Abstract
Chronic myeloid leukemia (CML) is a stem cell disease, in which the BCR/ABL oncoprotein is considered essential for abnormal growth and accumulation of neoplastic cells. During the past 10 years, the BCR/ABL tyrosine kinase inhibitor imatinib (STI571) has successfully been introduced in the treatment of the disease. However, intrinsic as well as acquired resistance against the drug have been described and have been recognized as an emerging problem and challenge in clinical practice, and a key issue in CML research. Most of the respective concepts focus on imatinib-resistant mutants of BCR/ABL that are detectable in a high proportion of cases. However, other factors also contribute to resistance against imatinib, including the genetic background, the biologic features of CML stem cells, gene amplifications, silencing of tumor suppressor genes, and various pharmacologic aspects. In this article, the mechanisms of resistance against imatinib and other BCR/ABL tyrosine kinase inhibitors in CML are discussed together with strategies to overcome and to prevent resistance with available drugs or with novel antileukemic approaches.
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disease characterized by the t(9; 22) and the related oncogene, BCR/ABL (CitationNowell and Hungerford 1960; CitationRowley 1973; Citationde Klein et al 1982). The respective fusion gene product, BCR/ABL, is a cytoplasmic 210 kDa protein that is considered essential for growth and survival of leukemic cells (CitationDaley et al 1990; CitationLugo et al 1990; CitationGishizky and Witte 1992; CitationWetzler et al 1993; CitationBiernaux et al 1995; CitationRen 2005). BCR/ABL displays constitutive tyrosine kinase (TK) activity and triggers a number of downstream signalling molecules including phosphoinositide 3-kinase (PI3K), mitogen-activated protein (MAP) kinase, nuclear factor-κB (NFκB), RAS, and signal transducer of activation and transcription 5 (STAT5) (CitationPendergast et al 1993; CitationPuil et al 1994; CitationSkorski et al 1997; CitationSillaber et al 2000; CitationSattler and Griffin 2003; CitationMelo and Deininger 2004; CitationVan Etten 2007). These signalling molecules and pathways supposedly act together to promote malignant transformation, to enhance genetic instability, and to suppress apoptosis in leukemic cells (CitationHoover et al 2001; CitationMelo and Deininger 2004; CitationVan Etten 2007).
The (natural) clinical course in CML can be divided into a chronic (early) phase (CP), in which cellular differentiation and maturation are largely preserved, an accelerated phase (AP) of the disease, and a terminal (=blast) phase of CML (CML-BP), which resembles acute leukemia (CitationCortes and Kantarjian 2003; CitationGiles et al 2004; CitationCortes et al 2006). In addition, based on the detection of BCR/ABL in apparently healthy subjects, a prephase of CML (with normal leukocyte counts), in which clonal BCR/ABL+ stem cells expand and generate subclones (CitationBiernaux et al 1995; CitationBose et al 1998), has been postulated (). What hits drive BCR/ABL-positive cells (subclones) from a prephase into overt CML, remains at present unknown. It also remains uncertain whether a ‘pre-BCR/ABL-phase’ of CML exists, in which monoclonal but preleukemic stem cell clones develop and expand to provide a suitable cellular background for the establishment of a BCR/ABL+ clone (). This hypothesis has been based on rare cases of BCR/ABL-negative but apparently monoclonal populations of leukemic cells (subclones) that may develop in CML patients during treatment with imatinib. All in all, BCR/ABL is considered a most critical factor, but may per se not be sufficient for disease-initiation. Also, whereas in CP, BCR/ABL is considered to play a predominant role for leukemia cell survival, additional pro-oncogenic molecules and pathways may become (more) important and contribute to malignant growth and thus disease-progression in advanced CML (AP, BP) (CitationShet et al 2002; CitationSattler and Griffin 2003; CitationCalabretta and Perrotti 2004; CitationMelo and Barnes 2007) ().
Figure 1 Evolution of CML with prephases—a proposed hypothesis.
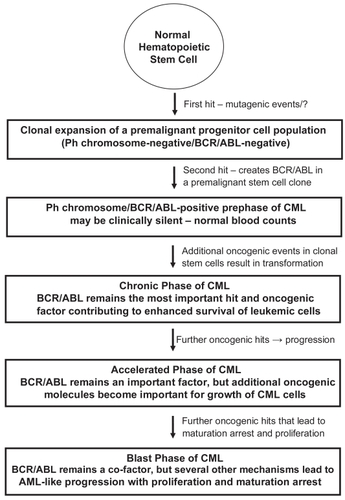
The leukemic clone in CML is organized hierarchically, with more mature cells that have a limited capacity to divide and to survive, and cells with unlimited capacity to divide and to self-renew, so-called leukemic stem cells (CitationEaves et al 1993, Citation1998; CitationHolyoake et al 2000, Citation2001; CitationEisterer et al 2005; CitationElrick et al 2005). Taking this concept into consideration, it seems clear that the clinically relevant portion of MRD and any resulting relapse derives from CML stem cells, and that therapy is curative only when eradicating these cells. During disease evolution and probably even before overt disease is diagnosed (prephase of CML), CML stem cells may acquire multiple (transforming) hits, resulting in subclone-formation (CitationHolyoake et al 2002; CitationJiang et al 2007a). Therefore, the CML clone supposedly is composed of several different subclones at diagnosis in most (if not all) patients, a hypothesis that explains the ‘occurrence’ of drug-resistant BCR/ABL-mutants during therapy through subclone-selection (CitationRoche-Lestienne et al 2002; CitationJiang et al 2007a). An unresolved question is why wild type (wt) BCR/ABL-bearing cells have a growth advantage over subclones exhibiting BCR/ABL-mutants. In fact, in most patients, the mutant subclone is only detectable after initiation of BCR/ABL-targeting therapy. A related question is how the disease can suppress growth of normal hematopoietic stem cells. Here, one hypothesis is, that stem cell-derived negative growth-regulators (chalones) such as lipocalin, suppress growth of normal (stem) cells through a specific receptor, whereas CML stem cells are resistant, as they display only low amounts or lack lipocalin-binding sites (CitationDevireddy et al 2005; CitationLin et al 2005). Whether mutant BCR/ABL-bearing subclones are also suppressed by leukemic cells displaying wt BCR/ABL through chalone-dependent inhibition or other mechanisms, remains unknown.
The BCR/ABL kinase inhibitor imatinib has successfully been introduced in the treatment of CML. Thus, imatinib induces major cytogenetic responses in a majority of all patients with CP CML (CitationDruker et al 2001a; CitationKantarjian H et al 2002; CitationBarbany et al 2002; CitationO’Brien et al 2003). Responses are also seen in (some) patients with AP or BP (CitationDruker et al 2001b; CitationTalpaz et al 2002; CitationKantarjian et al 2002; CitationSawyers et al 2002). However, despite overwhelming initial data and high expectations, little is known about long-term effects of imatinib (CitationDruker et al 2006). An apparent result from follow up studies is that imatinib is unable to eradicate all neoplastic stem cells (subclones) in CML. Rather, many patients develop overt resistance against imatinib during therapy, which is often associated with the outgrowth of subclones bearing mutations in BCR/ABL (CitationBranford et al 2002, Citation2003; CitationKantarjian et al 2006a; CitationHochhaus et al 2007a). For such patients, treatment options are usually limited. In fact, many of them are in AP or BP, and only a subgroup of them are eligible for stem cell transplantation (SCT).
Therefore, a number of attempts have been made to identify new drugs that act antileukemic in imatinib-resistant CML (CitationShah et al 2004; CitationWeisberg et al 2005, Citation2007b; CitationO’Hare et al 2005; CitationMartinelli et al 2005). Such drugs are directed against BCR/ABL and its mutants, but may also be directed against other (BCR/ABL-independent) molecules that play a role in malignant transformation (CitationMartinelli et al 2005; CitationWeisberg et al 2007b). Thus, molecular resistance against imatinib may not only be caused by changes in BCR/ABL, but also by other pro-oncogenic molecules (CitationKantarjian et al 2006a; CitationHochhaus et al 2007a). Therefore, less specific targeted drugs and combinations of targeted drugs have been proposed, and are currently applied in clinical trials to overcome resistance. Some of the emerging TK inhibitors act on BCR/ABL as well as on other key signalling targets, such as Lyn or/and other Src kinases (CitationShah et al 2004; CitationMartinelli et al 2005; CitationKimura et al 2005; CitationWeisberg et al 2007b).
Apart from molecular resistance against imatinib, other mechanisms that cause resistance in CML, have also been described. First, immature leukemic cells (stem cells) may exhibit intrinsic (BCR/ABL-independent) resistance (CitationBarnes and Melo 2006; CitationJiang et al 2007b). Second, a number of cellular molecules involved in the regulation of drug-uptake, drug-metabolism or drug-efflux, may influence the bio-availability of imatinib (CitationHerweijer et al 1990; CitationIllmer et al 2004; CitationThomas et al 2004; CitationWang et al 2007; CitationBrendel et al 2007). Lastly, more and more data suggest that imatinib is not capable of entering all organ-compartments in vivo. Likewise, imatinib is unable to cross the blood-brain barrier in amounts sufficient to reach a pharmacologic drug concentration in the central nervous system (CNS) (CitationTakayama et al 2002; CitationSenior 2003; CitationDai et al 2003; CitationWolff et al 2003). Correspondingly, CNS relapses are increasingly described in CML patients treated with imatinib (CitationAbruzzese et al 2003; CitationLeis et al 2004; CitationBornhäuser et al 2004; CitationRajappa et al 2004; CitationRytting and Wierda 2004; CitationMatsuda et al 2005; CitationPavlu et al 2005; CitationKim et al 2006; CitationAichberger et al 2007).
