Abstract
COPD is a widespread inflammatory respiratory disorder characterized by a progressive, poorly reversible airflow limitation. Currently available therapies are mostly based on those used to treat asthma. However, such compounds are not able to effectively reduce the gradual functional deterioration, as well as the ongoing airway and lung inflammation occurring in COPD patients. Therefore, there is an urgent need to improve the efficacy of the existing drug classes and to develop new treatments, targeting the main cellular and molecular mechanisms underlying disease pathogenesis. These therapeutic strategies will be highlighted in the present review.
Introduction
COPD is a chronic respiratory disease characterized by a progressive, not fully reversible airflow limitation, which is associated with an abnormal inflammatory response of the lungs to noxious particles or gases (CitationCelli et al 2004). In particular, the main risk factor for COPD is tobacco smoking, whose pathogenic action may be potentiated by other harmful agents such as air pollution. However, because about 20% of COPD patients are not smokers and only 15%–20% of smokers develop COPD, a genetically based individual susceptibility also plays an important role in disease pathogenesis. Indeed, in addition to the well known defects occurring in the α 1-antitrypsin gene, which are responsible for a very small percentage of COPD cases, several different polymorphisms involving other genes encoding tumor necrosis factor-α (TNF-α ), matrix metalloproteinases (MMPs), antioxidants, and other molecules have also been detected (CitationMolfino 2004). These complex interactions between environmental and genetic factors make COPD one of the leading causes of death worldwide, and the current epidemiological projections predict further increases in its prevalence and mortality during the next decades.
The main cellular elements of the inflammatory process that characterizes COPD include neutrophils, macrophages, and predominantly cytotoxic (Tc) CD8+ T lymphocytes (CitationLacoste et al 1993; CitationSaetta et al 1999; CitationShapiro 1999). Migration and activation of these cells are mediated by a complex network of cytokines and chemokines synthesized by both inflammatory and resident cells, the latter including fibroblasts, and epithelial, endothelial, and airway smooth muscle cells. In COPD, airway and lung inflammation is associated with pulmonary structural changes, arising from a relative imbalance between elastolytic proteinases and their inhibitors (CitationShapiro and Ingenito 2005). Therefore, the resulting altered turnover of extracellular matrix leads to the parenchymal destruction and loss of elastic recoil typical of emphysema. Conversely, collagen deposition and fibrosis characterize the small airways, thereby thickening their wall and further contributing to airflow limitation in COPD. On the other hand, the same proteinases that cause elastolysis such as MMP-9, may also activate fibrogenic growth factors like transforming growth factor-β (TGF-β ), which is in turn involved in MMP-9 activation thus triggering a positive, feed-forward pathogenic circuit (CitationBarnes et al 2003). Moreover, oxidative stress is heavily implicated in both the inflammatory and tissue-damaging processes detectable in COPD patients, who are subjected to a remarkable oxidant burden due to cigarette smoking, air pollution, and intense metabolic activity of inflammatory and structural cells (Maselli and Pelaia 2005).
Currently available therapies for COPD are mostly based on those used to treat asthma, mainly including bronchodilators and anti-inflammatory drugs such as corticosteroids. In COPD, however, these compounds are less effective than in asthma. With regard to bronchodilators such as long-acting β 2-adrenergic agonists and anticholinergics, though airflow limitation in COPD is characterized by poor reversibility, when given to COPD patients these drugs seem to be quite effective in improving lung function, exercise tolerance, and health status, as well as in reducing symptoms and frequency of exacerbations (CitationCazzola and Donner 2000; CitationCasaburi et al 2002). Much more evident is the relatively low sensitivity of COPD patients to inhaled corticosteroids, possibly for several reasons including corticosteroid-dependent reduction of neutrophil apoptosis, putative resistance of CD8+ T cells to corticosteroids and, most importantly, oxidative stress-induced inhibition of expression and activity of histone deacetylases (HDACs) in alveolar macrophages (CitationIto et al 2005); HDACs are indeed essential for corticosteroid action in that these enzymes play a key role in remodeling chromatin structure and switching off pro-inflammatory genes.
Therefore, there is an urgent need to achieve a much better control of COPD, and ongoing pharmacological research is at least in part focused on improving the efficacy of currently available drug classes, but it especially aims to develop novel therapeutic strategies, specifically targeting the main cellular and molecular mechanisms underlying disease pathogenesis. This review will outline the most promising approaches under development for COPD treatment, including improved bronchodilators and corticosteroids, as well as new compounds such as selective phosphodiesterase inhibitors, leukotriene modifiers, adhesion molecule blockers, tachykinin antagonists, inhibitors of cytokines and growth factors, chemokine inhibitors, anti-inflammatory cytokines, inhibitors of transcription factors and intracellular signal transduction, proteinase inhibitors, and antioxidants ().
