Abstract
In this review we consider the therapeutic potential of targeting Akt for the treatment of COPD. Akt is a serine/threonine protein kinase that functions as a signaling intermediate linked to multiple signaling programs involved in survival, inflammation, and growth. Akt is closely associated with key membrane-bound receptors and represents a convergent integration point for multiple stimuli implicated in COPD pathogenesis. Persistent activation of Akt secondary to somatic mutations in regulatory oncogenes, such as PTEN, may explain why inflammation in COPD does not resolve when smoking is ceased. Akt is also implicated in the systemic manifestations of COPD such as skeletal muscle wasting and metabolic disturbances. Furthermore, targeting Akt may provide a useful means of limiting the severity and duration of disease exacerbations in COPD. As such, Akt represents a particularly attractive therapeutic target for the treatment of COPD. Interestingly, current knowledge suggests that both inhibitors and activators of Akt may be useful for treating different clinical subpopulations of COPD patients.
Keywords:
Chronic obstructive pulmonary disease will be the third most common cause of death worldwide by 2020 (CitationMurray and Lopez 1996), and costs the global healthcare system tens of billions of dollars annually. For reasons that are largely unknown, COPD is only marginally responsive to all contemporary drugs, even powerful antiinflammatory glucocorticosteroids (CitationKeatings et al 1997; CitationBarnes 2000). COPD is diverse and encompasses emphysema, the proteolytic destruction of alveolar units; bronchitis, associated with massive goblet cell and mucous gland proliferation; and bronchiolitis, an inflammatory condition of small airways associated with fibroblast proliferation and fibrosis. The cause of most COPD is cigarette smoking, but the molecular pathogenesis of COPD is obscure. Inhaled smoke or irritants are thought to trigger alveolar macrophages and the epithelium to secrete tumor necrosis factor-alpha (TNF-α), interleukin 8 (IL-8), and chemokines such as macrophage inflammatory proteins (MIPs). These factors are chemotactic and activating factors for neutrophils, macrophages, and other inflammatory cells. Over time the lung also accumulates increased numbers of CD8+ lymphocytes, which are capable of triggering macrophage-dependent lung proteolysis. Emphysema results from destruction of alveolar units by proteases such as neutrophil elastase (NE; also a potent goblet cell secretagogue), macrophage metalloelastases like MMP-12 (CitationFinkelstein et al 1995; CitationHautamaki et al 1997), and possibly also by apoptosis of alveolar wall cells.
In the small airways, fibroblast proliferation and collagen deposition cause fixed airway obstruction (CitationHogg et al 2004). The resulting airflow limitation is compounded in many patients by mucus hypersecretion and inflammation. Lung destruction in COPD is well correlated with the intensity of inflammation and once inflammation is established in COPD, removing the provocative stimulus through smoking cessation does not resolve disease (CitationTurato et al 1995). Furthermore, it is unknown why COPD is associated with a very high prevalence of both viral and bacterial exacerbations (known triggers of the innate immune system, specifically macrophages and natural killer cells), prompting further damage to the lungs. It is believed that much of the deterioration that accompanies exacerbations is due to flaring of inflammation. This interpretation is supported by spikes in inflammatory markers during exacerbations measured in sputum and in breath condensates.
Although there remains much to be understood, our current understanding of molecular pathways in COPD pathogenesis implicates Akt as a central regulator. Akt, (also previously referred to as protein kinase B [PKB]), is an intracellular serine/threonine protein kinase that is activated by a broad range of cytokines (eg, TNFα) (CitationMurao et al 2000), growth factors (eg, PDGF, GM-CSF, CTGF) (CitationKlein et al 2000; CitationRauch et al 2000; CitationCrean et al 2002), and cigarette smoke components, including nicotine (CitationNakayama et al 2002; CitationWest et al 2003). In particular, Akt is a major target of PI3-kinase (PI3K) dependent signaling pathways (). On activation, Akt is recruited to membrane associated signaling complexes and activated by phosphorylation. In addition to Akt, PI3K activates multiple signaling kinases (PKC, MAPK, Btk, ILK) involved in key processes. Hence, targeting PI3K directly may be detrimental due to its pleiotropic activities.
