as a result of AKT phosphorylation.
The Rho proteins are Ras-related GTP-binding proteins that coordinate growth factor-induced assembly of intracellular focal adhesions and actin stress fiber formation (CitationRidley and Hall 1992). Posttranslational prenylation occurs in these proteins, leading to both farnesylated and geranylgeranylated forms. The RhoB isoform is expressed at highest levels during the S-phase of the cell cycle. This small GTPase is involved in regulation of endosomal traffic (CitationEllis and Mellor 2000). FTI treatment leads to a shift toward geranylgeranylation of RhoB and altered cellular localization and function. As such, Rho may be a relevant target for FTI therapy. The geranylgeranylated form of RhoB accounts for up to 70% of the total RhoB protein in untreated cells. In the presence of FTIs, all of the cellular RhoB is geranylgeranylated (CitationLebowitz et al 1997). Inhibition of RhoB farnesylation may contribute to the antitumor activity of FTIs. Recently, it has been found that the geranylgeranylated form of RhoB inhibits expression from the cyclin B promotor and that under low serum conditions FTI treatment reduced cyclin B levels, leading to cell cycle arrest and apoptosis (CitationKamasani et al 2004).
Alternative targets
While it is becoming clear that FTIs only partly target Ras, how these agents selectively target leukemic cells has emerged as an important question. In the proteome, there are close to 300 proteins with a CAAX motif that are potentially farnesylated and over 20 proteins have been proven to be farnesylated (CitationKho et al 2004). Proteins that are not geranylgeranylated in FTI-treated cells are more likely to play a critical role in the biological response to FTIs.
CENP-E and CENP-F
CENP-E is a centromere-associated kinesin motor protein that functions in microtubule attachment to kinetochores, involved in the segregation of sister chromatids during mitosis. CENP-E is also essential for positioning chromosomes at the metaphase plate (CitationYao et al 1997). CENP-F is a cell cycle-regulated chromosome passenger protein that functions in mitosis (CitationLiao et al 1995). FTIs may interfere with bipolar spindle formation during transition from prophase to metaphase in mitosis. Centromere proteins CENP-E and CENP-F are substrates for FTase but not geranylgeranyl transferase I (CitationAshar et al 2000). This mechanism contributes to the observed enhancement of antineoplasic activity of the combination of tipifarnib with antimicrotubule agents (CitationZhu et al 2005).
Rheb
Another potential target for FTI action include Rheb, a GTPase with two forms Rheb1 and Rheb2. Downstream from AKT in the signaling pathway, the target of rapamycin occupies a central position. AKT can inhibit the tuberous sclerosis TSC1/TSC2 (hamartin/tuberin) complex that turn off Rheb, which can activate the mamalian target of rapamycin/S6 kinase signaling (mTOR signaling). Rheb proteins are farnesylated. Rheb1 and Rheb2 are in vitro substrates for Ftase, but not GGTase-1, and treatment of cells with FTIs completely inhibits prenylation of Rheb. Activation of Rheb stimulates growth and cell cycle progression (CitationAspuria and Tamanoi 2004; CitationGau et al 2005). Inhibition of Rheb farnesylation may play a role in FTI enhancement of the antitumor response to other chemotherapeutics (CitationBasso et al 2005). FTIs lead to a more complete of the Rheb signaling pathway than do mTOR/Raptor inhibitors such as rapamycin. FTIs inhibit signaling downstream of Rheb, including both mTOR/Raptor, which regulates growth, and mTOR/Rictor, which regulates the actin cytoskeleton.
RhoB
Rho proteins are involved in remodeling of actin cytoskeleton and in integrin-mediated cell adhesion: RhoA plays a role in the regulation of actomyosin contractility, RhoB regulates cytokine trafficking, and RhoC may be important in cell locomotion (CitationWheeler and Ridley 2004). RhoB can be prenylated. When farnesylation is blocked by FTIs, a shift of RhoB to RhoB geranylgeranylated occurs (CitationDu and Prendergast 1999; CitationLiu et al 2001). It has been suggested that the short half-life of RhoB corresponds to the rapid morphological reversion observed in some FTI-treated cells (CitationLebowitz et al 1997).