Table 1 New therapeutic interventions for COPD
Improvements in existing classes of drugs
Improved long-acting β 2-adrenoceptor agonists
Currently used long-acting β 2-adrenoceptor agonists such as salmeterol and formoterol are, like all other marketed compounds of this drug class, racemic mixtures of both R(right)- and S(sinister)-isomers. However, only R-enantiomers are pharmacologically active, whereas S-isomers are essentially inert (CitationHandley et al 2000). Therefore, the newly developed pure R-form of formoterol (arformoterol) appears to be quite promising for a better control of COPD patients (CitationMolfino 2005). Moreover, the pharmacological industry has tried to combine the therapeutic advantages of R-enantiomers with a once-daily dosing approach, thus developing a non-catechol, pure (R,R)-isomer and 24-hour-lasting β 2-agonist named carmoterol (TA-2005) (CitationKikkawa et al 1994; CitationCazzola et al 2005). This drug has been shown to have a high binding affinity for β 2-adrenergic receptors, and to be more potent than formoterol, salmeterol, salbutamol, procaterol, and isoprenaline. The carmoterol-derivative indacaterol (QAB-149) produces a very long bronchodilation, which lasts more than 24 hours, and preliminary clinical data suggest that such a compound is strongly efficacious in COPD (CitationCazzola et al 2005). It can be thus argued that COPD patients will probably benefit from these further advances in β 2-agonist pharmacology.
Improved anticholinergics
In addition to the long-acting tiotropium bromide, which dissociates slowly from M3 and M1 muscarinic receptors with respect to the inhibitory, pre-junctional M2 autoreceptors, more selective M1/M3 antagonists are in clinical development. For instance, the M3 anti-muscarinic compound LAS34273 is a long-acting anticholinergic characterized by a rapid and significant bronchodilatory action, coupled with a good tolerability pattern in both healthy subjects and COPD patients (CitationSchelfhout et al 2003). Revatropate is an M1/M3 antagonist, with no effect on M2 autoreceptors and available for inhalation, which has been shown to be an effective and well-tolerated bronchodilator in COPD patients (CitationAlabaster 1997).
Improved corticosteroids
As already mentioned, corticosteroids are quite unable to suppress airway and lung inflammation in COPD, mainly because of the inhibitory effect exerted by cigarette smoke and oxidative stress on histone deacetylation, which is required for the repressive action of corticosteroids on pro-inflammatory gene expression. However, the new, non-halogenated, inhaled glucocorticoid pro-drug ciclesonide is under development for both asthma and COPD (CitationDent 2002). This compound, which is converted into a ciclesonide-active principle (CIC-AP) in the lung, seems to be more potent than the currently available corticosteroid agents.
Improved theophyllines
The current renewed interest for theophylline, whose clinical use has been quite neglected during recent years, arises from its suggested anti-inflammatory action, detectable even at low plasma concentrations (CitationBarnes 2003). These newly recognized pharmacological properties are likely independent from either phosphodiesterase (PDE) inhibition or adenosine antagonism, and appear to be due to the ability of theophyllyne to activate HDACs (CitationBarnes et al 2005). The latter mechanism may be very useful to restore HDAC activity impaired by oxidative stress, thus resensitizing COPD patients to the anti-inflammatory effects of corticosteroids. Therefore, there is a rational basis for a future design of novel theophylline-like molecules with improved HDAC-activating properties and devoid of the side-effects mainly caused by adenosine receptor antagonism. Such types of drugs could be indeed usefully utilized alone or in combination with corticosteroids, thereby increasing significantly the therapeutic potential of these anti-inflammatory agents, rather limited in COPD at present.