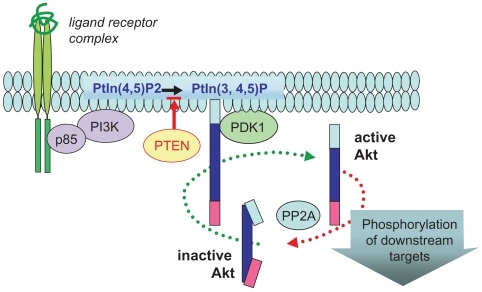
Figure 1 Ligand-targeted activation of Akt. Ligand-mediated activation of a broad range of receptors promotes recruitment of PI3K (p85 and p110 complex) to the plasma membrane, where this lipid kinase catalyzes the production of phosphatidylinositol-3,4,5-phosphate (PtIn3,4,5)-P. PTEN (lipid phosphatase) limits this reaction by reverting PtIns(3,4,5)-P to PtIns(3,4)-P. This phospholipid acts as a docking molecule for both Akt and its activator PDK-1, which activates Akt by direct phosphorylation of the critical T(activation)-loop residue (Thr-308). Once active, Akt is released from the membrane to target multiple cellular substrates and is subsequently inactivated by protein phosphatase2A (PP2A) dephosphorylation.
There are three known homologs of Akt that display a high level of homology at the amino acid level (). The most characterized isoform, Akt1, is expressed in various tissues including lung, and targets diverse substrates involved in critical cellular events such as cell survival, proliferation, and transcription.
Table 1 Mammalian Akt homologs
Hence, the multifunctional activities of Akt have the potential to coordinate cellular mechanisms that are strongly implicated in the destructive pathogenesis of COPD (). As Akt is a serine/threonine kinase it is amenable to selective pharmacological inhibition (CitationGlazer 1998).
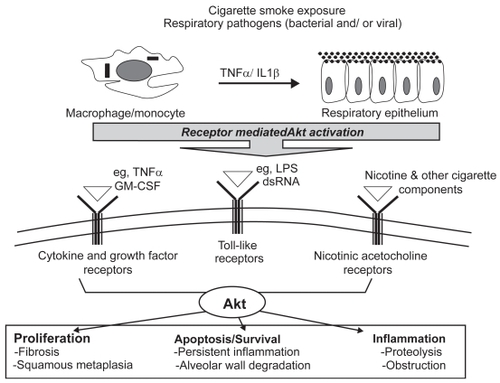
Figure 2 Akt is a molecular mediator of cellular processes central to COPD pathogenesis. Cell surface receptors on alveolar macrophages and the bronchial epithelium recognize and respond to cigarette smoke components and respiratory pathogens. These unique signaling modules converge onto and activate multiple pathways including Akt. Once active, Akt contributes to key cellular processes including proliferation, apoptosis, and inflammation. Therefore, dysregulated activation of Akt in a chronically inflamed environment has the potential to disrupt homeostasis, leading to an altered pathology responsible for dramatic and progressive decline in lung function.