PTP-CAAX/PRL
The PTP-CAAX or PRL family of protein tyrosine phosphatases plays a role in regulating cell growth and mitosis. The PRL family includes three members, all of which are farnesylated proteins (CitationCates et al 1996). Cells ectopically expressing not prenylated PRL display defects in mitosis and cytokinesis characterized by chromosome bridges and lagging chromosomes. Farnesylated PRL is required for proper spindle dynamics. Inhibition of PRL farnesylation may in part account for FTI-induced accumulation of cells in the G2/M phase of the cell cycle (CitationWang et al 2002).
HDJ
Human DnaJ homologs include HDJ2, HSJ1, and HDJ1/Hsp40. These proteins serve as co-chaperones and stimulate the ATPase activity of Hsp70 (CitationNagata et al 1998). They increase the level of Hsp70 in its ADP-bound form that as greater affinity for unfolded polypeptide subtrates. This facilitates the folding of proteins and Hsp70-mediated nuclear trafficking. Although the functional significance of HDJ2 farnesylation remains unclear, HDJ2 prenylation status has been used as a marker for FTase inhibition in clinical trials. Studies suggest that farnesylation may regulate the activity, localization, or complex formation that is required for YDJ1 (the Saccharomyces cerevisiae homolog of HDJ2) function (CitationCaplan et al 1992). There is little work reported to date on the functional consequences of blocking HDJ2 farnesylation in human cells.
Nuclear lamins
The nuclear lamina consists of lamin proteins that are required for nuclear envelope assembly. Lamin B was one of the first proteins shown to be modified by prenylation. Lamin A is also farnesylated. Although the functional role of lamin farnesylation remains unclear, it is though to play a role in targeting prelamin A to the nuclear membrane, where mature lamin A is released by the action of a protease (CitationSinensky et al 1994). The accumulation of unfarnesylated prelamin A can be detected and provides another marker for FTase inhibition. The functional role of lamin farnesylation remains unclear.
Additional farnesylated proteins
A number of other farnesylated proteins have been identified that may contribute to the biological activity of FTIs. These include other small GTPases: RhoD, RhoE, Rho6, Rho7, TC10, all of which have been demonstrated to be substrates for alternative prenylation by GGTase-1 (CitationBasso et al 2006). Additional farnesylated proteins include the GTPase RRP22 and the prostacyclin receptor (CitationElam et al 2005).
Effects on cell cycle progression and on apoptosis
The inhibition of protein farnesylation interrupts the functions of diverse proteins that help to move the cell through its division cycle. FTIs may also impede the farnesylation and function of the kinetochore-binding centromeric proteins (CENPs) E and F, which exert their maximal effets in the G2 and M phases of the cell cycle (CitationAshar et al 2000). FTIs block the growth of a variety of leukemia cell lines both in vitro and when grown as xenografts in vivo. Tumor growth is inhibited by tipifarnib in these models over a dose range of 25–100 mg/kg twice daily (bid) (CitationEnd et al 2001). FTIs induce G2/M accumulation and G1 arrest (CitationSuzuki et al 1998) in both a p53-dependent and -independent fashion. FTIs are potent activators of apoptosis in Ras-transformed cells if attachment to substratum is prevented. This apoptotic response is blocked by BCL-XL and inhibited by a myristylated form of RhoB (CitationLebowitz et al 1997b).