New therapeutic strategies
Selective phosphodiesterase inhibitors
At least 11 PDE families are currently known (CitationSoderling and Beavo 2000), among which PDE4 is the predominant isoenzyme expressed in neutrophils, CD4+/CD8+ T cells, and monocytes/macrophages, as well as in airway epithelial and smooth muscle cells. Therefore, PDE4 is a suitable therapeutic target for treatment of COPD (CitationSturton and Fitzgerald 2002). Specific PDE4 inhibitors such as cilomilast and roflumilast are active in some animal models of neutrophil inflammation and have been tested in COPD patients (CitationBarnes and Hansel 2004). A 6-week study performed in patients with moderate to severe COPD showed that cilomilast improved lung function (CitationCompton et al 2001), and this drug was also able to reduce airway inflammation in biopsy samples (CitationGamble et al 2003). Furthermore, TNF-α release from peripheral blood monocytes was significantly inhibited by roflumilast (CitationReid 2002), which in a placebo-controlled trial also increased FEV1 in COPD patients (CitationMolfino 2005). This latter compound is better tolerated than cilomilast, especially with regard to gastrointestinal side-effects like vomiting, due to inhibition of the PDE4D gene preferentially expressed in the chemosensitive trigger zone in the brain stem (CitationLamontagne et al 2001). Cilomilast is indeed a selective PDE4D inhibitor whereas roflumilast does not target any specific member of the PDE4 family, thus exhibiting a more favorable therapeutic index that could be further improved by the future availability of selective inhibitors of the PDE4B isoform, which appears to play a prominent role in inflammatory cells (CitationBarnes and Stockley 2005).
Other PDE4 inhibitors in clinical development for COPD and asthma include ONO-6126, arofylline, and AWD-12-281. Oral administration of ONO-6126 has been reported to inhibit both airway inflammation and bronchoconstriction (CitationMolfino 2005). Inhaled arofylline, tested for 3 months in a double-blind, placebo-controlled Phase III trial involving 141 subjects with moderate to severe COPD, improved FEV1 and reduced the incidence of disease exacerbations (CitationMolfino 2005). AWD-12-281 is a new compound, already available as oral and inhaled formulations, which has the potential for inhibiting the release of pro-inflammatory mediators such as histamine, TNF-α, and granulocyte-macrophage colony stimulating factor (GM-CSF), and appears to be effective also in in vivo models of airway diseases (CitationKuss et al 2003). More recently, a selective PDE7 inhibitor named BRL 50481 has been developed (CitationSmith et al 2004) that targets the PDE7 isoform expressed by monocytes/macrophages, CD8+ T-cells, and neutrophils, thus potentiating the anti-inflammatory actions of PDE4 inhibitors.
Leukotriene modifiers
Differently from the pathogenic mechanisms underlying asthma, which are characterized by an important role played by cysteinyl leukotrienes (LTC4, D4, E4), LTB4 is the leukotriene mostly involved in the inflammatory process associated with COPD. In fact, LTB4 concentrations are markedly increased in both sputum and exhaled breath of patients with COPD (CitationHill et al 1999; CitationMontuschi et al 2003), especially during exacerbations. LTB4 interacts with the high-affinity BLT1 receptor expressed by immune/inflammatory cells, thus exerting a powerful chemoattractant effect particularly on neutrophils, but also on CD8+ T-cells (CitationMartin et al 1989; CitationGoodarzi et al 2003).
Several selective BLT1 antagonists are currently under investigation for their potential therapeutic use in COPD. LY293111 and SB225002 are able to inhibit the neutrophil chemotactic activity of sputum from COPD patients (CitationCrooks et al 2000; CitationBeeh et al 2003). A powerful and long-lasting activity characterizes the pharmacological profile of the LTB4 receptor antagonist amelubant (BIIL-284), which is in clinical development for treatment of COPD, asthma, and cystic fibrosis (CitationBirke et al 2001). BIIL-284 is an orally active pro-drug that is converted by ubiquitously expressed esterases into two active metabolites (BIIL-260 and BIIL-315). A more limited therapeutic potential seems to characterize LTB 019, another LTB4 receptor antagonist that was tested in COPD patients, but did not elicit significant changes in FEV1 as well as in induced sputum levels of neutrophils, TNF-α, and interleukin-8 (IL-8) (CitationGronke and Beeh 2002). Since LTB4 is synthesized by 5′-lipoxygenase (5-LO), some 5-LO inhibitors such as CJ-13610 and BAYx1005 have been evaluated in COPD patients without showing marked activity in reducing sputum concentrations of LTB4 and neutrophil expression of activation markers (CitationGompertz and Stockley 2002). It is thus likely that more potent 5-LO inhibitors are still needed.
Adhesion molecule blockers
Airway and lung infiltration by inflammatory cells depends on their adhesion to vascular endothelium and the subsequent migration into the respiratory system. In this regard a key role is played by the expression on neutrophils, monocytes, CD8+ T-lymphocytes, and endothelial cells, of several different families of adhesion molecules, which represent another potential target for anti-COPD therapies (CitationMatera and Cazzola 2004). For instance, rolling of inflammatory cells along vascular endothelium is mediated by P-selectin, whose function may be inhibited by an antagonist (TBC1269) of its sialyl-LewisX ligand, which preferentially blocks neutrophil adhesion (CitationDavenpeck et al 2000). Another selectin inhibitor, the imidazole compound OC 229648, is under evaluation for treatment of COPD and asthma (CitationRomano and Slee 2001). The Mac-1 (CD11b/CD18) integrin might be a further interesting target because this adhesion molecule is overexpressed on neutrophils from COPD patients (CitationNoguera et al 1998), and it is also present on the surface of monocytes and macrophages. However, these therapeutic approaches raise some concerns due to the possible risk of infections caused by a defective neutrophil migration.