In the following sections, various types of resistance against imatinib are discussed together with possibilities to prevent or to overcome resistance with currently available drugs, combination strategies employing drugs and SCT, or future therapeutic approaches such as siRNA or immunotherapies.
Leukemic stem cells exhibit ‘intrinsic resistance’
Most patients in CML CP enter a complete cytogenetic response (CCR) during treatment with imatinib (CitationDruker et al 2001a; CitationKantarjian H et al 2002; CitationBarbany et al 2002; CitationO’Brien et al 2003). In many of these patients, BCR/ABL-transcripts decrease to low or even undetectable levels over time (CitationBarbany et al 2002; CitationHughes et al 2003; CitationDruker et al 2006). However, discontinuation of imatinib is usually followed by a cytogenetic and hematologic relapse (CitationBreccia et al 2006; CitationRousselot et al 2007). Based on this observation and other studies, it has been hypothesized that MRD in imatinib-treated patients contains leukemic stem cells (clinically relevant subclones), and that these residual stem cells exhibit imatinib-resistance (CitationGraham et al 2002; Bhatia et al 2003; CitationMichor et al 2005; CitationGoldman and Gordon 2006; CitationDeininger 2007). A remarkable aspect is that the relapsing subclones that reappear after discontinuation of imatinib usually display wt BCR/ABL. Therefore, apart from well known molecular mechanisms (BCR/ABL mutations) leading to resistance, stem cell resistance against imatinib in CML is also considered to result from stem cell-related (intrinsic) mechanisms. The exact molecular basis of intrinsic stem cell resistance against imatinib is not well understood. The different hypotheses that have been raised are summarized in . Apart from stem cell quiescence and overexpression of BCR/ABL, these cells may also utilize BCR/ABL-independent survival mechanisms (CitationCopland et al 2005; CitationCopland et al 2006; CitationBarnes and Melo 2006; CitationJiang et al 2007b). In addition, it has been hypothesized that imatinib-resistance in CML stem cells may be associated with reduced drug-uptake and increased drug-efflux. In particular, compared to more mature clonal cells, CML stem cells (CD34+/CD38−/Lin−) apparently display decreased levels of organic cation transporter-1 (OCT-1), a surface transporter involved in the uptake of imatinib, and increased levels of drug-efflux-related surface molecules including the multi-drug resistance protein-1 (MDR-1), known to mediate the efflux of imatinib (CitationJiang et al 2007b). Efflux mechanisms may also contribute to resistance against other drugs including new BCR/ABL TK inhibitors, such as nilotinib (AMN107) (CitationBrendel et al 2007). More recently, it has been described, that dasatinib may act better on immature CML (stem) cells compared to imatinib, but still may not be capable of killing all leukemic stem cells (subclones) (CitationCopland et al 2006). An interesting approach to measure the response to imatinib on a qualitative basis and to possibly predict the response (and relapse) in progenitor compartments, are recently proposed mathematical models (CitationMichor et al 2005; CitationRoeder et al 2006). It may be an interesting idea to employ such models in forthcoming clinical trials studying new TK inhibitors or combination therapies.
Table 1 Resistance of CML stem cells against imatinib: Proposed hypothesesTable Footnote*
As outlined above, stem cell resistance in CML is an emerging issue and major focus in clinical and preclinical research, and although it is difficult to purify CML stem cells for in vitro investigations, the availability of sensitive MRD parameters offers a valuable basis for the design of clinical trials examining the effects of novel drugs and drug-combinations on residual leukemic stem cells. For the near future, one of the most important questions will be whether any of the new TK inhibitors, like dasatinib, nilotinib, INNO-406, or others, can induce long-lasting CCR and consecutive cure through eradication of all relevant CML stem cell subclones in CP. Respective clinical trials employing dasatinib or nilotinib as frontline therapy in CML CP are in progress. These trials should reveal the exact curative potential of these drugs and thus will answer the question as to whether they can overcome ‘intrinsic stem cell resistance’. It should be mentioned here that not all CML stem cell subclones may be of clinical relevance (causing relapse), and that some of these patients may stay in complete hematologic remission even if a BCR/ABL+(sub)clone is detectable (CitationGriswold et al 2006; CitationKhorashad et al 2006; CitationGoldman and Gordon 2006).
The next important question would be whether combinations of targeted drugs may (better) overcome stem cell resistance against imatinib. First, some of these combinations may facilitate the uptake of imatinib or other BCR/ABL TK inhibitors in CML stem cells, or may prevent enhanced drug efflux from these cells (CitationMahon et al 2003; CitationThomas et al 2004; CitationIllmer et al 2004). Likewise, a number of MDR-1 blockers (cyclosporin-A, verapamil, others) are available, and it may be an interesting approach to combine such inhibitors with imatinib or other BCR/ABL TK inhibitors to enhance intracellular drug levels in CML stem cells. More recently, it has been described that combinations of TK inhibitors with each other may also enhance intracellular levels of individual drugs and thereby may lead to cooperative (synergistic) antileukemic effects (CitationWhite et al 2007). Indeed, most of the BCR/ABL TK inhibitors exert synergistic antileukemic effects on CML cells (CitationWeisberg et al 2007a).
Another important aspect is that conventional antileukemic drugs such as interferon-alpha, may have a more pronounced effect on CML progenitor cells compared with imatinib (CitationAngstreich et al 2005; CitationVerbeek et al 2006). Therefore, several trials employ combinations between interferon-alpha and BCR/ABL TK inhibitors. Another important aspect is that many of the novel inhibitors are less specific drugs that do not only recognize BCR/ABL, but also (many) other key kinase-targets (CitationShah et al 2004; CitationMartinelli et al 2005; CitationKimura et al 2005; CitationWeisberg et al 2007b). The differential target profiles of TK inhibitors may also explain why several of them, when combined, produce synergistic antileukemic effects (CitationWeisberg et al 2007a). In this regard it may be of great importance to learn which kinase-targets and related pathways play a predominant role in the biology and growth of CML stem cells. An important consideration in this regard is that the biology, function, and target expression profiles of CML stem cells may be similar but not identical to that of normal stem cells, and that the profile may change during disease evolution, ie, progression to AP or BP (CitationZheng et al 2006; CitationRadich et al 2006; CitationVilluendas et al 2006; CitationDiaz-Blanco et al 2007).
Lastly, it has to be emphasized that the only established stem cell eradicating (=curative) therapy in CML remains SCT, and that SCT may also work in a group of patients with advanced CML (CitationGoldman et al 1986; CitationSilberman 1994; CitationGratwohl et al 1998; CitationDutcher and Wiernik 2000; CitationDeininger 2007). It also has been described that pre-transplant therapy with imatinib may be a reasonable approach in advanced CML (CitationGiralt et al 2007; CitationWeisser et al 2007). Moreover, SCT should remain an important option and major decision-point in treatment algorithms in imatinib-resistant CML (CitationJabbour et al 2006). Depending on the clinical situation and other factors, such therapy (SCT) may be combined with BCR/ABL TK inhibitors (CitationMenzel et al 2007). Likewise, patients with imatinib-resistant CML in AP or BP who are young and have a suitable donor, may benefit from targeted therapy with a second generation BCR/ABL TK inhibitor inducing remission or at least disease-reduction (debulking), followed by allogeneic SCT (CitationJabbour et al 2007a; CitationMenzel et al 2007). As to whether such patients should also be treated with the same BCR/ABL TK inhibitors after SCT (maintenance, prophylaxis) remains at present unknown. At least for patients with detectable BCR/ABL after SCT, maintenance therapy should be considered. Patients who fail SCT or relapse after SCT may also benefit from new TK inhibitors. Whether such patients may even have a better outcome when receiving combination therapy (drug-combinations or donor lymphocytes plus a TK inhibitor) remains to be defined.