Preclinical and biological studies of tipifarnib in leukemia
The preclinical studies of FTI activities against a wide range of tumor cell types have demonstrated that the action of FTIs is not dependent upon the presence of ras mutations. In most preclinical models, FTIs induce tumor growth inhibition, when used as monotherapy (CitationEnd et al 2001; CitationCox and Der 2002; CitationLancet and Karp 2003). Several genes involved in FTI biology were identified as being modulated following treatment with tipifarnib in addition to pathways involved with cytoskeletal organization, cell signaling, immunity, and apoptosis (CitationRaponi et al 2004). Microarray analysis of pre-treatment bone marrow samples from patients with relapsed and refractory AML identified 8 genes that were associated with response to tipifarnib (CitationRaponi et al 2007). The most robust marker was the guanine nucleotide exchange factor AKAP13, which showed increased expression in patients who were resistant to tipifarnib.
Clinical trials in acute myeloid leukemia
First trials in advanced cancer
Several single-agent phase I studies with tipifarnib have been reported. In an initial study, tipifarnib was dosed orally bid. for 5 days every two weeks. The maximum tolerated dose with this schedule was identified as 500 mg bid. Adverse events included nausea, vomiting, and fatigue. Myelosuppression was mild (CitationZujewski et al 2000). Another study explored a more prolonged schedule of 28 days of treatment followed by 1–2 weeks of rest. The maximum tolerated dose was 300 mg bid. and myelosuppression was dose-limiting (CitationPunt et al 2001). A third study used tipifarnib at 300 mg bid. as continuous treatment. Myelosuppression and neurotoxicity were dose-limiting, but one partial response was observed (CitationCrul et al 2002).
Phase I and phase II studies with tipifarnib as a single agent in AML
The most promising clinical results with tipifarnib have been observed in hematologic malignancies, including AML. In a phase I study of 35 adults with refractory or relapsed acute leukemia, 10 patients responded including 2 with complete responses. Patients were treated for 21 days, and dose limiting toxicities, including neurotoxicity, nausea and myelosuppression, were observed at the 1,200 mg bid. dose (CitationKarp et al 2001). Responses were independent of ras mutational status. The optimal inhibition of protein farnesylation was obtained at 300 mg bid. for 21 days of a 28 day cycle (CitationZimmerman et al 2004). Clinical activity was confirmed in myelodysplastic syndromes (MDS), a pre-leukemic disease. In 21 patients with MDS, tipifarnib given at 300 mg bid. (3 weeks on/1 week off) resulted in one complete response, 2 partial responses, and 3 hematologic improvements (CitationKurzrock et al 2003). Responses were also observed when using a 4 week on/2 week off schedule (CitationKurzrock et al 2004). Intermittent dosing (bid. on alternative weeks) has also been shown more effective, in that higher doses (600 mg) can be given without toxicities (CitationLara et al 2005). In the presence of poor risk features, including older age, unfavorable cytogenetics, antecedents of MDS, and properties of multidrug resistance, the likelihood of disease-free survival (DFS) at one year is less than 20% (CitationLeith et al 1997; CitationLancet et al 2000). In a phase 2 study, 82 patients with poor-risk MDS received tipifarnib (300 mg twice daily) for the first 21 days of each 28-day cycle. Twenty-six patients (32%) responded and 37 (45%) had stable disease. The median response duration was 11.5 months, and the median time to progression was 12.4 months (CitationFenaux et al 2007).
In patients older than 75 years, survival was in the order of 3 to 4 months, clearly highlighting the need for novel therapies in this group of patients (CitationAppelbaum et al 2006; CitationKantarjian et al 2006). Activity has been reported with single-agent tipifarnib in poor-risk AML patients (CitationLancet et al 2007). In this study, a majority of patients had antecedents of MDS. Complete remission was achieved in 22 patients (14%) and partial remission or hematologic improvement occurred in 15 patients, for an overall response rate of 23%. Achievement of CR appeared to impart a major survival benefit. The median of DFS was 7.3 months and the median survival of complete responders was 18 months (). Tipifarnib was relatively well tolerated. Drug-related nonhematologic serious adverse events were observed in 47% of cases. The more frequently encountered were infection, gastrointestinal disturbances, renal insufficiency, and skin rash. Neurologic toxicity was rare and non-disease-related early mortality rate was only 7%, that compares favorably with death rates observed with induction chemotherapy in the elderly. Furthermore, because tipifarnib was administered orally, the median number of days spent in the hospital was low. Adverse karyotype and poor performance status correlated negatively with survival. Inhibition of farnesylation of the surrogate protein HDJ-2 occurred in a large majority of cases. Baseline levels of phosphorylated mitogen-activated protein kinase and AKT did not correlate with clinical response.