Tachykinin antagonists
Antagonists of NK-1, NK-2, and NK-3 tachykinin receptors, stimulated by substance P (SP) and neurokinins A (NKA) and B (NKB), respectively, have been suggested to be potentially beneficial in both asthma and COPD (CitationJoos and Pauwels 2001). Therefore, some single (NK-3), dual (NK-1/NK-2), or even triple (NK-1/NK-2/NK-3) orally active, NK receptor antagonists such as CS-003, DNK-333, and SB-223412 (talnetant), are in clinical development for COPD (CitationMolfino 2005).
Inhibitors of cytokines and growth factors
TNF-α inhibitors
TNF-α is a pro-inflammatory cytokine involved in activation and migration of neutrophils, monocytes/macrophages, and T lymphocytes. In mice models, TNF-α is implicated in the pathogenesis of airway inflammation sustained by neutrophils and macrophages, as well as in cigarette smoke-induced emphysema (CitationChurg et al 2002). In COPD patients, high concentrations of this protein have been detected in induced sputum and plasma, and increased amounts of TNF-α-mRNA have also been found in skeletal muscles (CitationKeatings et al 1996; Citationde Boer 2005). On the other hand, the severe wasting occurring in many patients with advanced COPD might be caused by TNF-α through the induction of skeletal muscle cell apoptosis. In support of this hypothesis, some TNF-α gene polymorphisms appear to be associated with a poorer disease prognosis (CitationKeatings et al 2000).
Therefore, TNF-α seems to be a suitable target for the development of anti-COPD drugs, and different approaches have been tried to inhibit either the production or functions of this cytokine. Many of these compounds are in clinical trial for COPD and other chronic disorders such as asthma, Crohn’s disease, rheumatoid arthritis, and psoriasis. TNF-α inhibitors include monoclonal, non-human or chimeric anti-TNF-α antibodies (infliximab, afelimomab, and CytoTab), humanized antibodies (adalimumab, CDP-571, CDP-870), human soluble TNF receptors (onercept) or TNF receptor fusion proteins (etanercept), and antisense oligonucleotides (ISIS-104838) that inhibit TNF-α mRNA translation into pre-TNF-α proteins (Citationde Boer 2005). The aim of humanization is to reduce the immunogenicity of therapeutic antibodies and the consequent side-effects. The latter cannot, however, be completely avoided, in that local reactions around the site of injection and, less frequently, a delayed hypersensitivity-like reaction and a new onset of autoimmunity have also been reported (Citationde Boer 2005). Very recently, the results of the first Phase II clinical trial evaluating the effects of infliximab in COPD patients have been published (Citationvan der Vaart et al 2005). According to this double-blind, placebo-controlled study involving 22 current smokers with mild to moderate COPD, 14 of which received 3 infusions of infliximab (5 mg/kg) at weeks 0, 2, and 6, respectively, such a treatment did not induce any change in respiratory symptoms, FEV1, quality of life, and percentage of inflammatory cells in induced sputum. Anyway, larger and longer-term trials are still needed to better understand whether infliximab may have a place in COPD management. Another anti-TNF-α strategy can be achieved by inhibiting TNF-α converting enzyme (TACE), which cleaves pre-TNF-α into the mature cytokine. Furthermore, TACE inhibitors (marimastat, BMS-561392, or DPC-33) may also reduce MMP activity and mucin 5AC production (CitationShao et al 2004), thereby providing potential additional benefits for COPD patients. However, anti-TNF-α therapies are only beginning to be evaluated for COPD treatment and, therefore, it is too early to predict the future impact of the above mentioned experimental approaches.
