Abstract
After the increasing rate of deaths observed during the 1980s due to human immunodeficiency virus (HIV) infection, the health-related quality of life and life expectancy of persons with hemophilia have improved, mainly due to the progresses of replacement therapy and antiviral drugs and to the improvement of the global comprehensive care provided by specialized centers. As a consequence, an increasing number of hemophiliacs have reached an older age and nowadays physicians in hemophilia centers find that they must handle age-related clinical problems never previously observed in this population. The management of elderly persons with congenital hemophilia is discussed in the first part of this review. The second part describes the general aspects of acquired hemophilia due to anti-factor VIII autoantibodies, focusing on the clinical management of elderly patients, one of the groups most frequently affected by this acquired bleeding disorder.
Introduction
Since the early 1970s there have been dramatic improvements in the availability and quality of treatment for persons with hemophilia (CitationMannucci and Tuddenham 2001). As a result of this progress, barring the consequences of the human immunodeficiency virus (HIV) epidemics in the 1980s, the life span of hemophiliacs has progressively become similar to that of males in the general population, at least in more developed countries (CitationMejia-Carvajal et al 2006). Accordingly, a considerable number of hemophiliacs now reach old age: in Italy, for instance, nearly 8% of persons with severe hemophilia A or B are 65 years old or older (CitationTagliaferri et al 2006). However, with age, persons with hemophilia develop medical and surgical diseases (eg, cardiovascular diseases, prostatic hypertrophy, cancers, renal disease) not previously seen in this group. This aspect, together with the management of these patients, is discussed in the first part of this review. The second part addresses the main features and clinical management of acquired hemophilia due to anti-factor VIII antibodies, an acquired disease that may occur in elderly people.
Management of congenital hemophilia in the elderly
Life-expectancy, causes of death and quality of life
A number of studies have analyzed life expectancy and causes of death in hemophiliacs. However, the vast majority of them have evaluated only subgroups of patients, not the whole population. The mortality of patients infected by the hepatitis C virus (HCV) or HIV has been specially analyzed. For example, CitationDarby et al (1997) analyzed mortality in hemophiliacs in the United Kingdom infused with blood products contaminated with HCV. Other studies evaluated the mortality rate among HIV-infected patients in Spain (Citationdel Amo et al 2006), the United Kingdom (CitationDarby et al 2004a) and Canada (CitationArnold et al 2006). Finally, another study investigated the effect of factor VIII and IX inhibitors on mortality in a hemophilia population (CitationDarby 2004b).
Although only a small number of studies have so far evaluated mortality in the whole population of hemophiliacs (CitationStreet et al 2006), these are interesting because they cover the whole history of the clinical-therapeutic approach to hemophilia, from the dramatic increase in life expectancy between the 1940s and 1980s with the advent of replacement therapy with plasma-derived clotting factor to the catastrophic contamination of the plasma pool by HIV and HCV in the 1980s (CitationStreet et al 2006). In parallel with the therapeutic progresses, the causes of death have changed in hemophilia, hemorrhage being replaced by the acquired immunodeficiency syndrome (AIDS) as the leading cause of death (CitationMejia-Carvajal et al 2006). Indeed, the mortality curve in hemophiliacs over the years has a biphasic form with two peaks: one before the 1960s (before the widespread availability of factor replacement therapy) and the other in the 1980s–1990s (after the occurrence of AIDS and of the clinical manifestations of earlier HCV infections). This trend has been confirmed by a recent survey that analyzed the mortality rate and causes of death in 967 Dutch hemophiliacs (CitationPlug et al 2006). During the period of the study (1992–2001) the mortality rate was 2.3 times higher in patients with hemophilia than in the general male population. Moreover, comparing these data with those previously reported by the same group (CitationRosendaal et al 1989; CitationTriemstra et al 1995), the life expectancy of patients with severe hemophilia decreased from 63 years in the period 1973–1986 to 61 and 59 years in the period 1986–1992 and 1992–2001, respectively. However, the exclusion of virus-related deaths resulted in a life expectancy at birth of 74 years, similar to that of the unaffected Dutch male population. Similar trends were observed in studies involving Scottish (CitationLudlam 2000) and Canadian (CitationWalker 1998) hemophiliacs. From 1900 to 1942 the life expectancy of severe hemophiliacs in Sweden was 16.5 years; this increased to 23.2 years between 1943 and 1957 (CitationRamgren 1962) and to 50 years between 1957 and 1980 (CitationLarsson and Wiechel 1983). For comparison, the life expectancy of unaffected Swedish men during the latter period was 75 years. Another study analyzed 163 patients with severe hemophilia A living in Finland in the period between 1930–79 (CitationIkkala 1982). During 50 years of observation, the mean age at death increased from 7.8 years in 1930–39 to 25.5 years in 1970–79. The effect of HIV infection on age and causes of death among persons with hemophilia A was analyzed in a study conducted in the United States (CitationChorba et al 2001). The median age at death decreased from 55 years in 1979–1982 to 40.5 years in 1987–1990 and increased to 46 years in 1995–1998, although the median age at death of patients with hemophilia A and HIV-related disease in the latter period was 33 years, compared to 72 years for those without HIV-related disease. During the same period, hemophilia A-associated deaths decreased by 41%, with a 78% decrease among those who had HIV-related disease. This decline in HIV mortality was consistent with that observed in the general population of HIV-infected persons and reflected improvements in anti-retroviral therapy. On the other hand, another study that analyzed the causes of death among hemophiliacs not infected by HIV found that the leading cause of death was hemorrhage, followed by liver disease (hepatitis C and B), stroke and cancer. summarizes the literature data on outcomes in hemophilia patients.