Rationale for inhibiting Akt activity in COPD
Role in cell survival
Apoptosis governs the persistence and severity of inflammation. Akt promotes inflammatory cell survival in vivo. More specifically, oxidative stress and/or GM-CSF- mediated activation of Akt enhances cell survival of macrophages, neutrophils, and T lymphocytes (CitationJones et al 2000; CitationKlein et al 2000; CitationWang et al 2000). Akt inhibits apoptosis by four main mechanisms: (1) phosphorylation and inactivation of BAD (CitationDatta et al 1997, Citation1999), which normally binds to and inactivates antiapoptotic Bcl2 and BclXL, a critical step in the activation of the caspase protease pathway. However, since Akt displays pro-survival properties in tissues that do not express detectable levels of BAD, alternative mechanisms must exist. Akt also promotes survival by: (2) phosphorylation of lytic pro-caspase-9 (CitationCardone et al 1998). This event blocks the activity of pro-caspase-9 by altering the conformation of its substrate-binding pocket, thereby inhibiting apoptosis. Apoptosis can also be induced by expression of key genes involved in cell death. For example, withdrawal of survival factors promotes transcription of the Fas ligand gene involved in positively regulating the caspase protease pathway. Akt controls their expression by: (3) maintaining Forkhead family of transcriptional factors (AFX) in a transcriptionally silent form (CitationBiggs et al 1999; CitationBrunet et al 1999). In the absence of Akt signaling, AFX members are directed to the nucleus where they bind to insulin-responsive sequence (Irs) found in a number of promoters responsible for controlling apoptosis gene expression. When Akt activity is maintained for prolonged periods, Forkhead members are sequestered away from the nucleus by Akt-mediated phosphorylation. Phosphorylated AFX members are kept transcriptionally silent due to a physical interaction with the structural cytoplasmic protein, 14-3-3. (4) More recently, Akt has been shown to promote activation of the transcription factor CREB, which enhances transcription of cell survival genes such as the Bcl-2 family member, Mcl-1 (CitationWang et al 1999). Therefore, in a chronically inflamed COPD lung that is constantly exposed to cigarette smoke and airborne pathogens, Akt activity will be maintained for prolonged periods. The persistence of Akt-mediated survival of inflammatory cells may prove to be an integral process responsible for the accumulation of macrophages, neutrophils, and T lymphocytes in the airways, parenchyma, and pulmonary vasculature. A paradox that cannot be explained with current knowledge is that the epithelium and T cells from COPD patients have a somewhat increased rate of apoptosis (CitationHodge et al 2005). CitationMajo et al (2001) have reported enhanced parenchymal apoptosis in COPD in association with lymphocytic infiltration. The increased rate of apoptosis needs to be balanced against evidence for enhanced proliferation (CitationYokohori et al 2004). CitationTomita and colleagues (2002) found that alveolar macrophage apoptosis was decreased in macrophages from smokers in association with increased p21(Cip/Waf) BCLxL expression. Neutrophil apoptosis is markedly reduced during COPD exacerbations (CitationPletz et al 2004). It has also been suspected for some time that vascular endothelial cell death may be a pathogenic contributor to the development of emphysema in COPD (CitationKasahara et al 2000).
Role in proliferation
Akt may promote in situ macrophage and fibroblast proliferation. Macrophage proliferation in COPD has been recognized for several decades (CitationBitterman et al 1984) notwithstanding recent evidence suggesting increased apoptotic rates. Most in vitro evidence suggests that smoke directly suppresses fibroblast proliferation in COPD but the pattern of growth factor expression in thickened small airways suggests the opposite occurs in vivo. The in vivo response of parenchymal fibroblasts is less certain. Macrophage and fibroblast proliferation in COPD worsens inflammation and promotes airway fibrosis, which leads to fixed obstruction in small airways. Inhaled smoke and pathogen products released during exacerbations cause lung tissue damage, which triggers tissue repair mechanisms releasing multiple growth factors including PDGF, TGFβ, IGF1, EGF, CTGF, and bFGF, all of which activate Akt.
Recently, Akt has been implicated in the regulation of cellular proliferation and cell cycle control, a function more usually associated with MAP kinases. Akt inactivates the cell cycle regulator GSK3 by phosphorylation (CitationShaw et al 1997), promoting cell cycle entry by targeting D-type cyclins for proteolysis (CitationDiehl et al 1998). Hence, mitogen-induced activation of cyclin D ultimately leads to the G1→S transition of the cell cycle. Furthermore, by phosphorylating and inactivating AFX transcription factor, Akt also blocks expression of the cell cycle inhibitor p27kip1, whose expression is AFX dependent (CitationMedema et al 2000). This prevents p27kip1 from binding to the cyclinD/CDK2 complex and derepressing cell cycle arrest.