Interleukin-1 inhibitors
Cigarette smoke and lipopolysaccharide (LPS) stimulate macrophages and epithelial cells to produce interleukin-1 (IL-1), which in turn induces TNF-α expression in macrophages (Citationde Boer 2005). When compared with healthy subjects, IL-1 concentrations have been reported to be increased in induced sputum from patients with mild COPD (CitationDignetti et al 2002). Anti-IL-1 strategies include the use of endogenous, soluble IL-1 receptor antagonists (sIL-1Ra), anti-IL-1 receptor antibodies, IL-1 binding proteins such as IL-1 Trap, anti-IL-1β antibodies (CDP 484), and inhibitors of IL-1β converting enzyme (ICE). A recombinant IL-1 receptor antagonist (anakinra) has been already tested in subjects with rheumatoid arthritis, improving both symptoms and tissue damage typical of this chronic disease (CitationOlsen and Stein 2004). Preliminary data suggest that administration of anakinra to patients with COPD may increase the risk of bacterial lung infections (Citationde Boer 2005). However, despite the apparently disappointing results of these early clinical studies, the various approaches aimed at inhibiting IL-1 functions and/or synthesis might be potentially beneficial in COPD, also because of the consequent reduction of TNF-α expression.
Interleukin-6 inhibitors
In the exhaled breath condensate, as well as in serum during disease exacerbations, COPD patients exhibit elevated levels of interleukin-6 (IL-6) (CitationWedzicha et al 2000; CitationBucchioni et al 2003), a pleiotropic cytokine released by several different cells and capable of stimulating the generation of cytotoxic T lymphocytes. Anti-IL-6 antibodies are currently in clinical development for many inflammatory disorders (CitationNishimoto and Kishimoto 2004), and they should thus be experimented also for COPD treatment.
TGF-β inhibitors
In patients with COPD, high expression levels of TGF-β are detectable in airway epithelium and macrophages of small airways (CitationTakizawa et al 2001). TGF-β 1 is a potent fibrogenic agent which plays a pivotal role in inducing the fibrosis and narrowing of peripheral airways that characterize obstructive bronchiolitis in COPD (CitationHogg et al 2004). As already mentioned, TGF-β also contributes to proteinase imbalance by activating MMP-9. Furthermore, TGF-β has been shown to promote apoptosis of both bronchial and alveolar epithelial cells (CitationHagimoto et al 2002; CitationPelaia et al 2003), which may represent a very initial event in COPD pathogenesis (CitationShapiro and Ingenito 2005). Therefore, the currently available low-molecular-weight inhibitors of TGF-β receptor kinase (CitationYakymovych et al 2002) could be used to evaluate their effects in COPD patients.
Chemokine inhibitors
Among the four known families of chemokines, CC and CXC chemokines, synthesized by both inflammatory and structural cells, are those mostly involved in regulating the traffic of macrophages, neutrophils, and CD8+ T lymphocytes, thus significantly contributing to COPD pathogenesis.
Inhibitors of monocyte/macrophage migration
Migration of monocytes/macrophages is mainly mediated by CC chemokines including monocyte chemoattractant protein 1 (MCP-1 or CCL2), acting on its CCR2 receptor, and macrophage inflammatory proteins 1α (MIP-1α or CCL3) and 1β (MIP-1β or CCL4), interacting with their CCR5 receptor. Both CCL2 and CCR2 proteins are highly expressed on macrophages from COPD patients (Citationde Boer et al 2000; CitationTraves et al 2002), thereby representing attractive targets for the development of small molecule antagonists (). The humanized mouse monoclonal anti-CCL2 antibody MLN1202 and the selective CCR2 antagonist INCB3284 have been taken into consideration for treatment of rheumatoid arthritis (Citationde Boer 2005). Bindarit is another CCL2 antagonist that was shown to inhibit monocyte production of CCL2 and TNF-α, being also capable of reducing monocyte number in animal models of arthritis (CitationSironi et al 1999; CitationGuglielmotti et al 2002). Binding of CCL2 to its CCR2 receptor can also be blocked by RS-504393, which is able to inhibit macrophage infiltration and activation (CitationKitagawa et al 2004). Therefore, the potential utility of these compounds for COPD therapy should be explored.
Figure 1 Main chemokine receptors expressed by macrophages.
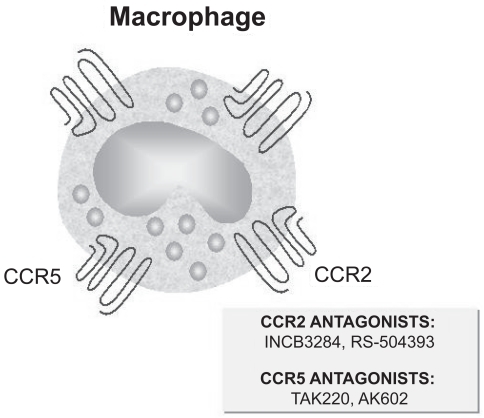
With regard to the interactions between CCR5 receptor and its CCL3/CCL4 ligands, whose expression is increased in COPD, some CCR5 antagonists such as TAK220 and AK602 are now available () (Citationde Boer 2005). However, these drugs are unable to significantly reduce macrophage chemoattraction because they inhibit only CCL3, but not CCL4 binding to CCR5, thus probably not being of particular interest for COPD treatment.