Table 1 Analysis of the literature data on life expectancy of persons with hemophilia
Currently, children with hemophilia look forward to a normal life expectancy and excellent health-related quality of life (CitationGringeri et al 2006). The most important factors contributing to the improved quality of life, reduced morbidity and increased life expectancy are the availability for replacement therapy of high-quality factor concentrates (ie, virus inactivated plasma-derived clotting factor concentrates and recombinant products), insights into the management of hemophilic arthropathy and liver disease (ie, surveillance of patients with chronic hepatitis, especially with respect to cancer and liver failure, and newer treatment options such as antiviral treatment against HIV and HCV and liver transplantation) and the improved medical management provided by specialized hemophilia treatment centers. High-quality factor concentrates are important not only for reducing the likelihood of death from hemorrhage but also for improving the quality of life, especially through prophylactic regimens and home treatment. Secondary prophylaxis, in particular, is becoming frequently prescribed among hemophiliacs, although only few data are available in this setting (CitationTagliaferri et al 2006a). The impact of comprehensive care has been demonstrated by Soucie et al (2006) who found that hemophiliacs who had received care in a specialized hemophilia treatment center had a significantly decreased risk of death. The impact of comprehensive care is further exemplified by the analysis of 164 patients treated at the International Haemophilia Training Centre at Bangkok with blood and blood components from 1971 to 2000. The estimated probability of survival beyond 13 years of age of patients with severe hemophilia increased from 0.85 during the first decade of observation to 0.94 and 1 in the second and third decades (CitationChuansumrit et al 2004). The mortality rate fell from 30% to 14% and 5% over the three decades of observation. Despite the lack of treatment with high quality clotting factor concentrates in this center, the patients’ outcome improved with the development of a comprehensive approach to management.
On behalf of the Italian Association of Hemophilia Centers (AICE), we are conducting a multicenter study to evaluate the health status and health-related quality of life (HR-QoL) of severely affected Italian hemophiliacs of 65 years or older, recruited from Italian hemophilia centers (CitationTagliaferri et al 2006b). Another primary goal of the study is to provide information on whether or not there are differences in the global health status and HR-QoL between healthy elderly individuals and persons with hemophilia, who started their lives in the first part of the last century when the availability and quality of treatment were poor. This study will also allow us to establish whether or not the burden of diseases other than hemophilia (including cognitive impairment measured using instruments of geriatric medicine) is similar or different in cases and controls, and to understand which factors help people with severe hemophilia to cope with their disease and reach old age.
Clinical management of congenital hemophilia in elderly patients
Several clinical problems have become evident in hemophiliacs with increasing age. Hemophilia is associated with bleeding episodes, which often occur into the joints or muscles. Over time, recurrent bleeding into joints and muscles can cause permanent problems such as arthritis, chronic pain and joint damage resulting in poor control of balance and falls which ultimately worsen quality of life. Thus joint disease remains the leading cause of morbidity in those elderly hemophiliacs who had little or no access to prophylactic regimens and home treatment when they were younger. However, there is a paucity of studies investigating these aspects in hemophiliacs. An ongoing Australian study is evaluating the degree of joint-related dysfunction in persons with hemophilia older than 30 years and the feasibility of home exercise programs targeting balance training (CitationStreet 2006). We have recently reported the case of a 72-year-old hemophiliac with severe joint bleeding in whom treatment was converted from ‘on-demand’ to a secondary prophylaxis regimen (CitationTagliaferri et al 2006a). The latter significantly reduced the number of bleeding episodes in target joints, slowing the progression of the arthropathy and improving the patient’s perceived well-being. However, further studies, dealing with the approach to replacement therapy and the development of preventive physical therapy programs for elderly patients, are warranted.