Table 1 Studies of phase I and II with tipifarnib as a single agent in acute myeloid leukemia and myelodysplastic syndrome
Another large study assessed the efficacy of tipifarnib as single-agent but in refractory or relapsing AML (CitationHarousseau et al 2007) and confirmed the antileukemic activity initially observed in the phase I setting (CitationKarp et al 2001). In this study, only 11 of 252 patients (4%) achieved complete remission (9 patients) or complete remission with incomplete platelet recovery (2 patients). Nineteen patients (8%), including those who achieved complete remission, achieved a reduction in bone marrow blasts to less than 5%. Bone marrow blasts were reduced by 50% in an additional 8 patients (CitationHarousseau et al 2007) (). Despite a disappointingly low response rate, it was especially interesting to note the observation of complete remissions in cases presenting unfavorable cytogenetics. Myelosuppression was the most common adverse event. No new side effects related to tipifarnib treatment were identified. Treatment-related mortality was lower than that associated with standard induction chemotherapy. Results from this study suggest the consideration of combining tipifarnib with other antileukemic therapies.
Preclinical work showed that farnesyltransferase remained inhibited for seven days after tipifarnib, suggesting an alternate week dosing schedule. In a phase I dose escalation trial in AML on a week on week off schedule, 30 patients were accrued (CitationKirschbaum et al 2006). It was showed that greater than two fold increase in tipifarnib dosing can be tolerated on this dosing schedule with enhanced efficacy. Similar results were observed in MDS (CitationKurzrock et al 2006).
A recent study has shown that the median overall survival of older patients treated with tipifarnib was in fact longer than that of patients treated with the combination of idarubicin with cytarabine or idarubicin with other agents (CitationEstey et al 2006). Despite these initial encouraging results, in 2005 the Oncology Drugs Advisory Committee rejected the approval of tipifarnib for the treatment of elderly patients with newly diagnosed poor-risk AML, citing an insufficient complete response rate. The relatively low toxicity profile may allow for extended therapy to maintian disease control, quality of life, and survival, even if full response is not achieved. The phase III trial (R115777-AML-301 trial) comparing tipifarnib with best supportive care may provide the necessary confirmatory data for approval. Tipifarnib in maintenance therapy during minimal residual disease has also been explored (R115777-INT-21 trial). Oral tipifarnib (400 mg bid for 2/3 weeks) was begun after start of final consolidation cycle and given for up to 36 cycles to 36 adults with poor risk AML (CitationKarp et al 2006). Tipifarnib was well tolerated. However, dose reductions for myelosupression occurred in 53% and 6% needed platelet transfusions. A total of 15 patients progressed while on tipifarnib at median of 6.5 months from complete remission. There was no negative impact on reinduction chemotherapy at relapse.
Tipifarnib combined with other agents in AML
Trials that combine tipifarnib with other agents, most particularly cytotoxics, are ongoing. Indeed, pre-clinical data suggest that tipifarnib may be synergistic with some chemotherapeutic agents. In a phase I/II study in 74 adults with previously untreated AML or high-risk MDS, tipifarnib (200 mg bid or 300 mg bid for 21 days every 28 days) was combined with idarubicin (12 mg/m2/day for 3 days) and cytarabine (1.5 mg/m2/day for 4 days) (CitationAlvarez et al 2006). A high rate of response was observed in AML (77%; 65% achieved complete remission and 12% complete remission with incomplete platelet recovery), with, however, an increased incidence of diarrhea and hyperbilirubinemia. Response by cytogenetics was 86% for diploid, 76% for monosomy 5 or 7, and 68% for other abnormalities. Patients achieving complete remission received 5 courses of consolidation with idarubicin (8 mg/m2/day for 2 days), cytarabine (0.75 mg/m2/day for 3 days), and tipifarnib (300 mg bid for 14 days) every 4–6 weeks. Maintenance was with tipifarnib 300 mg bid for 21 days every 4–6 weeks for 6 months.