Inhibitors of neutrophil chemotaxis
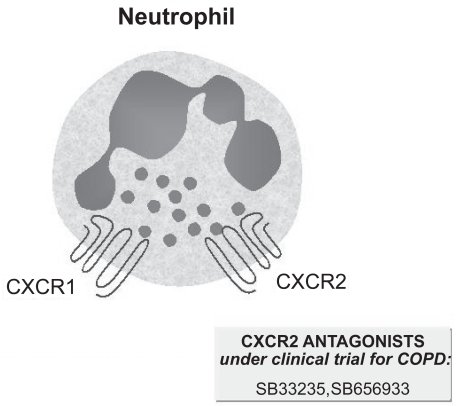
The CXC chemokines IL-8 (CXCL8) and growth-related oncoprotein-α (GRO-α or CXCL1) play a key role in neutrophil chemotaxis via stimulation of their CXCR1 and CXCR2 receptors (). IL-8 is overexpressed in COPD, thus being considered as an important target for therapeutic intervention (Citationde Boer 2003; CitationFuke 2004). In a recent study, COPD patients received 3 injections (800 mg at baseline and 400 mg in the 2 subsequent months) of a monoclonal antibody (ABX-IL-8) against IL-8, which attenuated the severity of dyspnea without, however, improving lung function, quality of life, and performance of the 6-minute walking test (CitationMahler et al 2004). CXCL1- and IL-8-mediated neutrophil chemotaxis can be inhibited by the non-peptide CXCR2 antagonist SB33235, whose safety profile and pharmacokinetic characteristics are not yet known (Citationde Boer 2005). SB656933 is another CXCR2 antagonist, currently under clinical investigation for COPD treatment, that was shown to impair neutrophil activation and recruitment into the bronchoalveolar lavage fluid (BALF) from rats exposed to aerosolized LPS (CitationCarpenter et al 2004; CitationSalmon et al 2004).
Figure 2 Main chemokine receptors expressed by neutrophils.
Inhibitors of CD8+ T cell recruitment
It has been recently suggested that COPD can be considered as an immunological disease featured by a predominant Tc1/Th1-mediated response (CitationDi Stefano et al 2004). In fact, a significant up-regulation of both interferon-inducible protein-10 (IP-10 or CXCL10) and its CXCR3 receptor, which is preferentially expressed by CD8+ Tc1 and CD4+ Th1 cells, was found in peripheral airways of COPD patients (CitationSaetta et al 2002). Specific inhibitors of CXCL10/CXCR3 interaction might therefore be useful for COPD treatment. In this regard, an anti-CXCL10 antibody has been recently developed and shown to inhibit CXCR3+ T cell recruitment in an experimental model of lung inflammation (CitationHildebrandt et al 2004).
Anti-inflammatory cytokines
In contrast to the pro-inflammatory cytokines implicated in COPD pathophysiology, interleukin-10 (IL-10) may exert some anti-inflammatory actions that could be exploited for therapeutic purposes. In particular, a recombinant human IL-10 (ilodecakin) is currently under evaluation for treatment of Crohn’s disease, rheumatoid arthritis, psoriasis, and hepatitis C (CitationAsadullah et al 2003). Administered to healthy volunteers before being exposed to endotoxin, IL-10 reduced pulmonary neutrophilia and was also capable of inhibiting the expression of TNF-α, IL-6, IL-8, CCL2, CCL3, and CCL4 (CitationPaikrt et al 1997). Repression of these pro-inflammatory genes is probably due to the inhibitory effect of IL-10 on activation of the transcription factor nuclear factor-κ B (NF-kB) (CitationDriessler et al 2004). Clinical studies performed in patients with psoriasis showed that subcutaneous injections of IL-10 improved skin lesions and reduced the expression of Th1 cytokines (CitationAsadullah et al 2003), thus suggesting an IL-10-induced shift from Th1 to Th2 immune response. This hypothesis appears to be confirmed by trials involving patients with chronic hepatitis C infection, treated for up to 1 year with IL-10, who exhibited an improvement in liver histology associated with a shift from interferon (IFN)γ +CD8+ T cells towards a Th2 secretory pattern (CitationNelson et al 2003). However, subjects with either rheumatoid arthritis or Crohn’s disease did not experience significant clinical improvements after treatment with IL-10, that surprisingly stimulated IFNγ expression and IFNγ-dependent production of CXCL10 (CitationAsadullah et al 2003), which may thus result in increased inflammation associated with CXCR3+ T lymphocytes, as occurring in COPD. On the other hand, it has been recently reported that inhaled corticosteroid therapy may promote in COPD patients the production of IL-10 from alveolar macrophages, though this effect was not accompanied by significant FEV1 increases (Citationde Boer 2005).