Role in inflammation
Akt is directly activated by gram-negative bacterial cell wall lipopolysaccharide (LPS) (CitationBozinovski et al 2002, Citation2004) and other innate immunity triggers implicated in COPD exacerbations (CitationSalh et al 1998). In addition, Akt is activated by oxidative stress, in part via the EGF receptor (EGFR) (CitationWang et al 2000), both of which are strongly implicated in COPD (CitationMacNee 2000). Akt selectively triggers NFκB p65/RelA heterodimer activation to coordinate monocyte and neutrophil recruitment in response to smoke and infection. The transduction pathways and inflammatory responses activated by cigarette smoke and infectious pathogen products such as LPS trigger innate immunity-mediated exacerbations in a similar manner, as inferred by coordinated expression of chemoattractant factors following activation of NFκB. This is not just because LPS is a biologically active component of cigarette smoke occurring in concentrations known to cause lung disease (CitationHasday et al 1999). It is also because cigarette smoke consists of multiple components capable of stimulating signal transduction pathways that are similar to those triggered by receptors of the innate immune system. Bacterial endotoxins stimulate lung epithelial cells to release neutrophil and monocyte chemotactic peptides (CitationKoyama et al 1999, Citation2000) by coordinated activation of Toll-like receptors (TLRs) that respond to LPS (gram-negative cell wall component; TLR4) and lipoteichoic acid (gram-positive cell wall component; TLR2) (CitationBrightbill et al 1999; CitationTakeuchi et al 1999; CitationUnderhill et al 1999). Consistent with LPS, cigarette smoke extract also promotes release of chemotactic factors (CitationMasubuchi et al 1998). Binding sites for NFκB are present in the promoter region of many inflammatory mediators including GM-CSF (CitationThomas et al 1997), MCP-1 (CitationTakaya et al 2000), and IL-8 (CitationOzes et al 1999); and NFκB cooperates as part of a higher order complex to promote transcription. NFκB is normally sequestered in the cytoplasm through an association with inhibitor of NFκB (IκB). When cells are exposed to activation signals such as LPS, oxidative stresses, and TNFα, IκB is phosphorylated and targeted for proteolytic destruction. This process is promoted by Akt, which triggers IκB kinase alpha (IKKa) to phosphorylate IκB (CitationOzes et al 1999). Akt can also enhance the DNA binding activity of NFκB by targeting the transactivation domain of the p65 subunit for phosphorylation (CitationSizemore et al 1999; CitationMadrid et al 2000).
Akt, lung cancer, and acquired somatic mutations of the lung epithelium
Lung cancer is the most frequent cause of tumor-associated deaths in the US and is strongly associated with chronic smoking. Lung cancer arises from a series of pathological changes linked to both gross genetic alterations and quite discrete acquired gene changes known as somatic mutations. Airflow obstruction is commonly present in patients with bronchial carcinoma (CitationCongleton and Muers 1995), and the presence of moderate to severe airflow obstruction is a significant predictor of incident lung cancer (CitationMannino et al 2003). Therefore, a logical and widely accepted corollary is that chronic inflammation predisposes to the development of cancer (CitationMarx 2004). Since the acquisition of somatic mutations predisposes to lung cancer, we have previously proposed that non-inherited genetic abnormalities also contribute to the pathogenesis of COPD (CitationAnderson and Bozinovski 2003). As only 15% of smokers develop COPD, much recent research has focused on identifying inherited genetic risk factors such as α1-antitrypsin deficiency associated with early onset COPD. However, less then 1% of COPD patients display α1-antitrypsin deficiencies, and searches for other heritable mutations including TNFα, IL-10, MMPs, and IL-8 have yielded limited success. We have proposed the novel hypothesis that a fundamental determinant of COPD is the acquisition of mutations (caused by mutagens in cigarette smoke) in regulatory oncogenes that control Akt activation status in the epithelium. This hypothesis is supported by cytogenetic studies on the molecular pathogenesis of lung cancer that have unequivocally demonstrated very frequent loss of heterozygosity (LOH) somatic mutations in preneoplastic/dysplastic epithelium of smokers (CitationWistuba et al 1997; Citation2002).