In another phase I trial tipifarnib was combined with oral etoposide (CitationKarp et al 2006b). This was conducted in an attempt to increase complete remission rates in elderly AML patients aged more than 70. Both tipifarnib and etoposide were given with escalating doses (300 mg to 600 mg bid for tipifarnib, and 100 mg to 200 mg/day for 6 days for etoposide) and 14 versus 21 days of tipifarnib every 28–63 days. While 43% of patients required hospitalization during the first cycle for a median of 7 days, overall hospitalization rate for all cycles was 28%. Complete remission was achieved in 21% of cases and partial remission or hematologic improvement in 20%. All patients who achieved complete remission have done so within two cycles. The oral regimen was therefore tolerable and feasible on an outpatient basis, with the suggestion of improvement of response rates over tipifarnib alone. Direct comparisons with chemotherapy alone are warranted in randomized trials.
Conclusion
FTIs exhibit encouraging signs of clinical activity in patients with AML. However, standard response criteria, which have proven valuable in the clinical development of cytotoxic agents, cannot be applied to FTIs, as with other new classes of targeted signal transduction inhibitors. This suggests that aggressive disease may not be appropriate settings to explore activity of FTIs. Greater activity is anticipated in the maintenance setting. New settings in which single-agent FTI therapy should be investigated include previously untreated AML and minimal residual disease. AML patients who are older than age 60 experience extremely poor long-term outcomes. New targeted agents like the FTIs offer the potential of increased therapeutic index, an important consideration in older patients with AML. One asset of this class of agents for this patient population is a toxicity profile that is very acceptable. FTIs might be administered on a chronic dosing schedule, either alone or in combination with other pharmacologic compounds, with the aim of maintaining the underlying disease in a clinically controlled state. On another front, FTIs seem optimal as postremission therapy for AML in elderly patients or those with other poor-risk features. Although there are limited data to suggest synergistic or additive effects, the combination of FTIs with chemotherapy represents another avenue of exploration.
Based on promising preliminary evidence of tolerability, biologic activity, and clinical response in some patients with AML, further studies with tipifarnib (as with other agent in this class) are warranted. The challenge will be to define the use of these new agents – when in the course of disease they should be administered, and in what combinations with other therapies. In this setting, novel agents with nonoverlapping mechanisms of action are to be combined and represent a potential step toward increasing favorable results in AML. A related challenge remains identifying a sensitive sub-type of leukemia based on a specific genetic profile. A common set of genes that were regulated by tipifarnib have been found. Expression of these candidate genes might be used as surrogate markers of drug activity. In addition, recent work has identified genes that may be predictive of resistance or response to tipifarnib. While these markers need to be further validated, their identification represent an important advance in the ability to stratify patients based on their likelihood of response.