Cardiovascular events are other clinical problems which increase with age. Attempts have been made to determine whether or not hemophilia protects from the development of atherosclerosis and cardiovascular events, but the results are contradictory. Studies of carotid and femoral artery intimalmedial thickness as a surrogate of atherosclerosis burden showed that hemophilia does not protect from atherosclerosis (CitationSramek et al 2001). On the other hand, hemophiliacs have a decreased mortality rate due to ischemic heart disease, supporting the views that they are protected from arterial thrombosis (CitationSramek et al 2003). Cases of myocardial infarction are rare in hemophiliacs (CitationFranchini 2004). Recently, CitationGirolami et al (2006) reviewed reported cardiovascular events in patients with hemophilia A. Among the 42 cases published so far, nine occurred in individuals aged 65 or over. Analyzing the literature data, the authors concluded that the bleeding diathesis did not help to prevent the occurrence of arterial occlusions. Thus factors which are known to predispose to atherothrombosis (eg, hypercholesterolemia, smoking, diabetes, hypertension, aging) appear to overcome the coagulation defect. Concomitant replacement therapy, especially with bypassing agents (ie, activated or non-activated prothrombin complex concentrates and recombinant activated factor VII), was identified as having a predominant role in thrombotic manifestations in the majority of the cases (CitationAledort 2004).
A number of successful cardiovascular interventions in elderly hemophiliacs have been carried out (CitationBovenzi et al 2003; CitationDe Bels et al 2004). However, these cases raise the question of how to conduct anticoagulant therapy in such naturally anticoagulated patients. The management of cardiovascular events in elderly hemophiliacs necessarily differs from that of non-hemophilic patients. Thus, while the treatment of acute coronary syndromes in hemophiliacs requires a combination of antithrombotic and replacement therapies, thrombolytic therapy for myocardial infarctions is unjustified in these patients due to the unacceptable risk of hemorrhage and should be substituted by alternative techniques such as percutaneous coronary angioplasty (CitationMafrici et al 2003). Similarly, long-term anticoagulation should be avoided whenever possible by using, for example, a bioprosthetic valve in valve surgery.
Elderly people with hemophilia are also at increased risk of chronic renal disease (CitationKulkarni et al 2003). This is probably due to chronic renal bleeding associated with the inherited coagulopathy which leads to structural renal abnormalities. Moreover, the concomitant presence of viral infections (eg, HIV and HCV) or exposure to nephrotoxic antiviral therapy may increase the incidence of renal dysfunction over time. Chronic renal disease is responsible for hypertension (CitationScully et al 2005), which is observed more frequently in elderly hemophiliacs than in the general population and is a risk factor not only for cardiac disease but also for cerebral hemorrhage.
Another important issue is the treatment of elderly patients with inhibitors. Although most inhibitors occur at a young age in patients with severe hemophilia, patients with mild hemophilia may develop inhibitors at an advanced age when they receive intensive factor VIII replacement therapy on the occasion of a surgical or invasive procedure for age-related problems such as prostatic hypertrophy, cardiovascular disease or cancer (CitationFranchini et al 2006a). Indeed, there are some concerns on the use of bypassing agents in those patients considered at higher risk of developing thrombotic complications. However, Leebek et al (2006) have recently reported the safe and effective use of recombinant activated factor VII in three elderly patients with mild hemophilia A and high-titer inhibitors.
Management of acquired hemophilia in the elderly
General aspects of acquired hemophilia
Acquired hemophilia is an uncommon but potentially life-threatening clinical syndrome characterized by the sudden onset of bleeding in patients with a negative family and personal history. Acquired hemophilia is caused by auto-antibodies that, in the vast majority of cases, are directed against functional epitopes of factor VIII causing its neutralization and/or accelerated clearance from the plasma (CitationDelgado et al 2003).