Indeed it is now apparent that the epithelium can become a mosaic of epithelial lineages, each carrying one or more somatic mutations. Although histological abnormalities in the epithelium associated with smoking can resolve after cessation, these somatic mutations persist as much as inflammation persists in the mucosa of ex-smokers (CitationWistuba et al 1997). Multiple gene mutations appear necessary to develop lung cancer, but single gene loci changes may be sufficient to trigger COPD. Of the known oncogenic mutations in the epithelium, three of the most frequent directly promote Akt activation. These are: (1) oncogenic mutations of Ras (p21); a GTP bound form of Ras interacts with PI3K, that in turn activates Akt. Ras mutations occur in approximately 30% of all human malignancies with a >30% mutation frequency in the bronchial epithelium of human lung cancer patients (CitationScott et al 1997). Ras-transformed adherent epithelial cells display elevated levels of Akt activity, and inhibition of this activity blocks the transforming potential of Ras by inducing cell death (CitationKhwaja et al 1997). (2) The lipid phosphatase tumor suppressor PTEN degrades phospholipid products generated by PI3K, thereby negatively regulating the activity of Akt. PTEN is mutated in a large fraction of all human cancers, with a mutation frequency approaching that of p53. More recently, PTEN mutations have been found in 6/15 (40%) of small cell lung carcinomas (SCLC) (CitationKohno et al 1998). Loss of PTEN activity attributed to homozygous deletions of the PTEN gene correlate with elevated levels of Akt activity, and reconstitution of PTEN restores normal Akt regulation (CitationStambolic et al 1998). (3) A constitutively active form of PI3K has been reported to exist in a small-scale screen of SCLC cell lines. Of the five cell lines tested, all exhibited high basal constitutive PI3K activity, which resulted in elevated basal Akt activity (CitationMoore et al 1998).
Preneoplastic changes also occur more commonly in smokers and may manifest as a phenotypic abnormality in the bronchial epithelium known as squamous metaplasia. Here, ciliated epithelial cells that normally trap and remove airborne particulates and mucus are replaced by flat, non-ciliated cells. The consequences of this altered phenotype are believed to include airway obstruction mediated by mucus accumulation. In addition, impaired pathogen clearance has been implicated in lower airway bacterial colonization and is prominent in advanced COPD. The mechanism that drives this altered phenotype is obscure and thought to be associated with an ongoing damage/repair cycle triggered by chronic smoking; however, Akt may prove to be an important molecular regulator of this process. Conceptually, sustained activation of Akt in the bronchial epithelium as a consequence of exposure to cigarette smoke favors an ongoing proliferative process that prevents differentiation into normal epithelial cells. Consistent with this hypothesis, nicotine has been shown to modulate the phenotype of normal airway epithelial cells via an Akt dependant mechanism (CitationNakayama et al 2002; CitationWest et al 2003).
Rationale for selective restoration of Akt activity in COPD
Akt and apoptosis-mediated emphysema
The long-standing paradigm for alveolar septal wall destruction in emphysema involves an imbalance between proteases and antiproteases that results in digestion of elastin and other structural proteins. However, alternate mechanisms may also exist as alveolar matrix destruction is not prevalent in cystic fibrosis, acute asthma, and other lung diseases associated with excessive inflammation and proteolysis. A recent model for the loss of lung tissue in emphysema implicates enhanced apoptosis as supported by animal models and a small number of human studies. Targeting disruption of vascular endothelial growth factor (VEGF) in mice results in increased alveolar and bronchial cell apoptosis, airspace enlargement, and persistent changes in lung elastic recoil (CitationTuder et al 2000, Citation2003; CitationTang et al 2004). Akt plays a critical role in VEGF-mediated cell survival (CitationGerber et al 1998) and, hence, would most likely prevent apoptosis in VEGF depleted lungs, although this hypothesis has yet to be tested. Even though these findings are quite striking in animal models, the role of VEGF in human COPD disease has not been clearly established. Despite there being no strong evidence implicating a defect in VEGF expression in emphysema, oxidative stress has been shown to impair VEGF survival signaling via inhibition of PI3K/Akt (Citationel-Remessy et al 2005). More recently, marked alveolar apoptosis has been observed in end-stage emphysema patients (CitationYokohori et al 2004; CitationCalabrese et al 2005). Selective restoration of Akt in VEGF dependent endothelial lung cells would therefore prevent accelerated alveolar apoptosis associated with cellular destruction in emphysema.