References
- AlvarezRHKantarjianHGarcia-ManeroG2006Farnesyl transferase inhibitor (tipifarnib, Zarnestra; Z) in combination with standard chemotherapy with idarubicin (Ida) and cytarabine (ara-C) for patients (pts) with newly diagnosed acute myeloid leukemia (AML) or high-risk myelodysplastic syndrome (MDS)Blood108565a566a
- AppelbaumFRGundackerHHeadDR2006Age and acute myeloid leukemiaBlood1073481516455952
- AsharHRJamesLGrayK2000Transferase inhibitors block the farnesylation of CENP-E and CENP-F and alter the association of CENP-E with the microtubulesJ Biol Chem27530451710852915
- AspuriaPJTamanoiF2004The Rheb family of GTP-binding proteinsCell signal1611051215240005
- BassoADMirzaALiuG2005The farnesyl transferase inhibitor (FTI) SCH66336 (lonafarnib) inhibits Rheb farnesylation and mTOR signaling. Role in FTI enhancement of taxane and tamoxifen anti-tumor activityJ Biol Chem28031101816006564
- BassoADKirschmeierPBishopWR2006Farnesyl transferase inhibitorsJ Lipid Res47153116278491
- BoguskiMSMcCormickF1993Proteins regulating Ras and its relativesNature366643548259209
- BonniABrunetAWestAE1999Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanismsScience28613586210558990
- BosJL1989Ras oncogenes in human cancer: a reviewCancer Res49468292547513
- BrunnerTBHahnSMGuptaAK2003Farnesyltransferase inhibitors: An overview of the results of preclinical and clinical investigationsCancer Res6356566814522880
- BurgeringBMCofferPJ1995Protein kinase B (c-Akt) in phosphatidylinositol-3-kinase signal transductionNature3765996027637810
- CaplanAJCyrDMDouglasMG1992YDJ1p facilitates polypeptide translocation across different intracellular membranes by a conversed mechanismCell711143551473150
- CatesCAMichaelRLStayrookKR1996Prenylation of oncogenic human PTP (CAAX) protein tyrosine phosphatasesCancer Lett11049559018080
- CoxADToussaintLGIIIFiordalisiJJSebtiSMHamiltonAD2001Farnesyltransferase and geranylgeranyltransferase inhibitors – The saga continuesFarnesyltransferase inhibitors in cancer therapyTotowa, NJHumana Press Inc25573
- CoxADDerCJ2002Farnesyltransferase inhibitors: promises and realitiesCurr Opin Pharmacol23889312127871
- CrulMde KlerkGJSwartM2002Phase I clinical and pharmacologic study of chronic oral administration of the farnesyl protein transferase inhibitor R115777 in advanced cancerJ Clin Oncol2027263512039935
- DaviesRJ1995Transcriptional regulation by MAP kinasesMol Reprod Dev42459678607977
- DuWPrendergastGC1999Geranylgeranylated RhoB mediates suppression of human tumor cell growth by farnesyltransferase inhibitorsCancer Res595492610554025
- ElamCHessonLVosMD2005RRP22 is a farnesylated, nucleolar, Ras-related protein with tumor suppressor potentialCancer Res6531172515833841
- EllisSMellorH2000Regulation of endocytic traffic by Rho family GTPasesTrends Cell Biol1085810675900
- EndDW1999Farnesyl protein transferase inhibitors and other therapies targeting the Ras signal transduction pathwayInvest New Drugs172415810665477
- EndDWSmetsGToddAV2001Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitroCancer Res61131711196150
- EsteyEHThallPFWangXM2006Comparison of survival times after administration of tipifarnib or Ara-C-containing therapy, to older patients with newly-diagnosed acute myeloid leukemiaBlood108217b
- FenauxPRazaAMuftiGJ2007A multicenter phase 2 study of the farnesyltransferase inhibitor tipifarnib in intermediate- to high-risk myelodysplastic syndromeBlood10941586317264294
- GauCLKato-StankiewiczJJiangC2005Farnesyl-transferase inhibitors reverse altered growth and distribution of actin filaments in Tsc-deficient cells via inhibition of both rapamycin-sensitive and -insensitive pathwaysMol Cancer Ther49182615956249
- HarousseauJLLancetJEReiffersJ2007A phase 2 study of the oral farnesyltransferase inhibitor tipifarnib in patients with refractory or relapsed acute myeloid leukemiaBlood1095151617351110