Pharmacologic aspects and pharmacologic resistance
Orally administered imatinib is rapidly (within 1–2 hours) and completely absorbed, with a bioavailability of >95%, and a peak plasma concentration reached after 2–4 hours (CitationCohen et al 2002; CitationPeng et al 2005). The pharmacologic half-life of the drug is approximately 18 hours (CitationCohen et al 2002; Citationle Coutre et al 2004; CitationPeng et al 2004a, Citation2005). At a daily dose of 400 mg, imatinib plasma concentrations peak to about 2–3 μg/mL, with a trough level of approximately 1 μg/mL (Citationle Coutre et al 2004; CitationPeng et al 2004a, Citation2005), which exceeds imatinib-doses required for complete inhibition of wt BCR/ABL TK activity (0.5 μg/mL = 1 μM). Imatinib is >95% bound to human plasma proteins, mainly albumin and alpha1-acid glycoprotein (CitationCohen et al 2002; CitationPeng et al 2005). The drug is eliminated predominantly via the bile in form of metabolites (CitationCohen 2002; CitationGschwind et al 2005; CitationPeng et al 2005). One of these metabolites, CGP74588, exhibits pharmacological activity comparable to the parent-drug (CitationCohen et al 2002). Imatinib is metabolized via cytochrome (CYP) P450 isoenzymes, primarily CYP3A4, but also by other CYP 450 species (CYP3A5, CYP2D6). Therefore, imatinib can competitively inhibit the metabolism of drugs that are substrates of CYP P450 isoenzymes (CitationCohen et al 2002; CitationO’Brien et al 2003). Vice versa, drugs that are metabolized via or are inducers of these CYP enzymes may influence the bioavailability of imatinib, and thus lead to changes (eg, decrease) in imatinib plasma concentrations (CitationCohen et al 2002; CitationBolton et al 2004; CitationDutreix et al 2004; CitationFrye et al 2004). Also, hepatic and renal dysfunction may result in slight changes in imatinib concentrations in biological fluids and tissues (CitationPeng et al 2005; CitationPappas et al 2005; CitationEckel et al 2005). However, these changes usually are mild and do not require dose-adjustments. Age, race, sex, and bodyweight have no documented influence on the pharmacokinetics or pharmacodynamics of imatinib (CitationPeng et al 2005).
From experience in clinical trials, patients with CML are judged to be imatinib-resistant when response is lost or is not seen with a daily dose of >400 mg imatinib (CitationKantarjian et al 2003; CitationBaccarani et al 2006). Studies on pharmacokinetics and pharmacodynamics of imatinib in CML suggest that a minimum dose of imatinib of 350–400 mg daily is required to reach a constant effective drug concentration in plasma, that would block wt BCR/ABL (CitationPeng et al 2004b). However, no detailed studies on tissue-concentrations of imatinib in various organs have been presented so far, and some of the tissues and organ-compartments (brain) may not be reached sufficiently by imatinib. In addition, a number of genetic and other factors may influence the bioavailability of the drug (CitationPeng et al 2005; CitationPappas et al 2005; CitationEckel et al 2005). Moreover, the expression of drug transporters and drug-efflux pumps, that are expressed in the apical membrane of the small intestine and the bile canalicular membrane, have been implicated in pharmacologic resistance (CitationBurger and Nooter 2004; CitationBurger et al 2005). All in all, a number of factors may influence the plasma- and tissue levels of imatinib, and under certain circumstances, may contribute to pharmacologic resistance.
More recent data suggest that pharmacologic resistance may indeed be of clinical relevance. In fact, it has been described that the trough plasma level of imatinib is associated with the rate of CCR and of major molecular responses (MMR) in patients with CML (CitationPicard et al 2007). In particular, significantly higher trough levels were found in patients with CCR and MMR (often >1 μg/ml) compared to those without CCR or MMR (often <1 μg/ml) (CitationPicard et al 2007). An unresolved question is whether the different trough levels in the two groups of patients resulted from a primary defect(s) in bioavailability (true pharmacologic resistance) or from massive drug-uptake by residual CML cells in less well responding patients. Whatever the reason is, the observation of different trough levels may be of clinical significance, and it seems appropriate to recommend that plasma trough levels are measured in patients with otherwise unexplained suboptimal response (or no response) to imatinib.
A number of different strategies have been proposed to overcome pharmacologic resistance against imatinib. Suspicion for pharmacologic resistance must be raised when cytogenetic (and molecular) response is lost or not achieved, no BCR/ABL mutations and no signs of clonal evolution are found, and trough imatinib levels are low. It is then important to ask for possible drug-interactions, patient’s complience, and concomitant disorders. After having excluded such causes, dose adjustments (increase) should be considered and may lead to a better response (CitationKantarjian et al 2003). Another potential strategy, that may become subject of future studies, would be to try to increase the imatinib uptake in the intestinal wall (and in other critical target cell populations), and thus bioavailability of the drug, or by imatinib with modulators of transport-proteins (CitationBreedveld et al 2006).
‘Anatomic resistance’ against imatinib
A special problem with imatinib is its marginal accumulation in the central nervous system (CNS) which is caused by low uptake via the blood-brain barrier (CitationTakayama et al 2002; CitationSenior 2003; CitationDai et al 2003; CitationWolff et al 2003). The biochemical basis of poor uptake is not well understood. One hypothesis is that the abundant expression of MDR-1 (P-glycoprotein) in cells forming the blood-brain barrier is associated with constant drug-efflux (CitationDai et al 2003; CitationBreedveld et al 2005; CitationBreedveld et al 2006). Clinically, the poor uptake into the CNS is reflected by CNS relapses that occur in imatinib-treated patients (CitationAbruzzese et al 2003; CitationLeis et al 2004; CitationBornhäuser et al 2004; CitationRajappa et al 2004; CitationRytting and Wierda 2004; CitationMatsuda et al 2005; CitationPavlu et al 2005; CitationKim et al 2006). This is a well known problem in lymphoid leukemias and in the lymphoid blast phase of CML. However, more recently, myeloid CNS relapses have also been described (CitationRytting and Wierda 2004; CitationAichberger et al 2007). Some of these CNS relapses occur even in patients with CCR (CitationBornhäuser et al 2004; CitationAichberger et al 2007).
A number of strategies have been proposed to treat and to prevent CNS relapses in CML. Once diagnosed, local therapy of the CNS relapse with intrathecal cytostatic drugs (cytarabine and others) and/or radiation seems an appropriate therapeutic maneuver (CitationAbruzzese et al 2003; CitationLeis et al 2004; CitationBornhäuser et al 2004; CitationRajappa et al 2004; CitationRytting and Wierda 2004; CitationMatsuda et al 2005; CitationPavlu et al 2005; CitationKim et al 2006; CitationAichberger et al 2007). In those with a concomitant systemic relapse, the additional replacement of imatinib by a second generation BCR/ABL inhibitor must be considered (CitationAbdelhalim et al 2007; CitationAichberger et al 2007). Interestingly, for some of these emerging drugs (dasatinib, INNO-406), it has been described that they can cross the blood-brain barrier quite effectively in animal models (CitationWild et al 2004; CitationYokota et al 2007), and the same may hold true for patients with CML in CNS relapse (CitationAbdelhalim et al 2007; CitationAichberger et al 2007). Therefore, it seems logic to consider the use of such new TK inhibitors as prophylaxis of CNS relapses as well. In case that the frequency of reported CNS relapses will further increase, such prophylactic therapy must be regarded as a mandatory approach. An alternative approach might be to increase the uptake of imatinib by applying modulators of drug transporters (eg, MDR-1).
BCR/ABL mutations
The predominant molecular defect that causes resistance against imatinib are point mutations in the BCR/ABL oncogene (CitationGorre et al 2001; Citationvon Bubnoff et al 2002; CitationBarthe et al 2002; CitationBranford et al 2002, Citation2003; CitationShah et al 2002; CitationHochhaus et al 2002). The respective BCR/ABL mutants retain their kinase activity and their oncogenic potential, but usually display impaired or absent drug-binding capacity (CitationRoumiantsev et al 2002; CitationAzam et al 2003; CitationCowan-Jacob et al 2004; CitationWeisberg et al 2007b). Other mutants may be less oncogenic and may not play an important role in disease evolution (CitationGriswold et al 2006; CitationKhorashad et al 2006). Most of the relevant mutations cluster within or in next vicinity to the imatinib-binding site, or are located in BCR/ABL domains critical to the topography and tertiary structure of the imatinib/ATP-binding site, with consecutive steric hindrance of drug-binding (CitationBranford et al 2002, Citation2003; CitationShah et al 2002; CitationHochhaus et al 2002; CitationRoumiantsev et al 2002; CitationAzam et al 2003; CitationCowan-Jacob et al 2004; CitationO’Hare et al 2005, CitationWeisberg et al 2007b). Examples of BCR/ABL residues that (when derived from mutated genes) directly inhibit imatinib-binding, are Thr315 and Phe317 (CitationCowan-Jacob et al 2004; CitationWeisberg et al 2007b). Other BCR/ABL mutations destabilize the inactive conformation of the nucleotide-binding loop (P-loop) or the DFG motif that binds to imatinib, thereby reducing imatinib-binding affinity (CitationRoumiantsev et al 2002; CitationCowan-Jacob et al 2004; CitationWeisberg et al 2007b). Residues affecting imatinib-binding through destabilization of the inactive conformation include Glu255, Tyr253, and Gly250 in the P-loop of ABL (CitationCowan-Jacob et al 2004; CitationWeisberg et al 2007b).
More than 50 different mutations in BCR/ABL have been described (Citationvon Bubnoff et al 2002; CitationShah et al 2002; CitationHochhaus et al 2002; CitationCowan-Jacob et al 2004; CitationO’Hare et al 2005; CitationWeisberg et al 2007b). These mutations cluster in four major regions of the oncogene, namley the phosphate-binding (P-loop) domain (examples: M224V, L248V, G250E/R, Q252R/H, Y253F/H, E255K/V), the imatinib-binding domain (F311L/I, T315I, F317L), the catalytic domain (M351T, E355G/D), and the activation loop domain (V379I, F382L, L387M, H396R/P) (CitationShah et al 2002; CitationBranford et al 2003; CitationHochhaus et al 2002). shows BCR/ABL mutations frequently detected in patients with imatinib-resistant CML.