The incidence of acquired hemophilia has been estimated to be 0.2–1.0 cases per 1 million persons per year, with a mortality rate estimated to be in the range of 8 to 22% (CitationFranchini et al 2005a). Most hemorrhagic deaths occur within the first few weeks after presentation. The age distribution of autoantibodies is typically biphasic, with a small peak between 20 and 30 years (mainly postpartum inhibitors) and a larger peak in patients aged 68–80 years. Factor VIII inhibitors are distributed equally between sexes, although females predominate in the younger age group because of the association with pregnancy while males constitute the majority of patients with inhibitors over the age of 60 (CitationBoggio and Green 2001). In approximately 50% of cases factor VIII autoantibodies occur in patients without concomitant diseases (spontaneous antibodies), and in nearly 10% of cases autoantibodies appear during the postpartum period, usually in primiparous women within 3 months of delivery (CitationGreen and Lechner 1981; CitationSolymoss 1998). However, several other conditions and diseases (eg, autoimmune disorders, malignancies and drugs) have been associated with the development of factor VIII inhibitors (CitationBossi et al 1998).
The clinical picture of acquired hemophilia differs from that of “classical” hereditary hemophilia A. More than 80% of patients with factor VIII autoantibodies bleed into the skin, muscles or soft tissues (including retroperitoneal hematoma) and mucous membranes (eg, epistaxis, gastrointestinal and urological bleeds), whereas hemarthroses, a typical manifestation of congenital factor VIII deficiency, are unusual. Not rarely the hemorrhages in acquired hemophilia are serious or life-threatening, as in the case of cerebral hemorrhage or rapidly progressive retroperitoneal hematomas (CitationFranchini et al 2005a).
The diagnosis of acquired hemophilia is based on the demonstration of an isolated prolongation of the activated partial thromboplastin time, not corrected by incubating the patient’s plasma with equal volumes of normal plasma (mixing study), associated with low factor VIII levels and evidence of a factor VIII inhibitor (which can be titrated in Bethesda units (BU)/mL), in a patient with no previous personal or family history of bleeding (CitationCohen and Kessler 1996). reports the laboratory algorithm for the diagnosis of acquired hemophilia.
Figure 1 Diagnostic algorithm of acquired hemophilia.
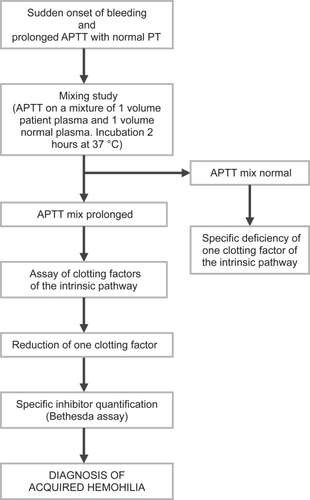
The treatment of acquired hemophilia is classically divided into two phases: acute and chronic. Acute phase treatment is focused on the bleeding symptoms (CitationLudlam et al 1994). Efficient hemostasis can be achieved with a variety of methods such as correction of FVIII deficiency (plasma-derived or recombinant FVIII concentrates, desmopressin) or bypassing the inhibitor (activated prothrombin complex concentrate [APCC], recombinant activated FVII [rFVIIa]) (CitationRoberts 1999). The choice of antihemorrhagic therapy depends mainly on the site and the entity of the bleeding. In patients with a high titer of inhibitor and severe hemorrhage, extracorporeal removal of the autoantibody by therapeutic plasmapheresis or immunoadsorption of immunoglobulins to staphylococcal protein A may be used (CitationGuillet 2001). The aim of the chronic phase of treatment is to eradicate the inhibitor, which is achieved by using immunosuppressive agents including corticosteroids and such drugs as cyclophosphamide, azathioprine, 6-mercaptopurine and vincristine (CitationGreen et al 1993). High-dose immunoglobulins and cyclosporin have also been shown to be effective in acquired hemophilia and can be considered as second-line therapy for those patients who do not respond to standard immunosuppressive regimens (CitationSchwartz et al 1995; CitationPetrovic et al 2000). More recently, serotherapy with anti-CD20 monoclonal antibody (rituximab) has been successfully used in immune disorders resulting from autoantibody formation, including acquired hemophilia (CitationWiestner et al 2002). Finally, immune tolerance induction protocols have been proposed for the eradication of autoantibodies against coagulation factors (CitationNemes and Pitlik 2000).