- KamasaniUHuangMDuhadawayJB2004Cyclin B1 is a critical target of RhoB in the cell suicide program triggered by farnesyl transferase inhibitionCancer Res6483899615548709
- KantarjianHO’BrienSCortesJ2006Results of intensive chemotherapy in 998 patients aged 65 and older with acute myeloid leukemia or high-risk myelodysplastic syndrome: predictive prognostic models for outcomeCancer1061090816435386
- KarpJELancetJEKaufmannSH2001Clinical and biological activity of the farnesyltranferase inhibitor R115777 in adults with refractory and relapsed acute leukemias: a phase I clinical-laboratory correlative trialBlood973361911369625
- KarpJEGojoIGreerJ2006Tipifarnib (Zarnestra, R115777) as maintenance therapy for adults in complete remission (CR) following induction and consolidation therapies for poor-risk acute myelogenous leukemia (AML): a phase II trialBlood108780a781a
- KarpJEFeldmanEJMorrisL2006bActive oral regimen for elderly adults with newly diagnosed acute myelogenous leukemia (AML): phase I trial of oral tipifarnib (T) combined with oral etoposide (E) for adults ≥age 70 who are not candidates for traditional cytotoxic chemotherapy (TCC)Blood108130a
- KhoYKimSCJiangC2004A tagging-via-substrate technology for detection and proteomics of farnesylated proteinsProc Natl Acad Sci USA101124798415308774
- Khosravi-FarRCoxADKatoK1992Protein prenylation: key to ras function and cancer interventionCell Growth Differ346191419908
- KirschbaumMSelwyn SteinATuscanoJ2006A phase I study of the farnesyltransferase inhibitor tipifarnib in a week-on week-off dose schedule in acute myelogenous leukemiaBlood108551a16537813
- KlippelAEscobedoMAWachowiczMS1998Activation of phosphatidylinositol-3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformationMol Cell Biol1856997119742087
- KurzrockRKantarjianHMCortesJE2003Farnesyltransferase inhibitor R115777 in myelodysplastic syndrome: clinical and biologic activities in the phase I settingBlood10245273412947010
- KurzrockRAlbitarMCortesJE2004Phase II study of R115777, a farnesyl transferase inhibitor, in myelodysplastic syndromeJ Clin Oncol2212879215051776
- KurzrockRVerstovsekSWrightJJ2006Alternate week administration of the farnesyltransferase inhibitor tipifarnib (ZARNESTRA®, R115777) in patients with myelodysplastic syndrome: results of a phase I studyBlood108708a
- LancetJEWillmanCLBennettJM2000Acute myelogenous leukemia and aging: clinical interactionsHematol Oncol Clin North Am142516710680081
- LancetJEKarpJE2003Farnesyltransferase inhibitors in hematologic malignancies: new horizons in therapyBlood1023880912920034
- LancetJEGojoIGotlibJ2007A phase 2 study of the farnesyltransferase inhibitor tipifarnib in poor-risk and elderly patients with previously untreated acute myelogenous leukemiaBlood10913879417082323
- LaraPNLawLYJrWrightJJ2005Intermittent dosing of the farnesyl transferase inhibitor tipifarnib (R115777) in advanced malignant solid tumors: a phase I California Cancer Consortium trialAnticancer Drugs163172115711184
- LebowitzPFCaseyPJPrendergastGC1997Farnesyltransferase inhibitors alter the prenylation and growth-stimulating function of RhoBJ Biol Chem2721559149188444
- LebowitzPFSakamuroDPrendergastGC1997bFarnesyl transferase inhibitors induce apoptosis of Ras-transformed cells denied substratum attachmentCancer Res57708139044849
- LeithCPKopeckyKJGodwinJ1997Acute myeloid leukemia in the elderly: assessment of multidrug resistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably distinct responses to standard chemotherapy. A Southwest Oncology Group studyBlood89332399129038
- LiaoHWinkfeinRJMackG1995CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosisJ Cell Biol130507187542657
- LiuAXRaneNLiuJP2001RhoB is dispensable for mouse development, but it modifies susceptibility to tumor formation as well as cell adhesion and growth factor signaling in transformed cellsMol Cell Biol2169061211564874