In vitro, in addition to decreasing the synthesis of TNF-α and IL-8 by macrophages, IL-10 also induced the expression of tissue inhibitor of metalloproteinase-1 (TIMP-1), without affecting MMP-9 levels (CitationLim et al 2000). Such actions might be particularly beneficial for patients with COPD, who are characterized by an unbalanced proteinase/antiproteinase ratio, shifted towards lung tissue destruction. These patients also have, when compared with normal subjects, lower IL-10 concentrations in induced sputum (CitationTakanashi et al 1999). However, in order to better delineate the potential therapeutic use of IL-10, its pathogenetic role in COPD needs to be further elucidated. Moreover, recombinant IL-10 can cause adverse side-effects including anemia, headache, and immunological reactions (Citationde Boer 2005). Therefore, all these considerations at present preclude COPD patients from entering trials aimed at evaluating the clinical effects of IL-10.
Inhibitors of transcription factors
Among the transcription factors that regulate the expression of pro-inflammatory cytokines and chemokines, a prominent role is played by NF-κ B, which is activated in macrophages and epithelial cells of patients with COPD, especially during disease exacerbations (CitationDi Stefano et al 2002; CitationCaramori et al 2003). NF-κ B is a dimeric transcription factor which in its inactive form is bound in the cytoplasm to an inhibitory protein (inhibitor of NF-κ B: Iκ B), that upon Iκ B kinase (IKK)-dependent phosphorylation undergoes ubiquitin-mediated proteolysis thus resulting in the release of active NF-κ B, able to translocate into the nucleus and to interact with the NF-κ B binding sites of target genes (CitationBaldwin 1996). Translation of NF-κ B p65 subunit has been experimentally blocked in airway epithelial cells by small interfering (si)RNAs, which in vitro reduced the expression of both IL-6 and IL-8 (Citationde Boer 2005). Furthermore, an antisense antagonist for p65 is now in clinical trial for Crohn’s disease (CitationHibi et al 2003).
A more promising approach is based on the use of orally active, small-molecule IKK inhibitors including SPC600839 and BMS345541. In a mouse model of collagen-induced arthritis, BMS345541 was shown to suppress NF-κ B nuclear translocation as well as TNF-α and IL-1β expression, thereby improving clinical symptoms and joint lesions (CitationMcIntyre et al 2003). Therefore, such compounds might be beneficial in COPD by inhibiting the production of cytokines and chemokines by both inflammatory and structural cells. However, these therapeutic strategies should be considered with extreme caution in that a prolonged inhibition of NF-κ B, which is crucially involved in the control of innate and adaptive immune responses, could expose recipient patients to a high risk of infections.
Inhibitors of signal transduction pathways
Inhibitors of mitogen-activated protein kinases (MAPK)
Among the various MAPK families (), p38 is mostly implicated in cytokine biosynthesis as well as in recruitment of inflammatory cells (CitationPelaia et al, 2005). In particular, p38 MAPK significantly contributes to neutrophil recruitment by up-regulating the vascular expression of intercellular adhesion molecule-1 (ICAM-1) and the release of TNF-α into airspaces (CitationTamura et al 1998; CitationNick et al 2000). Moreover, monocyte differentiation and chemotaxis are regulated by p38, which also stimulates the release of TNF-α and macrophage inflammatory protein-2 (MIP-2) from human macrophages (CitationAyala et al 2000; CitationNick et al 2000). Several pyridinylimidazole inhibitors of p38 are now available, and the combination of mutagenesis studies, X-ray crystallography and mechanistic enzymology has enabled elucidation of the molecular basis of inhibitor specificity, as well as development of second-generation compounds with increased potency and selectivity (CitationWang et al 1998; CitationLee et al 2000; CitationEnglish and Cobb 2002). The first p38 inhibitors able to block the biosynthesis of pro-inflammatory cytokines included the bicyclic pyridinylimidazole SKF 86002 and the 2,4,5-triaryl imidazole SB 203580 (CitationLee et al 1993; CitationGallagher et al 1995). Subsequently, other compounds such as SB 220025, HEP 689, and VX-745 have been designed; more recently, combinatorial chemistry strategies have led to the identification of a new class of highly potent p38 inhibitors, characterized by the replacement of the pyrazole ring with either an isoxazole or a thiophene (CitationDumas et al 2000). These latter molecules are very promising, as shown by their ability to inhibit, within a submicromolar range of concentrations, IL-6 production induced by TNF-α and IL-1.