Akt and muscle wasting in COPD
COPD elicits a persistent local inflammatory process that has quite marked peripheral consequences such as the wasting syndrome seen in advanced COPD. In COPD, even clinically obese patients have loss of lean muscle mass identified on MRI imaging. Locally, cytokines such as IL-6, TNFα, and IL-1β are released in chronically inflamed COPD lungs, which can act on multiple organ systems, tissues, and cell types. Such inflammatory cytokines are known to impair activity of anabolic hormones such as insulin growth factor-1 (IGF-1) (CitationSpate and Schulze 2004). Akt is activated by IGF-1, an important mediator of signaling programs in skeletal muscle. For instance, increasing Akt activation has the potential to induce hypertrophic pathways while repressing atrophic processes (CitationBodine et al 2001; CitationRommel et al 2001). Akt promotes skeletal muscle hypertrophy via a number of downstream signaling pathways; direct and indirect targets include the mammalian target of rapamycin (mTOR), p70 S6 kinase (p70S6K), PHAS-1 (4EBP-1), and glycogen synthase kinase 3β (CitationGlass 2003). Reduced IGF-1 expression is observed in most wasting syndromes (CitationSpate and Schulze 2004), which will impact on Akt activity in muscle tissue. In rodents, selective overexpression of Akt1 in skeletal muscle cells causes hypertrophy and protects against denervation-induced atrophy, while mice with genetically disrupted Akt1 display growth defects (CitationChen et al 2001). Thus, the IGF-1/Akt signaling pathway provides a potential therapeutic target for promoting muscle hypertrophy in chronic diseases where wasting manifests. However, since Akt is ubiquitously expressed and promotes cell survival, proliferation, and inflammation, selective targeting of Akt in affected tissue would be most efficacious.
Summary
COPD is a heterogenous disease that is pathologically diverse, complex in nature, and is associated with defects in cell growth, survival, and inflammation. Consequently, developing an effective therapeutic strategy is difficult as multiple cellular processes need to be targeted. Dysregulation of Akt activity will impact on all these essential cellular processes, therefore implicating Akt as an attractive therapeutic target. The strategy of modulating Akt activity will develop as our understanding of COPD pathogenesis progresses, since there is evidence for either inhibiting or restoring Akt activity. The significance of Akt will become more apparent as we evolve a better understanding of the cellular processes that drive the altered phenotype that leads to reduced lung function. The challenge that lies ahead is that of generating highly specific therapies that either block or restore Akt activity in discrete tissue compartments.
References
- AndersonGPBozinovskiS2003Acquired somatic mutations in the molecular pathogenesis of COPDTrends Pharmacol Sci2471612559770
- BarnesPJ2000Chronic obstructive pulmonary diseaseN Engl J Med3432698010911010
- BiggsWHMeisenhelderJHunterT1999Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR 1Proc Natl Acad Sci U S A967421610377430
- BittermanPBSaltzmanLEAdelbergS1984Alveolar macrophage replication. One mechanism for the expansion of the mononuclear phagocyte population in the chronically inflamed lungJ Clin Invest7446096746904
- BodineSCStittTNGonzalezM2001Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivoNat Cell Biol310141911715023
- BozinovskiSJonesJBeavittSJ2004Innate immune responses to LPS in mouse lung are suppressed and reversed by neutralization of GM-CSF via repression of TLR-4Am J Physiol Lung Cell Mol Physiol286L8778514617520
- BozinovskiSJonesJEVlahosR2002Granulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappa B and AP-1 in vivoJ Biol Chem277428081412208854
- BrightbillHDLibratyDHKrutzikSR1999Host defense mechanisms triggered by microbial lipoproteins through toll-like receptorsScience285732610426995
- BrunetABonniAZigmondMJ1999Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factorCell968576810102273
- CalabreseFGiacomettiCBegheB2005Marked alveolar apoptosis/proliferation imbalance in end-stage emphysemaRespir Res61415705190
- CardoneMHRoyNStennickeHR1998Regulation of cell death protease caspase-9 by phosphorylationScience2821318219812896