Clinical management of acquired hemophilia in elderly patients
As reported above, with the exception of post-partum cases, a large number of patients with acquired hemophilia are elderly.
Although acquired hemophilia is considered a very rare occurrence, its true incidence may be underestimated. Indeed, patients may be seen by several specialists and undergo dangerous invasive investigations and interventions before the correct diagnosis is made. The high mortality rate reported previously is in part related to a delay in the diagnosis. Besides misdiagnoses, acquired hemophilia may be underdiagnosed as many cases of clinically silent low titer inhibitors may be unrecognized unless patients undergo surgery or trauma.
In approximately 50% of the elderly patients with acquired hemophilia, no underlying primary disorders are identified (idiopathic forms), while most of the remaining cases are associated with autoimmune diseases or cancer, which increase with age (CitationHauser et al 1999; CitationYee et al 2000). The prognosis of acquired hemophilia in elderly patients is strongly linked to the associated primary disorder, which should be always searched for and, if possible, cured. The knowledge of the underlying disorder has important therapeutic consequences. Idiopathic cases with a low inhibitor titer and mild bleeding may be managed with desmopressin alone and may not require eradicating therapy but only a “watch and wait” strategy. Similarly, drug-induced inhibitors may require nothing else than close observation, since these inhibitors tend to disappear spontaneously within a few months after drug discontinuation. On the other hand, it is unlikely that cases associated with autoimmune disorders will remit spontaneously and thus they require more aggressive eradication therapy with steroids, whether or not combined with cytotoxic agents such as cyclophosphamide (CitationFranchini et al 2005b). However, particular care must be taken when using these agents in the elderly and the choice of drugs and dosages should be tailored to the individual patient’s age. An analysis of the data from the European Acquired Hemophilia Registry (CitationBaudo et al 2004) showed that infections related to immunosuppressive therapy are the first cause of death in patients with acquired hemophilia. Thus, less toxic agents should be preferred for inhibitor eradication in elderly patients. In this context, the use of rituximab seems to be particularly promising. We have recently reviewed the literature data on the use of rituximab in acquired hemophilia and have collected 50 cases, including many elderly patients (CitationFranchini et al 2006b). Although most of the reports were isolated cases, there was a high%age of complete or partial responses (86%) when rituximab was used alone or in association with immunosuppressive therapy. Thus, if confirmed by further controlled studies, this agent could be very useful in the management of such patients.
The presence of an underlying malignancy was associated with a poorer prognosis in a recent meta-analysis by CitationDelgado et al (2003). However, CitationSallah et al (2001), in a previous review of literature data on 41 cases of patients with cancer-associated factor VIII inhibitors, including patients with solid and hematologic malignancies, found a 70% rate of complete responses to treatment of the inhibitor. Low titer inhibitors associated with early stage tumors were more likely to disappear after treatment than high titer inhibitors and the eradication of autoantibody was associated with a higher overall survival. The same authors also observed that 22% of responders achieved complete remission after treatment of the cancer. On the basis of this high response rate, it appears that the treatment of the primary malignancy in patients with cancer-associated factors VIII inhibitors is of great importance, because this facilitates eradication of the antibody. The presence of an underlying cancer is not a contraindication to the use of immunosuppressive therapy aimed at eradicating the autoantibody, so that these patients should be treated in the same manner as other patients with acquired hemophilia.
Although only a few thrombotic adverse events have been recorded in patients with acquired hemophilia following the use of bypassing agents (CitationGuillet et al 2002; CitationRoberts et al 2004), particular care should be exercised when these drugs are used in elderly patients who should be monitored not only for clinical evidence of hemorrhage but also for the possible onset of thrombotic complications. Concomitant cardiovascular diseases should be ruled out before using desmopressin in elderly patients with congenital or acquired hemophilia (CitationMannucci 1997).
Conclusions
With their greatly prolonged life expectancy, persons with congenital hemophilia are now developing medical and surgical conditions previously not seen in this group. The new challenge for physicians in hemophilia centers is to provide optimal care for this aging population of patients. Their optimal management requires a hemophilia team with close cooperation among physicians from different specialties such as hematology, oncology, cardiology, and nephrology.
The prognosis of acquired hemophilia in elderly people depends on the underlying primary disorder. Particular care must be taken when choosing the inhibitor eradication regimen, which should be tailored to the patient’s age.