Table 2 BCR/ABL mutations detectable in CML patients treated with imatinib
In most CML patients, BCR/ABL mutations may already be present in (stem cell) subclones before imatinib therapy is initiated (CitationRoche-Lestienne et al 2002, Citation2003; CitationKreuzer et al 2003; CitationJiang et al 2007a). However, in some patients, the BCR/ABL mutation may not simply be revealed through selection by drug-therapy, but may represent a newly occurring defect. An unresolved question in this regard is whether treatment with imatinib or other (targeted) drugs can modulate (increase) the BCR/ABL mutation rate (mutagenic potential of drug). The more likely scenario is that the rapid and sustained elimination of all (many) subclones by TK inhibitors is important and should counteract the development of new BCR/ABL mutations, because the size of the target cell population (CML stem cells) in which such mutations can develop, is constantly shrinking over time in responding patients.
Another unresolved question is how the wt BCR/ABL subclone is capable of suppressing (all) BCR/ABL mutants. This phenomenon may be explained by chalone-dependent inhibition or may be related to the different oncogenic potencies of the mutants. Clinically, this phenomenon is of diagnostic importance, as BCR/ABL mutations may not be detectable at diagnosis but only after drug-induced selection of stem cell subclones.
As mentioned above, the various BCR/ABL mutants display different oncogenic (transforming) potential (CitationGriswold et al 2006; CitationSkaggs et al 2006; CitationWeisberg et al 2007a). Taking their in vitro activity into consideration, the following rank order of (oncogenic) potency is found: Y253F = E255K > wt BCR-ABL > T315I > H396P > M351T > others. Thus, certain P loop mutations (Y253F, E255K) and the T315I mutation display a high oncogenic potential, which is consistent with the clinical observation of a poor outcome concerning overall and progression-free survival (CitationBranford et al 2003; CitationSoverini et al 2005; CitationNicolini et al 2006). However, not all P-loop mutations may be associated with a poor prognosis in CML (CitationJabbour et al 2006). In particular, several of the BCR/ABL mutations are far less oncogenic, and some of them may not even exhibit a proliferative advantage over normal (stem) cells, and thus may not even cause overt CML (CitationKhorashad et al 2006). These mutations should not count in the evaluation of drug resistance and the consecutive treatment plan in the same way as clinically relevant (oncogenic) mutations.
A number of different strategies have been proposed to treat patients with imatinib resistant CML, in whom BCR/ABL mutations are detected. Treatment in these patients depends on several different factors, including the type (oncogenic potential) of the mutation, phase of disease, presence of other pro-oncogenic disease-features (clonal evolution), age, co-morbidity, overall status of the patient, and availability of a SCT donor in those who are eligible for high-dose therapy. With regard to BCR/ABL mutations, four categories are proposed and related to specific treatment recommendations: a) mutations that do not cause clinically overt resistance (recommendation: wait and watch if possible), b) mutants that have low oncogenic potential and may disappear (at least kept under control) upon dose-escalation (recommendation: increase imatinib from 400 to 600 or 800 mg/day), c) non-T315I-mutants that are not expected to disappear on imatinib dose-escalation (recommendation: switch from imatinib to a second generation BCR/ABL inhibitor: nilotinib or dasatinib), and d) the T315I mutant as well as a few other mutants that are also resistant against dasatinib and nilotinib (recommendation: high-dose chemotherapy or experimental drugs and proceed to SCT if possible). The majority of all imatinib-resistant patients are in group b and c. Therefore, recent efforts have focused on the development of new, more effective BCR/ABL TK inhibitors that can overcome resistance. Among these are nilotinib (AMN107), dasatinib (BMS354825), INNO-406, and several others (). These drugs act on various imatinib-resistant BCR/ABL mutants and can produce complete hematologic and cytogenetic responses in patients with imatinib-resistant disease (CitationTalpaz et al 2006; CitationKantarjian et al 2006; CitationWeisberg et al 2006; CitationQuintas-Cardama et al 2007; CitationHochhaus et al 2007b; CitationCortes et al 2007; CitationGuilhot et al 2007). Encouraging results have particularly been obtained in CP, but hematologic and sometimes cytogenetic or molecular responses may also be seen in AP or BP. However, as stated above, not all BCR/ABL mutants are responsive to these inhibitors, and the relative potencies vary among drugs. Unfortunately, patients with the T315I mutant of BCR/ABL are clinically resistant against nilotinib and dasatinib, and also against most other available TK inhibitors (CitationTalpaz et al 2006; CitationKantarjian et al 2006; CitationWeisberg et al 2006). As mentioned above, for these patients, alternative therapies have to be considered. One possibility are novel kinase inhibitors or drugs that act independent of BCR/ABL (Martinelli et al 2005; CitationGumireddy et al 2005; CitationTauchi and Ohyashiki 2006; CitationJabbour et al 2007b) (). Another option is SCT with or without a second generation BCR/ABL inhibitor (CitationJabbour et al 2007a; CitationMenzel et al 2007).
Table 3 Novel pharmacologic inhibitors proposed for imatinib-resistant CML
An interesting aspect is that BCR/ABL TK inhibitors, when applied in combination, may produce antileukemic effects on CML cells exhibiting BCR/ABL T315I, even if leukemic cells are resistant against single agents (CitationWhite et al 2007; CitationWeisberg et al 2007a). This phenomenon may be explained by additional drug targets expressed in these cells, by cooperative effects at BCR/ABL epitopes, or by increased drug accumulation in target cells (CitationWhite et al 2007; CitationWeisberg et al 2007a). Whether combinations of TK inhibitors will also induce long lasting remission in (all) patients with TK-inhibitor resistant CML, remains at present unknown. It also remains unknown which of the new drugs, that have been described to counteract in vitro growth of leukemic cells exhibiting BCR/ABL T315I (CitationTseng et al 2005; CitationCarter et al 2005; CitationGiles et al 2007; CitationCheetham et al 2007; CitationRahmani et al 2007), will induce complete (and long lasting) cytogenetic remissions in vivo in these patients.
An important therapeutic consideration is prevention of occurrence (selection) of subclones carrying imatinib-resistant BCR/ABL mutants. One approach may be to combine TK inhibitors in an early phase of disease, similar to the situation in HIV-positive patients, where early intervention is performed using multiple drugs. Another strategy may be to combine novel TK inhibitors with a stem cell-attacking approach, like SCT or high-dose chemotherapy, or with stem cell-suppressing maintenance therapy (eg, interferon-alpha).
Finally, several treatment concepts focus on the mobilization of the immune system, with the ultimate goal to target residual leukemic (stem) cells (MRD) in CML (CitationLi et al 2005; CitationWestermann et al 2007; CitationVolpe et al 2007; CitationPeng et al 2007). In most instances, immunotherapy is combined with a BCR/ABL TK inhibitor. Whether such intervention may lead to the eradication of (all) relevant CML stem cell subclones remains to be elucidated.
Other BCR/ABL defects
Apart from BCR/ABL mutations, other defects in BCR/ABL may also contribute to resistance against imatinib. Such alternative defects include BCR/ABL gene duplications and -amplifications (Citationle Coutre et al 2000; CitationWeisberg and Griffin 2000; CitationGorre et al 2001; CitationNguyen Khac et al 2002; CitationCampbell et al 2002; CitationMorel et al 2003; CitationGargallo et al 2003; CitationGadzicki et al 2005). These defects may be associated with (multiple) cytogenetic abnormalities (CitationGadzicki et al 2005; CitationPienkowski-Grela et al 2007), and several of these patients are in an accelerated phase or blast phase of CML (CitationTanaka et al 2000; CitationGadzicki et al 2005; CitationPienkowski-Grela et al 2007). Therefore, in most cases, the contribution of amplified BCR/ABL in the malignant process (progression) and in drug resistance remains uncertain. Nevertheless, some of these patients respond to elevated doses (or even standard doses) of imatinib, suggesting that the BCR/ABL defect may have pharmacologic and clinical impact.
BCR/ABL-independent molecular resistance
During disease, the CML clone may acquire additional BCR/ABL-independent molecular (genetic) defects and pro-oncogenic hits in stem cell subclones, which may lead to disease-progression. Such clonal evolution is often accompanied by the occurrence of cytogenetic defects. Leukemic cells in these patients are frequently resistant against imatinib, and may exhibit aneuploidy, sometimes in form of a second Ph chromosome or trisomy 8 (+8) (CitationHochhaus et al 2002). Other cytogenetic defects that have been described in imatinib-resistant CML include trisomy 6 (+6), +9, +12, +18, and monosomy 7 (−7) (CitationHochhaus et al 2002; CitationCortes et al 2003; CitationMarktel et al 2003; CitationO’Dwyer et al 2004). Most cytogenetic defects are considered to be of prognostic significance concerning survival in imatinib-treated patients (CitationCortes et al 2003; CitationMarktel et al 2003; CitationO’Dwyer et al 2004). However, not all cytogenetic defects may lead to imatinib-resistance. Especially isolated chromosome defects may disappear or persist at stable (low) level without loss of hematologic response during therapy. In other patients, resistance may develop within short time.