- ChenWSXuPZGottlobK2001Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 geneGenes Dev152203811544177
- CongletonJMuersMF1995The incidence of airflow obstruction in bronchial carcinoma, its relation to breathlessness, and response to bronchodilator therapyRespir Med8929167597269
- CreanJKFinlayDMurphyM2002The role of p42/44 MAPK and protein kinase B in connective tissue growth factor induced extracellular matrix protein production, cell migration, and actin cytoskeletal rearrangement in human mesangial cellsJ Biol Chem277441879412218048
- DattaSRBrunetAGreenbergME1999Cellular survival: a play in three AktsGenes Dev1329052710579998
- DattaSRDudekHTaoX1997Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machineryCell91231419346240
- DiehlJAChengMRousselMF1998Glycogen synthase kinase-3 beta regulates cyclin D1 proteolysis and subcellular localizationGenes Dev1234995119832503
- el-RemessyABBartoliMPlattDH2005Oxidative stress inactivates VEGF survival signaling in retinal endothelial cells via PI 3-kinase tyrosine nitrationJ Cell Sci1182435215615788
- FinkelsteinRFraserRSGhezzoH1995Alveolar inflammation and its relation to emphysema in smokersAm J Respir Crit Care Med1521666727582312
- GerberHPMcMurtreyAKowalskiJ1998Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activationJ Biol Chem27330336439804796
- GlassDJ2003Signalling pathways that mediate skeletal muscle hypertrophy and atrophyNat Cell Biol5879012563267
- GlazerRI1998The protein kinase ABC’s of signal transduction as targets for drug developmentCurr Pharm Des42779010197043
- HasdayJDBascomRCostaJJ1999Bacterial endotoxin is an active component of cigarette smokeChest1158293510084499
- HautamakiRDKobayashiDKSeniorRM1997Requirement for macrophage elastase for cigarette smoke-induced emphysema in miceScience277200249302297
- HodgeSHodgeGHolmesM2005Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessationEur Respir J254475415738287
- HoggJCChuFUtokaparchS2004The nature of small-airway obstruction in chronic obstructive pulmonary diseaseN Engl J Med35026455315215480
- JonesRGParsonsMBonnardM2000Protein kinase B regulates T lymphocyte survival, nuclear factor kappaB activation, and Bcl-X(L) levels in vivoJ Exp Med19117213410811865
- KasaharaYTuderRMTaraseviciene-StewartL2000Inhibition of VEGF receptors causes lung cell apoptosis and emphysemaJ Clin Invest10613111911104784
- KeatingsVMJatakanonAWorsdellYM1997Effects of inhaled and oral glucocorticoids on inflammatory indices in asthma and COPDAm J Respir Crit Care Med15554289032192
- KhwajaARodriguez-VicianaPWennstromS1997Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathwayEmbo J162783939184223
- KleinJBRaneMJScherzerJA2000Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathwaysJ Immunol16442869110754327
- KohnoTTakahashiMMandaR1998Inactivation of the PTEN/MMAC1/TEP1 gene in human lung cancersGenes Chromosomes Cancer2215269598803
- KoyamaSSatoENomuraH1999The potential of various lipopolysaccharides to release monocyte chemotactic activity from lung epithelial cells and fibroblastsEur Respir J,145455210543273
- KoyamaSSatoENomuraH2000The potential of various lipopolysaccharides to release IL-8 and G-CSFAm J Physiol Lung Cell Mol Physiol278L6586610749742
- MacNeeW2000Oxidants/antioxidants and COPDChest117303S17S10843965
- MadridLVWangCYGuttridgeDC2000Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaBMol Cell Biol2016263810669740
- MajoJGhezzoHCosioMG2001Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysemaEur Respir J179465311488331
- ManninoDMAguayoSMPettyTL2003Low lung function and incident lung cancer in the United States: data From the First National Health and Nutrition Examination Survey follow-upArch Intern Med16314758012824098
- MarxJ2004Inflammation and cancer: the link grows strongerScience306966815528423
- MasubuchiTKoyamaSSatoE1998Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activityAm J Pathol1531903129846980
- MedemaRHKopsGJBosJL2000AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1Nature404782710783894
- MooreSMRintoulRCWalkerTR1998The presence of a constitutively active phosphoinositide 3-kinase in small cell lung cancer cells mediates anchorage-independent proliferation via a protein kinase B and p70s6k-dependent pathwayCancer Res585239479823338