References
- AledortLM2004Comparative thrombotic event incidence after infusion of recombinant factor VIIa versus factor VIII inhibitor bypass activityJ Thromb Haemost21700815456478
- ArnoldDMJulianJAWalkerIRAssociation of Hemophilia Clinic Directors of Canada2006Mortality rates and causes of death among all HIV-positive individuals with hemophilia in Canada over 21 years of follow-upBlood108460416551974
- BaudoFde CataldoF2004Acquired hemophilia: a critical bleeding syndromeHaematologica899610014754611
- BoggioLNGreenD2001Acquired hemophiliaRev Clin Exp Hematol538940411844135
- BossiPCabaneJNinetJ1998Acquired haemophilia due to factor VIII inhibitors in 34 patientsAm J Med10540089831424
- BovenziFDe LucaLSignoreN2003Abciximab for the treatment of an acute thrombotic coronary occlusion during stent implantation in a patient with severe hemophilia BItal Heart J47283014664288
- ChorbaTLHomanRCClarkeMJ2001Effects of HIV infection on age and cause of death for persons with hemophilia A in the United StatesAm J Haematol6622940
- ChuansumritAKrasaesubSAngchaisuksiriP2004Survival analysis of patients with haemophilia at the International Haemophilia Training Centre, Bangkok, ThailandHaemophilia10542915357781
- CohenAJKesslerCM1996Acquired inhibitorsBailleres Clin Haematol933154
- DarbySCEwartDWGiangrandePL1997Mortality from liver cancer and liver disease in haemophilic men and boys in UK given blood products contaminated with hepatitis C. UK Haemophilia Centre Directors’ OrganisationLancet3501425319371165
- DarbySCKanSWSpoonerRJ2004aThe impact of HIV on mortality rates in the complete UK haemophilia population. UK Haemophilia Centre Doctors’ OrganisationAIDS185253315090806
- DarbySCKeelingDMSpoonerRJ2004bThe incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977–99. UK Haemophilia Centre Doctors’ OrganisationJ Thromb Haemost210475415219185
- De BelsDDemeereJLDugauquierJ2004Continuous infusion of factor VIIIc during heart surgery in a patient with haemophiliaEur J Anaesthesiol21984615719864
- del AmoJPerez-HoyosSMorenoA2006Trends in AIDS and mortality in HIV-infected subjects with hemophilia from 1985 to 2003: the competing risks for death between AIDS and liver diseaseJ Acquir Immune Defic Syndr416243116652037
- DelgadoJYimenez-YusteVHernandez-NavarroF2003Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factorsBr J Haematol121213512670328
- FranchiniM2004Thrombotic complications in patients with hereditary bleeding disordersThromb Haemost9229830415269825
- FranchiniMGandiniGDi PaolantonioT2005aAcquired hemophilia A: a concise reviewAm J Hematol80556316138334
- FranchiniMGirelliDOlivieriO2005bClinical heterogeneity of acquired hemophilia A: a description of 4 casesHaematologica90ECR1615753057
- FranchiniMSalvagnoGLLippiG2006aInhibitors in mild/moderate haemophilia A: An updateThromb Haemost96113816894451
- FranchiniMVeneriDLippiG2006bThe efficacy of rituximab in the treatment of inhibitor-associated hemostatic disordersThromb Haemost961192516894452
- GirolamiARuzzonEFabrisF2006Myocardial infarction and other arterial occlusions in hemophilia A patientsActa Haematol116120516914907
- GreenDLechnerK1981A survey of 215 non-hemophilic patients with inhibitors to factor VIIIThromb Haemost4520036792737
- GreenDRademakerAWBrietE1993A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodiesThromb Haemost7075378128430
- GringeriAMantovaniLvon MackensenS2006Quality of life assessment in clinical practice in haemophilia treatmentHaemophilia12Suppl. 322916683993
- GuilletBKriaaFHuisseMG2001Protein A sepharose immunoadsorption: immunological and haemostatic effects in two cases of acquired haemophiliaBr J Haematol1148374411564072
- GuilletBPinganaudCProulleV2002Myocardial infarction occurring in a case of acquired haemophilia during the treatment course with recombinant activated factor VIIThromb Haemost88698912362252
- HauserILechnerK1999Solid tumors and factor VIII antibodiesThromb Haemost821005710494753