The molecular defects that accompany cytogenetic abnormalities and may contribute to resistance against imatinib, have not been defined yet. Therefore, at present, it is difficult to predict the clinical impact of isolated cytogenetic defects for imatinib-treated patients. A special situation is the occurrence of cytogenetic defects in Ph-negative subclones during imatinib therapy (CitationMedina et al 2003; CitationTerre et al 2004; CitationLoriaux and Deininger 2004; CitationLin et al 2006; CitationNavarro et al 2007; CitationJabbour et al 2007). One hypothesis is that these subclones derive from a very immature progenitor that was involved in a pre-Ph (pre-BCR/ABL) phase of CML (), and under certain circumstances can be activated (by additional hits) to transform into a secondary Ph-negative (but still monoclonal) neoplasm. Indeed, some of these patients may develop overt secondary disease (that may resemble a myelodysplastic syndrome, MDS or acute myeloid leukemia, AML), even if the Ph-positive (sub)clones are completely suppressed (CitationLoriaux and Deininger 2004; CitationLin et al 2006; CitationNavarro et al 2007; CitationJabbour et al 2007). The subclone hypothesis is supported by HUMARA analysis as well as the fact, that the karyotype abnormalities are the same as those detectable in Ph-positive subclones (CitationTerre et al 2004; CitationNavarro et al 2007; CitationJabbour et al 2007). An alternative hypothesis is that Ph-negative clones develop independent of the primary disease (unrelated clone). Such hypothesis would pose the question as to whether imatinib exhibits a substantial mutagenic potential and can attack normal stem cells similar to conventional cytostatic drugs. So far, no clear evidence for such hypothesis has been presented, although single case reports have suggested that even transplanted normal stem cells may undergo transformation and accumulate cytogenetic defects during treatment with imatinib (CitationAgis et al 2004). However, again, such additional clones may not be relevant clinically (in all patients), and these patients may still stay in a complete hematologic remission with normal blood counts over time (CitationAgis et al 2004).
As mentioned above, little is known so far about specific molecular defects and mechanisms underlying BCR/ABL-independent resistance to imatinib in CML, and especially about defects that can lead to malignant transformation in subclones. In fact, although an extensive number of (potentially deregulated) molecules and numerous mechanisms have been discussed, no specific recurrent gene defects that would explain transformation of CML into AP or BP have been identified (CitationCalabretta and Perrotti 2004; CitationMelo and Barnes 2007). General pathogenetic factors that have been discussed as being involved in disease progression in CML include activation of (mutation-induced) signal transduction molecules (by mutations or oncogene activation), differentiation arrest, genomic instability (deficiency of DNA repair, mutator phenotype), telomer shortening, and loss of tumor suppressor function (CitationCalabretta and Perrotti 2004; CitationMelo and Barnes 2007). Some of these defects may be triggered in part also by BCR/ABL. Likewise, BCR/ABL has been implicated in hypermethylation of the genome, in deactivation of tumor suppressors, and in the hypermutation-status of leukemic cells (CitationNeviani et al 2006; CitationMelo and Barnes 2007). However, most of the secondary transforming hits in CML may be BCR/ABL-independent events. The (numerous) candidate genes potentially involved in disease progression in CML, and their function, have been reviewed elsewhere (CitationCalabretta and Perrotti 2004; CitationMelo and Barnes 2007).
So far, it remains unknown which of these defects and deregulated molecules may contribute to resistance against imatinib in CML. Respective preclinical and clinical studies are in progress and hopefully will reveal new important therapeutic targets in the near future. Such studies focus primarily on genes involved in the differentiation block, in abnormal signalling, in abnormal DNA repair, and in the deactivation of tumor suppressors (CitationMartinelli et al 2005; CitationMelo and Barnes 2007). It is the hope for the future that these studies will lead to the development of new treatment strategies aimed at preventing disease-progression in CML. A likely scenario is that such novel therapies will then be combined with most effective BCR/ABL TK inhibitors.
Intolerance and side effects
An important aspect in the treatment of CML with imatinib or other BCR/ABL TK inhibitors, are side effects that may lead to dose-reductions and thus may predispose for the development of resistance. For imatinib, only a few major side effects have been reported, including transient edema formation and mild myelosuppression (CitationDruker et al 2001a; CitationKantarjian H et al 2002; CitationBarbany et al 2002; CitationTalpaz et al 2002; CitationSawyers et al 2002; CitationO’Brien et al 2003). Other side effects such as hepatic dysfunction or cardiac problems are uncommon. However, some of these side effects may lead to dose reductions or even to drug withdrawal. Nilotinib (AMN107) also exhibits a favorable toxicity profile, although rare adverse side effects such as an elevation in pancreatic enzymes, have been reported (CitationKantarjian et al 2006). With regard to dasatinib, a number of side effects have been reported using the proposed standard dose of 2 × 70 mg per os daily. These side effects include pleural and pericardial effusions and myelosuppression (CitationTalpaz et al 2006; CitationHochhaus et al 2007). Based on first observations in clinical trials and unpublished data, the frequency of side effects may be lower when the dose of dasatinib is reduced, which points to the question as to whether the standard dose should be reconsidered. Notably, dasatinib is a most potent inhibitor of leukemic cell growth in CML, and in many patients, the drug may still work at reduced dose levels (CitationTalpaz et al 2006; CitationHochhaus et al 2007). Some of the side effects may also be less frequent when the drug is administered once daily (1 × 140 mg instead of 2 × 70 mg).
For most other TK inhibitors, side effect profiles in CML patients remain to be established.
Clinical practice: Algorithm
Definitions for ‘suboptimal response’ and ‘drug-resistance’ in CML patients treated with imatinib are well established (CitationBaccarani et al 2006). It is also well established, that patients with drug resistance should undergo restaging and BCR/ABL mutation analysis. In addition, the availability of a SCT donor should be (re)explored. The final treatment plan will be based on a number of different variables, including disease-specific factors (phase of disease, presence and type of BCR/ABL mutation, presence and type of additional chromosomal abnormalities, extramedullary involvement, lymphoid versus myeloid blasts), patient-related factors (age, fitness, comorbidity, patients attitude, availability of a donor), and the overall situation in each case. After having collected all necessary information (including BCR/ABL mutations, and if required an imatinib trough level), a straight forward approach may be to estimate chances for long term disease-free survival (cure in young patients) with each therapeutic approach, and to weigh treatment-associated mortality and morbidity against the chances for cure (long term disease-free survival). Depending on mutations and other features of the clone, some patients may benefit from imatinib dose-escalation. In other cases, treatment has to be switched to dasatinib or nilotinib. Both drugs are registered and approved for treatment of imatinib-resistant CML. The decision to introduce such therapy should be based on a thorough investigation for BCR/ABL mutations, as treatment will fail when CML cells display the T315I mutant. For these patients, alternative treatment approaches have to be considered. In younger patients with a suitable donor who display BCR/ABL T315I or other highly resistant mutants, allogeneic SCT should be considered. When no donor is available or the patient is not considered fit enough for SCT, new experimental drugs, some of them known to target BCR/ABL T315I, or drug combinations, should be offered in clinical trials.
Summary and future perspectives
Resistance against imatinib is an emerging problem in the treatment of CML. Dose-adjustments, new BCR/ABL-targeting drugs, and other antileukemic approaches may be sufficient to overcome resistance in many cases. A specific challenge remains the T315I mutant of BCR/ABL that is resistant against most available TK inhibitors. Other specific challenges are the intrinsic resistance of CML stem cells, clonal evolution, involvement of BCR/ABL-independent signalling pathways, and poor accumulation of imatinib in the central nervous system. For the future, new more effective BCR/ABL TK inhibitors, drug combinations, and drugs entering the blood–brain barrier, may be straightforward approaches to improve anti-CML therapy. Such approaches will also aim at preventing the occurrence of drug resistance in an early phase of CML. For those patients who fail drug therapy and are eligible, allogeneic stem cell transplantation with or without additional TK inhibitors, will remain an alternative option of treatment. The value of new future treatment strategies (immunotherapies, siRNA) remains at present unknown.