- MuraoKOhyamaTImachiH2000TNF-alpha stimulation of MCP-1 expression is mediated by the Akt/PKB signal transduction pathway in vascular endothelial cellsBiochem Biophys Res Commun276791611027549
- MurrayCJLLopezAD1996The global burden of diseaseA comprehensive assessment of mortality and disability from diseases, injuries and risk factors in 1990 and projected to 2020Cambridge, Massachusetts GBD Series Volume IHarvard School of Public Health on behalf of the World Health Organization and the World Bank
- NakayamaHNumakawaTIkeuchiT2002Nicotine-induced phosphorylation of Akt through epidermal growth factor receptor and Src in PC12h cellsJ Neurochem831372912472891
- OzesONMayoLDGustinJA1999NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinaseNature40182510485710
- PletzMWIoanasMde RouxA2004Reduced spontaneous apoptosis in peripheral blood neutrophils during exacerbation of COPDEur Respir J23532715083750
- RauchBHWeberABraunM2000PDGF-induced Akt phosphorylation does not activate NF-kappa B in human vascular smooth muscle cells and fibroblastsFEBS Lett4813710984605
- RommelCBodineSCClarkeBA2001Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathwaysNat Cell Biol310091311715022
- SalhBWageyRMarottaA1998Activation of phosphatidylinositol 3-kinase, protein kinase B, and p70 S6 kinases in lipopolysaccharide-stimulated Raw 264.7 cells: differential effects of rapamycin, Ly294002, and wortmannin on nitric oxide productionJ Immunol1616947549862729
- ScottFMModaliRLehmanTA1997High frequency of K-ras codon 12 mutations in bronchoalveolar lavage fluid of patients at high risk for second primary lung cancerClin Cancer Res3479829815708
- ShawMCohenPAlessiDR1997Further evidence that the inhibition of glycogen synthase kinase-3beta by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216FEBS Lett416307119373175
- SizemoreNLeungSStarkGR1999Activation of phosphatidylinositol 3-kinase in response to interleukin- 1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunitMol Cell Biol19479880510373529
- SpateUSchulzePC2004Proinflammatory cytokines and skeletal muscleCurr Opin Clin Nutr Metab Care7265915075917
- StambolicVSuzukiAde la PompaJL1998Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTENCell9529399778245
- TakayaHAndohAShimadaM2000The expression of chemokine genes correlates with nuclear factor-kappaB activation in human pancreatic cancer cell linesPancreas21324010881930
- TakeuchiOHoshinoKKawaiT1999Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall componentsImmunity114435110549626
- TangKRossiterHBWagnerPD2004Lung-targeted VEGF inactivation leads to an emphysema phenotype in miceJ Appl Physiol97155966 discussion 154915208295
- ThomasRSTymmsMJMcKinlayLH1997ETS1, NFkappaB and AP1 synergistically transactivate the human GM-CSF promoterOncogene142845559190901
- TomitaKCaramoriGLimS2002Increased p21(CIP1/WAF1) and B cell lymphoma leukemia-x(L) expression and reduced apoptosis in alveolar macrophages from smokersAm J Respir Crit Care Med1667243112204872
- TuderRMKasaharaYVoelkelNF2000Inhibition of vascular endothelial growth factor receptors causes emphysema in ratsChest117281S10843952
- TuderRMZhenLChoCYTaraseviciene-StewartL2003Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockadeAm J Respir Cell Mol Biol29889712600822
- TuratoGDi StefanoAMaestrelliP1995Effect of smoking cessation on airway inflammation in chronic bronchitisAm J Respir Crit Care Med152126277551380
- UnderhillDMOzinskyASmithKD1999Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophagesProc Natl Acad Sci U S A96144596310588727
- WangJMChaoJRChenW1999The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREBMol Cell Biol19619520610454566
- WangXMcCulloughKDFrankeTF2000Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survivalJ Biol Chem275146243110799549
- WestKABrognardJClarkAS2003Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cellsJ Clin Invest111819012511591
- WistubaIILamSBehrensC1997Molecular damage in the bronchial epithelium of current and former smokersJ Natl Cancer Inst891366739308707
- WistubaIIMaoLGazdarAF2002Smoking molecular damage in bronchial epitheliumOncogene21729830612379874
- YokohoriNAoshibaKNagaiA2004Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysemaChest1256263214769747