- IkkalaEHelskeTMyllylaG1982Changes in the life expectancy of patients with severe hemophilia A in Finland in 1930–79Br J Haematol527126810913
- KulkarniRSoucieJMEvattBthe Hemophilia Surveillance System Project Investigators2003Renal disease among males with haemophiliaHaemophilia97031014750936
- LarssonSAWiechelB1983Deaths in Swedish hemophiliacs, 1957–80Acta Med Scand2141992066660026
- LeebeekFWGKappers-KlunneMCJieKSG2004Effective and safe use of recombinant factor VIIa (NovoSeven) in elderly mild haemophilia A patients with high-titre antibodies against factor VIIIHaemophilia10250315086322
- LudlamCAMorrisonAEKesslerC1994Treatment of acquired hemophiliaSem Hematol31169
- LudlamCALeeRJPrescottRJ2000Haemophilia care in central Scotland 1980–94. I. Demographic characteristics, hospital admission and causes of deathHaemophilia649450311012692
- MafriciABaudoF2003Hemophilia and percutaneous coronary interventionsItal Heart J4731314664289
- MannucciPM1997Desmopressin (DDAVP) in the treatment of bleeding disorders: the first 20 yearsBlood902515219326215
- MannucciPMTuddenhamEG2001The hemophilias – from royal genes to gene therapyN Engl J Med3441773911396445
- Mejia-CarvajalCCzapekEEValentinoLA2006Life expectancy in hemophilia outcomeJ Thromb Haemost4507916460431
- NemesLPitlikE2000New protocol for immune tolerance induction in acquired hemophiliaHaematologica8564811187874
- PetrovicMDeromEBaeleG2000Cyclosporine treatment of acquired hemophilia due to factor VIII antibodiesHaematologica85895610942956
- PlugIvan der BomJGPetersM2006Mortality and causes of death in patients with hemophilia, 1992–2001: a prospective cohort studyJ Thromb Haemost45101616460432
- RamgrenO1962A clinical and medico-social study of haemophilia in SwedenActa Med Scand17111190
- RobertsHR1999The use of agents that by-pass factor VIII inhibitors in patients with hemophiliaVox Sang77Suppl 1384110529686
- RobertsHSMonroeDMIIIHoffmanM2004Safety profile of recombinant factor VIIaSem Hematol1Suppl 11018
- RosendaalFRVarekampISmitC1989Mortality and causes of death in Dutch hemophiliacs, 1973–86Br J Haematol717162917132
- SallahSWanJY2001Inhibitors against factor VIII in patients with cancerCancer9110677411267950
- SchwartzRSGabrielDAAledortLM1995A prospective study of treatment of acquired (autoimmune) factor VIII inhibitors with high-dose intravenous gammaglobulinBlood867978047606010
- ScullyMFMacGregorDWalshM2005Preliminary results of a cross-sectional study of mild hemophilia A in Newfoundland, CanadaJ Thromb Haemost3Suppl. 1P1404
- SolymossS1998Postpartum acquired factor VIII inhibitors: results of a surveyAm J Haematol5914
- SoucieJMNussREvattB2000Mortality among males with hemophilia: relations with source of medical care. The Hemophilia Surveillance System Project InvestigatorsBlood964374210887103
- SramekAReiberJHCGerritsWBJ2001Decreased coagulability has no clinically relevant effect on atherogenesisCirculation104762711502699
- SramekAKriekMRosendaalFR2003Decreased mortality of ischaemic heart disease among carriers of haemophiliaLancet362351412907007
- StreetAHillKSussexB2006Haemophilia and ageingHaemophilia12Suppl. 381216683991
- TagliaferriARivoltaGFRossettiG2006aExperience of secondary prophylaxis in 20 adolescent and adult Italian hemophiliacsThromb Haemost96542317003937
- TagliaferriAFranchiniMvon MackensenS2006bHealth status and quality of life in elderly Italian patients with hemophiliaHaemophilia12Suppl. 2252 (abstract)
- TriemstraMRosendaalFRSmitC1995Mortality in patients with hemophilia: changes in a Dutch population from 1986 to 1992 and 1973 to 1986Ann Intern Med12382377486463
- WalkerIRJulianJAAssociation of Hemophilia Clinic Directors of Canada1998Causes of death in Canadians with haemophilia 1980–1995Haemophilia4714209873876
- WiestnerAChoHJAschAS2002Rituximab in the treatment of acquired factor VIII inhibitorBlood1003426812384448
- YeeTTTaherAPasiKJ2000A survey of patients with acquired hemophilia in a hemophilia centre over a 28-year periodClin Lab Haem222758