References
- AbruzzeseECantonettiMMorinoL2003CNS and cutaneous involvement in patients with chronic myeloid leukemia treated with imatinib in hematologic complete remission: two case reportsJ Clin Oncol214256814615464
- AbdelhalimABarcosMBlockAW2007Remission of Philadelphia chromosome-positive central nervous system leukemia after dasatinib therapyLeuk Lymphoma481053617487757
- AgisHMannhalterCSperrWR2004Detection of trisomy 8 in donor-derived Ph- cells in a patient with Ph+ chronic myeloid leukemia successfully treated with Imatinib (STI571) in relapse after allogeneic transplantationLeuk Lymphoma451453815359647
- AngstreichGRMatsuiWHuffCA2005Effects of imatinib and interferon on primitive chronic myeloid leukaemia progenitorsBr J Haematol1303738116042686
- AichbergerKJHerndlhoferSAgisH2007Liposomal cytarabine for treatment of myeloid central nervous system relapse in chronic myeloid leukemia occurring during imatinib therapyEur J Clin Invest378081317727673
- AzamMLatekRRDaleyGQ2003Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABLCell1128314312654249
- BaccaraniMSaglioGGoldmanJ2006Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNetBlood10818092016709930
- BarbanyGHoglundMSimonssonBSwedish CML Group2002Complete molecular remission in chronic myelogenous leukemia after imatinib therapyN Engl J Med3475394012181416
- BarnesDJMeloJV2006Primitive, quiescent and difficult to kill: the role of non-proliferating stem cells in chronic myeloid leukemiaCell Cycle52862617172863
- BartheCGharbiMJLagardeV2002Mutation in the ATP-binding site of BCR-ABL in a patient with chronic myeloid leukaemia with increasing resistance to STI571Br J Haematol1191091112358910
- BhatiaRHoltzMNiuNPersistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatmentBlood1014701712576334
- BiernauxCLoosMSelsAHuezGStryckmansP1995Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individualsBlood863118227579406
- BoltonAEPengBHubertM2004Effect of rifampicin on the pharmacokinetics of imatinib mesylate (Gleevec, STI571) in healthy subjectsCancer Chemother Pharmacol53102614605865
- BornhäuserMJenkeAFreiberg-RichterJ2004CNS blast crisis of chronic myelogenous leukemia in a patient with a major cytogenetic response in bone marrow associated with low levels of imatinib mesylate and its N-desmethylated metabolite in cerebral spinal fluidAnn Hematol83401214673623
- BoseSDeiningerMGora-TyborJ1998The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual diseaseBlood92336279787174
- BranfordSRudzkiZWalshS2002High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistanceBlood993472511964322
- BranfordSRudzkiZWalshS2003Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosisBlood1022768312623848
- BreedveldPBeijnenJHSchellensJH2006Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugsTrends Pharmacol Sci27172416337012
- BreedveldPPluimDCiprianiG2005The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patientsCancer Res6525778215805252
- BrecciaMDiverioDPaneF2006Discontinuation of imatinib therapy after achievement of complete molecular response in a Ph(+) CML patient treated while in long lasting complete cytogenetic remission (CCR) induced by interferonLeuk Res301577916630657
- BrendelCScharenbergCDohseM2007Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cellsLeukemia 20072112677517519960
- BurgerHNooterK2004Pharmacokinetic resistance to imatinib mesylate: role of the ABC drug pumps ABCG2 (BCRP) and ABCB1 (MDR1) in the oral bioavailability of imatinibCell Cycle31502515611623
- BurgerHvan TolHBrokM2005Chronic imatinib mesylate exposure leads to reduced intracellular drug accumulation by induction of the ABCG2 (BCRP) and ABCB1 (MDR1) drug transport pumpsCancer Biol Ther47475215970668
- CalabrettaBPerrottiD2004The biology of CML blast crisisBlood10340102214982876
- CampbellLJPatsourisCRayerouxKC2002BCR/ABL amplification in chronic myelocytic leukemia blast crisis following imatinib mesylate administrationCancer Genet Cytogenet13930312547154
- CarterTAWodickaLMShahNP2005Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinasesProc Natl Acad Sci USA10211011616046538
- CheethamGMCharltonPAGolecJM2007Structural basis for potent inhibition of the Aurora kinases and a T315I multi-drug resistant mutant form of Abl kinase by VX-680Cancer Lett251323917240048
- CohenMHWilliamsGJohnsonJR2002Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemiaClin Cancer Res89354212006504
- CoplandMHamiltonAElrickLJ2006Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fractionBlood1074532916469872
- CoplandMJorgensenHGHolyoakeTL2005Evolving molecular therapy for chronic myeloid leukaemia – are we on targetHematology103495916203604
- CortesJKantarjianH2003Advanced-phase chronic myeloid leukemiaSemin Hematol40798612563614
- CortesJETalpazMGilesF2003Prognostic significance of cytoge-netic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapyBlood101379480012560227
- CortesJRousselotPKimDW2007Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisisBlood10932071317185463
- CortesJETalpazMO’BrienS2006Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposalCancer10613061516463391
- Cowan-JacobSWGuezVFendrichG2004Imatinib (STI571) resistance in chronic myelogenous leukemia: molecular basis of the underlying mechanisms and potential strategies for treatmentMini Rev Med Chem42859915032675
- DaleyGQVan EttenRABaltimoreD1990Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosomeScience247824302406902
- de KleinAvan KesselAGGrosveldG1982A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemiaNature30076576960256
- DaiHMarbachPLemaireM2003Distribution of STI-571 to the brain is limited by P-glycoprotein-mediated effluxJ Pharmacol Exp Ther30410859212604685
- DeiningerMW2007Optimizing therapy of chronic myeloid leukemiaExp Hematol35S11445417379100
- DevireddyLRGazinCZhuXGreenMR2005A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptakeCell123129330516377569
- Diaz-BlancoEBrunsINeumannF2007Molecular signature of CD34(+) hematopoietic stem and progenitor cells of patients with CML in chronic phaseLeukemia2149450417252012
- DrukerBJGuilhotFO’BrienSG2006Five-year follow-up of patients receiving imatinib for chronic myeloid leukemiaN Engl J Med35524081717151364
- DrukerBJSawyersCLKantarjianH2001Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosomeN Engl J Med34410384211287973
- DrukerBJTalpazMRestaDJ2001Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemiaN Engl J Med3441031711287972
- DutcherJPWiernikPH2000Accelerated and blastic phase of chronic myeloid leukemiaCurr Treat Options Oncol1516212057061
- DutreixCPengBMehringG2004Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjectsCancer Chemother Pharmacol54290415138710
- EavesACBarnettMJPonchioLCashmanJDPetzerALEavesCJ1998Differences between normal and CML stem cells: potential targets for clinical exploitationStem Cells16S1778311012149
- EavesCUdomsakdiCCashmanJ1993The biology of normal and neoplastic stem cells in CMLLeuk Lymphoma11S1245538251904
- EckelFvon DeliusSMayrM2005Pharmacokinetic and clinical phase II trial of imatinib in patients with impaired liver function and advanced hepatocellular carcinomaOncology693637116319507
- EistererWJiangXChristO2005Different subsets of primary chronic myeloid leukemia stem cells engraft immunodeficient mice and produce a model of the human diseaseLeukemia194354115674418
- ElrickLJJorgensenHGMountfordJCHolyoakeTL2005Punish the parent not the progenyBlood1051862615528314
- FryeRFFitzgeraldSMLagattutaTF2004Effect of St John’s wort on imatinib mesylate pharmacokineticsClin Pharmacol Ther76323915470331
- GadzickiDvon NeuhoffNSteinemannD2005BCR-ABL gene amplification and overexpression in a patient with chronic myeloid leukemia treated with imatinibCancer Genet Cytogenet159164715899391
- GargalloPMCuelloMTArangurenPN2003Amplification of the BCR/ABL fusion gene clustered on a masked Philadelphia chromosome in a patient with myeloblastic crisis of chronic myelocytic leukemiaCancer Genet Cytogenet143140412781448
- GilesFJCortesJEKantarjianHMO’BrienSM2004Accelerated and blastic phases of chronic myelogenous leukemiaHematol Oncol Clin North Am187537415271404
- GilesFJCortesJJonesD2007MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutationBlood109500216990603
- GiraltSAAroraMGoldmanJM2007Impact of imatinib therapy on the use of allogeneic haematopoietic progenitor cell transplantation for the treatment of chronic myeloid leukaemiaBr J Haematol137461717459051
- GishizkyMLWitteON1992Initiation of deregulated growth of multipotent progenitor cells by bcr-abl in vitroScience25683691375394
- GoldmanJGordonM2006Why do chronic myelogenous leukemia stem cells survive allogeneic stem cell transplantation or imatinib: does it really matterLeuk Lymphoma471716321820
- GoldmanJMApperleyJFJonesL1986Bone marrow transplantation for patients with chronic myeloid leukemiaN Engl J Med31420273510388
- GorreMEMohammedMEllwoodK2001Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplificationScience2938768011423618
- GrahamSMJorgensenHGAllanE2002Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitroBlood993192511756187
- GratwohlAHermansJGoldmanJM1998Risk assessment for patients with chronic myeloid leukaemia before allogeneic blood or marrow transplantation. Chronic Leukemia Working Party of the European Group for Blood and Marrow TransplantationLancet3521087929798583