- LöwenbergBDowningJRBurnettA1999Acute myeloid leukemiaN Engl J Med41105162
- LowyDRWillumsenBM1993Function and regulation of rasAnnu Rev Biochem62851918352603
- MacKenzieKLDolnikovAMillingtonM1999Mutant N-ras induces myeloproliferative disorders and apoptosis in bone marrow repopulated miceBlood9320435610068678
- MazureNMChenEYLaderouteKR1997Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol-3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional elementBlood903322319345014
- MorganMAGanserAReuterCWM2003Therapeutic efficacy of prenylation inhibitors in the treatment of myeloid leukemiaLeukemia1714829812886235
- NagataHHansenWJFreemanB1998Mammalian cytosolic DnaJ homologues affect the hsp70 chaperone-substrate reaction cycle, but do not interact directly with nascent or newly synthesized proteinsBiochemistry376924389578579
- PuntCJvan MaanenLBolCJ2001Phase I and pharmacokinetic study of the orally administered farnesyl transferase inhibitor R115777 in patients with advanced solid tumorsAnticancer Drugs12193711290865
- RaponiMBellyRTKarpJE2004Microarray analysis reveals genetic pathways modulated by tipifarnib in acute myeloid leukemiaBMC Cancer4566815329151
- RaponiMHarousseauJLLancetJE2007Identification of molecular predictors of response in a study of tipifarnib treatment in relapsed and refractory acute myelogenous leukemiaClin Cancer Res1322546017404110
- RidleyAJHallA1992The small Gtp-binding protein Rho regulates the assembly of focal adhesions and action stress fibers in response to growth factorsCell70389991643657
- RowinskyEKWindleJJVon HoffDD1999Ras protein farnesyl-transferase: A strategic target for anticancer therapeutic developmentJ Clin Oncol1736315210550163
- RoweJM2000Treatment of acute myelogenous leukemia in older adultsLeukemia14480710720146
- SinenskyMFantleMKDaltonM1994An antibody which specifically recognizes prelamin A but not mature lamin A: application to detection of blocks in farnesylation-dependent protein processingCancer Res543229328205544
- SuzukiNUranoJTamanoiF1998Franesyltransferase inhibitors induce cytochrome c release and caspase 3 activation preferentially in transformed cellsProc Natl Acad Sci USA9515356619860973
- VenetMEndDAngibaudP1998Farnesyl protein transferase inhibitor ZARNESTRA R115777 – history of a discoveryCurr Top Med Chem3109510212769710
- WangJKirbyCEHerbstR2002The tyrosine phosphatase PRL-1 localizes to the endoplasmic reticulum and the mitotic spindle and is required for normal mitosisJ Biol Chem277466596812235145
- WheelerAPRidleyAJ2004Why three Rho proteins? RhoA, RhoB, RhoC, and cell motilityExp Cell Res30143915501444
- WhyteDBKirschmeierPHockenberryTN1997K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitorsJ Biol Chem27214459649162087
- YaoXAndersonKLClevelandDW1997The microtubule-dependent motor centromere-associated protein E (CENP-E) is an integral component of kinetochore corona fibers that link centromeres to spindle microtubulesJ Cell Biol139435479334346
- ZhangFLKirschmeierPCarrD1997Characterization of Ha-Ras, N-Ras, Ki-Ras4A, and Ki-Ras4B as in vitro substrates for farnesyl protein transferase and geranylgeranyl protein transferase type IJ Biol Chem2721023299092572
- ZhuKGerbinoEBeaupreDM2005Farnesyltransferase inhibitor R115777 (Zarnestra, Tipifarnib) synergizes with paclitaxel to induce apoptosis and mitotic arrest and to inhibit tumor growth of multiple myeloma cellsBlood10547596615728126
- ZimmermanTMHarlinHOdenikeOM2004Dose-ranging pharmacodynamic study of tipifarnib (R115777) in patients with relapsed or refractory hematologic malignanciesJ Clin Oncol2248162215570084
- ZujewskiJHorakIDBolCJ2000Phase I and pharmacokinetic study of farnesyl protein transferase inhibitor R115777 in advanced cancerJ Clin Oncol189274110673536