- GriswoldIJMacPartlinMBummT2006Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinibMol Cell Biol2660829316880519
- GschwindHPPfaarUWaldmeierFPokornyRSeiberlingMBen-AmMPengBGrossG2005Metabolism and disposition of imatinib mesylate in healthy volunteersDrug Metab Dispos3315031216006570
- GuilhotFApperleyJKimDW2007Dasatinib induces significant hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in accelerated phaseBlood10941435017264298
- GumireddyKBakerSJCosenzaSC2005A non-ATP-competitive inhibitor of BCR-ABL overrides imatinib resistanceProc Natl Acad Sci USA1021992715677719
- HerweijerHSonneveldPBaasFNooterK1990Expression of mdr1 and mdr3 multidrug-resistance genes in human acute and chronic leukemias and association with stimulation of drug accumulation by cyclosporineJ Natl Cancer Inst821133401972761
- HochhausAErbenPErnstTMuellerMC2007Resistance to targeted therapy in chronic myelogenous leukemiaSemin Hematol44S152417292737
- HochhausAKantarjianHMBaccaraniM2007Dasatinib induces notable hematologic and cytogenetic responses in chronic-phase chronic myeloid leukemia after failure of imatinib therapyBlood1092303917138817
- HochhausAKreilSCorbinAS2002Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapyLeukemia162190612399961
- HolyoakeTLHorrocksCThomasT2000Cell separation improves the sensitivity of detecting rare human normal and leukemic hematopoietic cells in vivo in NOD/SCID miceCytotherapy24112112044221
- HolyoakeTLJiangXDrummondMW2002Elucidating critical mechanisms of deregulated stem cell turnover in the chronic phase of chronic myeloid leukemiaLeukemia165495811960331
- HolyoakeTLJiangXJorgensenHG2001Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3Blood97720811157490
- HooverRRGerlachMJKohEYDaleyGQ2001Cooperative and redundant effects of STAT5 and Ras signaling in BCR/ABL transformed hematopoietic cellsOncogene2058263511593388
- HughesTPKaedaJBranfordS2003Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemiaN Engl J Med34914233214534335
- IllmerTSchaichMPlatzbeckerU2004P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylateLeukemia18401814724652
- JabbourECortesJKantarjianHM2006Allogeneic stem cell transplantation for patients with chronic myeloid leukemia and acute lymphocytic leukemia after Bcr-Abl kinase mutation-related imatinib failureBlood1081421316601247
- JabbourECortesJKantarjianH2007Novel tyrosine kinase inhibitor therapy before allogeneic stem cell transplantation in patients with chronic myeloid leukemia: no evidence for increased transplant-related toxicityCancer1103404417559140
- JabbourECortesJO’BrienS2007New targeted therapies for chronic myelogenous leukemia: opportunities to overcome imatinib resistanceSemin Hematol44S1S253117292738
- JabbourEKantarjianHMAbruzzoL2007Chromosomal abnormalities in Philadelphia chromosome-negative metaphases appearing during imatinib mesylate therapy in patients with newly diagnosed chronic myeloid leukemia in chronic phaseBlood
- JabbourEKantarjianHJonesD2006Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylateLeukemia2017677316855631
- JiangXSawKMEavesAEavesC2007Instability of BCR-ABL gene in primary and cultured chronic myeloid leukemia stem cellsJ Natl Cancer Inst996809317470736
- JiangXZhaoYSmithC2007Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapiesLeukemia219263517330101
- KantarjianHMCortesJO’BrienS2002Imatinib mesylate (STI571) therapy for Philadelphia chromosome-positive chronic myelogenous leukemia in blast phaseBlood9935475311986206
- KantarjianHGilesFWunderleL2006Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALLN Engl J Med35425425116775235
- KantarjianHSawyersCHochhausA2002Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemiaN Engl J Med3466455211870241
- KantarjianHMTalpazMGilesF2006New insights into the patho-physiology of chronic myeloid leukemia and imatinib resistanceAnn Intern Med1459132317179059
- KantarjianHMTalpazMO’BrienS2003Dose escalation of imatinib mesylate can overcome resistance to standard-dose therapy in patients with chronic myelogenous leukemiaBlood101473512393385
- KhorashadJSAnandMMarinD2006The presence of a BCR-ABL mutant allele in CML does not always explain clinical resistance to imatinibLeukemia206586316467863
- KimHJJungCWKimK2006Isolated blast crisis in CNS in a patient with chronic myelogenous leukaemia maintaining major cytogenetic response after imatinibJ Clin Oncol2440282916921058
- KimuraSNaitoHSegawaH2005NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib-resistant leukemiaBlood10639485416105974
- KreuzerKALe CoutrePLandtO2003Preexistence and evolution of imatinib mesylate-resistant clones in chronic myelogenous leukemia detected by a PNA-based PCR clamping techniqueAnn Hematol82284912692682
- le CoutrePKreuzerKAPurscheS2004Pharmacokinetics and cellular uptake of imatinib and its main metabolite CGP74588Cancer Chemother Pharmacol533132314658008
- le CoutrePTassiEVarella-GarciaM2000Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplificationBlood9517586610688835
- LeisJFStepanDECurtinPT2004Central nervous system failure in patients with chronic myelogenous leukemia lymphoid blast crisis and Philadelphia chromosome positive acute lymphoblastic leukemia treated with imatinib (STI-571)Leuk Lymphoma45695815160941
- LiZQiaoYLiuB2005Combination of imatinib mesylate with autologous leukocyte-derived heat shock protein and chronic myelogenous leukemiaClin Cancer Res114460815958631
- LinHMonacoGSunT2005Bcr-Abl-mediated suppression of normal hematopoiesis in leukemiaOncogene2432465615735695
- LinYBruyereHHorsmanDE2006Philadelphia-negative clonal hematopoiesis following imatinib therapy in patients with chronic myeloid leukemia: a report of nine cases and analysis of predictive factorsCancer Genet Cytogenet170162316965950
- LoriauxMDeiningerM2004Clonal cytogenetic abnormalities in Philadelphia chromosome negative cells in chronic myeloid leukemia patients treated with imatinibLeuk Lymphoma45219720315512807
- LugoTGPendergastAMMullerAJWitteON1990Tyrosine kinase activity and transformation potency of bcr-abl oncogene productsScience2471079822408149
- MahonFXBellocFLagardeV2003MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line modelsBlood10123687312609962
- MarktelSMarinDFootN2003Chronic myeloid leukemia in chronic phase responding to imatinib: the occurrence of additional cytogenetic abnormalities predicts disease progressionHaematologica88260712651263
- MartinelliGSoveriniSRostiG2005New tyrosine kinase inhibitors in chronic myeloid leukemiaHaematologica905344115820950
- MatsudaMMoritaYShimadaT2005Extramedullary blast crisis derived from 2 different clones in the central nervous system and neck during complete cytogenetic remission of chronic myelogenous leukemia treated with imatinib mesylateInt J Hematol81307915914360
- MedinaJKantarjianHTalpazM2003Chromosomal abnormalities in Philadelphia chromosome-negative metaphases appearing during imatinib mesylate therapy in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phaseCancer9819051114584073
- MeloJVBarnesDJ2007Chronic myeloid leukaemia as a model of disease evolution in human cancerNat Rev Cancer74415317522713
- MeloJVDeiningerMW2004Biology of chronic myelogenous leukemia – signaling pathways of initiation and transformationHematol Oncol Clin North Am185456815271392
- MenzelHvon BubnoffNHochhausA2007Successful allogeneic stem cell transplantation in second chronic-phase CML induced by the tyrosine kinase inhibitor nilotinib (AMN107) after blast crisis under imatinibBone Marrow Transplant4083417450179
- MichorFHughesTPIwasaY2005Dynamics of chronic myeloid leukaemiaNature43512677015988530
- MorelFBrisMJHerryA2003Double minutes containing amplified bcr-abl fusion gene in a case of chronic myeloid leukemia treated by imatinibEur J Haematol70235912656747
- NavarroJTFeliuEGrauJ2007Monosomy 7 with severe myelodysplasia developing during imatinib treatment of Philadelphia-positive chronic myeloid leukemia: Two cases with a different outcomeAm J Hematol828495117563075
- NevianiPSanthanamRTrottaR2006The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET proteinCancer Cell535568
- Nguyen KhacFWaillMCRomanaSP2002Identical abnormality of the short arm of chromosome 18 in two Philadelphia-positive chronic myelocytic leukemia patients with erythroblastic transformation, resulting in duplication of BCR-ABL1 fusionCancer Genet Cytogenet13822612419580
- NicoliniFECormSLeQH2006Mutation status and clinical outcome of 89 imatinib mesylate-resistant chronic myelogenous leukemia patients: a retrospective analysis from the French intergroup of CML (Fi(phi)-LMC GROUP)Leukemia201061616642048
- NowellPCHungerfordDA1960A minute chromosome in human granulocytic leukemiaScience1321497
- O’BrienSGGuilhotFLarsonRA2003Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemiaN Engl J Med348994100412637609
- O’BrienSGMeinhardtPBondE2003Effects of imatinib mesylate (STI571, Glivec) on the pharmacokinetics of simvastatin, a cytochrome p450 3A4 substrate, in patients with chronic myeloid leukaemiaBr J Cancer891855914612892
- O’DwyerMEMauroMJBlasdelC2004Clonal evolution and lack of cytogenetic response are adverse prognostic factors for hematologic relapse of chronic phase CML patients treated with imatinib mesylateBlood103451514512312
- O’HareTWaltersDKStoffregenEPJiaTManleyPWMestanJCowan-JacobSWLeeFYHeinrichMCDeiningerMWDrukerBJ2005In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutantsCancer Res654500515930265
- PappasPKaravasilisVBriasoulisE2005Pharmacokinetics of imatinib mesylate in end stage renal disease. A case studyCancer Chemother Pharmacol563586015883819
- PavluJCzepulkowskiBKaczmarskiRJan-MohamedR2005Early blastic transformation with CNS infiltration in a patient with chronic myeloid leukaemia treated with imatinibLancet Oncol612815683824
- PendergastAMQuilliamLACripeLD1993BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor proteinCell75175858402896
- PengBDutreixCMehringG2004Absolute bioavailability of imatinib (Glivec) orally versus intravenous infusionJ Clin Pharmacol441586214747424
- PengBHayesMRestaD2004Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patientsJ Clin Oncol229354214990650
- PengCBrainJHuY2007Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cellsBlood1106788517395781
- PengBLloydPSchranH2005Clinical pharmacokinetics of imatinibClin Pharmacokinet448799416122278
- PicardSTitierKEtienneG2007Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemiaBlood1093496917192396
- Pienkowska-GrelaBWoronieckaRSolarskaI2007Complete cytogenetic and molecular response after imatinib treatment for chronic myeloid leukemia in a patient with atypical karyotype and BCR-ABL b2a3 transcriptCancer Genet Cytogenet174111517452251
- PuilLLiuJGishG1994Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathwayEMBO J13764738112292
- Quintas-CardamaAKantarjianHJonesD2007Dasatinib (BMS-354825) is active in Philadelphia chromosome-positive chronic myelogenous leukemia after imatinib and nilotinib (AMN107) therapy failureBlood109497916990591
- RadichJPDaiHMaoM2006Gene expression changes associated with progression and response in chronic myeloid leukemiaProc Natl Acad Sci USA1032794916477019
- RahmaniMNguyenTKDentP2007The multikinase inhibitor sorafenib induces apoptosis in highly imatinib mesylate-resistant BCR/ABL+ human leukemia cells in association with STAT5 inhibition and MCL-1 downregulationMol Pharmacol727889517595328
- RajappaSUppinSGRaghunadharaoD2004Isolated central nervous system blast crisis in chronic myeloid leukaemiaHematol Oncol221798115995975
- RenR2005Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemiaNat Rev Cancer51728315719031
- Roche-LestienneCPreudhommeC2003Mutations in the ABL kinase domain pre-exist the onset of imatinib treatmentSemin Hematol40S280212783380
- Roche-LestienneCSoenen-CornuVGrardel-DuflosN2002Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatmentBlood1001014812130516
- RoederIHornMGlaucheIHochhausAMuellerMCLoefflerM2006Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights and clinical implicationsNat Med121181417013383
- RoumiantsevSShahNPGorreME2002Clinical resistance to the kinase inhibitor STI-571 in chronic myeloid leukemia by mutation of Tyr-253 in the Abl kinase domain P-loopProc Natl Acad Sci USA9910700512149456
- RousselotPHuguetFReaD2007Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 yearsBlood109586016973963
- RowleyJD1973A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa stainingNature24329034126434
- RyttingMEWierdaWG2004Central nervous system relapse in two patients with chronic myelogenous leukemia in myeloid blastic phase on imatinib mesylate therapyLeuk Lymphoma451623615370215
- SattlerMGriffinJD2003Molecular mechanisms of transformation by the BCR-ABL oncogeneSemin Hematol4041012783368
- SawyersCLHochhausAFeldmanE2002Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II studyBlood993530911986204
- SeniorK2003Gleevec does not cross blood-brain barrierLancet Oncol419812681250
- ShahNPNicollJMNagarB2002Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemiaCancer Cell21172512204532
- ShahNPTranCLeeFY2004Overriding imatinib resistance with a novel ABL kinase inhibitorScience30539940115256671
- ShetASJahagirdarBNVerfaillieCM2002Chronic myelogenous leukemia: mechanisms underlying disease progressionLeukemia1614021112145676
- SilbermanGCrosseMGPetersonEA1994Availability and appropriateness of allogeneic bone marrow transplantation for chronic myeloid leukemia in 10 countriesN Engl J Med331106378090167
- SillaberCGesbertFFrankDA2000STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cellsBlood9521182510706883
- SkaggsBJGorreMERyvkinA2006Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutantsProc Natl Acad Sci USA103194667117164333
- SkorskiTBellacosaANieborowska-SkorskaM1997Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathwayEMBO J166151619321394
- SoveriniSMartinelliGRostiG2005ABL mutations in late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are associated with a greater likelihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on Chronic Myeloid LeukemiaJ Clin Oncol234100915867198
- TakayamaNSatoNO’BrienSG2002Imatinib mesylate has limited activity against the central nervous system involvement of Philadelphia chromosome-positive acute lymphoblastic leukaemia due to poor penetration into cerebrospinal fluidBr J Haematol119106812358909
- TalpazMShahNPKantarjianH2006Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemiasN Engl J Med35425314116775234
- TalpazMSilverRTDrukerBJ2002Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 studyBlood9919283711877262
- TanakaKArifMKyoT2000Transposition of duplicated chromosomal segment involving fused BCR-ABL gene or ABL oncogene alone in chronic myelocytic leukemia and Ph chromosome-positive acute leukemia with complex karyotypesCancer Genet Cytogenet11981410812164
- TauchiTOhyashikiK2006The second generation of BCR-ABL tyrosine kinase inhibitorsInt J Hematol8329430016757427
- TerreCEclacheVRousselotP2004Report of 34 patients with clonal chromosomal abnormalities in Philadelphia-negative cells during imatinib treatment of Philadelphia-positive chronic myeloid leukemiaLeukemia181340615190256
- ThomasJWangLClarkREPirmohamedM2004Active transport of imatinib into and out of cells: implications for drug resistanceBlood10437394515315971
- TsengPHLinHPZhuJ2005Synergistic interactions between imatinib mesylate and the novel phosphoinositide-dependent kinase-1 inhibitor OSU-03012 in overcoming imatinib mesylate resistanceBlood1054021715665113
- Van EttenRA2007Oncogenic signaling: new insights and controversies from chronic myeloid leukemiaJ Exp Med204461517353369
- VerbeekWKonigHBoehmJ2006Continuous complete hematological and cytogenetic remission with molecular minimal residual disease 9 years after discontinuation of interferon-alpha in a patient with Philadelphia chromosome-positive chronic myeloid leukemiaActa Haematol1151091216424660
- VilluendasRSteegmannJLPollanM2006Identification of genes involved in imatinib resistance in CML: a gene-expression profiling approachLeukemia2010475416598311
- VolpeGCignettiAPanuzzoC2007Alternative BCR/ABL splice variants in Philadelphia chromosome-positive leukemias result in novel tumor-specific fusion proteins that may represent potential targets for immunotherapy approachesCancer Res675300717545610
- von BubnoffNSchnellerFPeschelC2002BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective studyLancet3594879111853795
- WangLGiannoudisALaneS2007Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemiaClin Pharmacol Ther10.1038/sj.clpt.6100268
- WeisbergEManleyPWBreitensteinW2005Characterization of AMN107, a selective inhibitor of native and mutant Bcr-AblCancer Cell71294115710326
- WeisbergEManleyPMestanJ2006AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABLBr J Cancer941765916721371
- WeisbergECatleyLWrightRD2007Beneficial effects of combining nilotinib and imatinib in preclinical models of BCR-ABL+ leukemiasBlood10921122017068153
- WeisbergEGriffinJD2000Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell linesBlood95349850510828035
- WeisbergEManleyPWCowan-JacobSW2007Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemiaNat Rev Cancer73455617457302
- WeisserMSchleuningMHaferlachC2007Allogeneic stem-cell transplantation provides excellent results in advanced stage chronic myeloid leukemia with major cytogenetic response to pre-transplant imatinib therapyLeuk Lymphoma4829530117325889
- WestermannJKoppJvan LessenA2007Vaccination with autologous non-irradiated dendritic cells in patients with bcr/abl+ chronic myeloid leukaemiaBr J Haematol13729730617408402
- WetzlerMTalpazMVan EttenRA1993Subcellular localization of Bcr, Abl, and Bcr-Abl proteins in normal and leukemic cells and correlation of expression with myeloid differentiationJ Clin Invest921925398408645
- WhiteDLSaundersVAQuinnSR2007Imatinib increases the intracellular concentration of nilotinib, which may explain the observed synergy between these drugsBlood10936091017409347
- WildRCastanedaSFleflehC2004BMS-354825, a dual SRC/ABL kinase inhibitor, displays potent anti-tumor activity in a model of intracranial CML growthBlood104549a
- WolffNCRichardsonJAEgorinMIlariaRL2003The CNS is a sanctuary for leukemic cells in mice receiving imatinib mesylate for Bcr/Abl-induced leukemiaBlood1015010312595307
- YokotaAKimuraSMasudaS2007INNO-406, a novel BCR-ABL/Lyn dual tyrosine kinase inhibitor, suppresses the growth of Ph+ leukemia cells in the central nervous system, and cyclosporine A augments its in vivo activityBlood1093061416954504
- ZhengCLiLHaakM2006Gene expression profiling of CD34+ cells identifies a molecular signature of chronic myeloid leukemia blast crisisLeukemia2010283416617318