Figure 3 Mitogen-activated protein kinases (MAPK) signalling pathways.
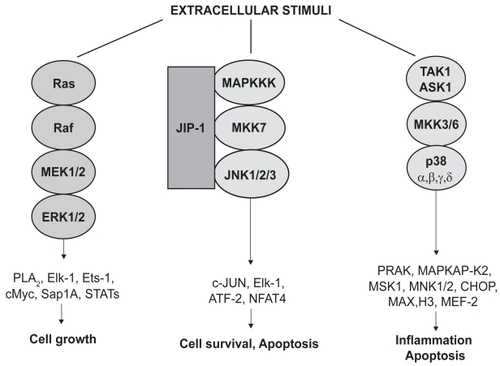
Over the past few years, the potential therapeutic role of p38 inhibitors has been tested in several different models of lung inflammation. SB 239063, a small- molecule p38 inhibitor, attenuated neutrophil infiltration induced by inhaled LPS and decreased IL-6 and MMP-9 levels in rat BALF (CitationUnderwood et al 2000), thus being possibly useful for COPD treatment. Another p38 inhibitor, SD-282, reduced LPS-induced production of TNF-α by human lung macrophages, without affecting the expression of IL-8 (Citationde Boer 2005). IL-8 release into cell-free supernatants of primary cultures of human bronchial epithelial cells, exposed to oxidative stress, was significantly inhibited by SB 203580 (CitationPelaia et al 2004). In these same cell cultures, SB 203580 completely prevented apoptosis elicited by TGF-β (CitationPelaia et al 2003). On the basis of all such in vitro findings, a p38 inhibitor named GSK-681323 has been developed for treatment of COPD, atherosclerosis, and rheumatoid arthritis, but clinical data are not yet available (CitationMolfino 2005). However, the pharmacological strategy of inhibiting p38 MAPK should be considered with caution as a treatment option in mammalian diseases, in that p38α knockout mice are characterized by a defective placental development (CitationAdams et al 2000; CitationMudgett et al 2000). An alternative therapeutic approach might thus consist of targeting the p38 substrate mitogen-activated protein kinase-activated protein kinase 2 (MAPKAP-K2) (), which mediates the stabilizing function exerted by p38 MAPK on many mRNAs encoding pro-inflammatory cytokines (CitationWinzen et al 1999). This strategy, however, also raises some concern because MAPKAP-K2 is probably required for the optimal effectiveness of immune responses; indeed, mice carrying a mutation in the gene encoding MAPKAP-K2 are highly susceptible to infections by intracellular pathogens such as Listeria monocytogenes (CitationLehner et al 2002). A more suitable approach could thereby be represented by the development of inhalant formulations of either p38 or MAPKAP-K2.
Inhibitors of phosphoinositide 3-kinase (PI-3K)
The PI-3K enzymatic pathway, leading to the generation of lipid second messengers, provides pro-inflammatory signals involved in recruitment and activation of neutrophils, monocytes, and T lymphocytes. Knockout of the PI-3Kγ is indeed responsible for inhibition of neutrophil migration and activation, as well as for impairment of T cell and macrophage functions (CitationSasaki et al 2000). Therefore, the selective small-molecule inhibitors of PI-3Kγ which are now in development may exert anti-inflammatory actions potentially useful for COPD therapy (CitationWard et al 2003; CitationBarnes and Stockley 2005).
Proteinase inhibitors
COPD is characterized by a relevant imbalance between proteinases (serine elastases, cysteine proteinases, MMPs), which degrade elastin and other structural proteins of lung parenchyma, and the protective array of antiproteinases (α 1-antitrypsin, elafin, secretory leukoprotease inhibitor, and tissue inhibitors of MMPs) (CitationBarnes et al 2003). In fact, smokers with a rapid decline in pulmonary function exhibit an increased urinary excretion of desmosine, a compound derived from elastin cross-links (CitationGottlieb et al 1996), which is a marker of connective tissue destruction. Neutrophil elastase is a powerful proteolytic enzyme that is predominantly inhibited by α 1-antitrypsin (α 1-AT). The latter is currently administered as an extracted protein to patients with genetically determined, low serum levels of α 1-AT and concomitant lung disease (CitationSandhaus 2004). In future, α 1-AT could be provided in recombinant form or delivered by viral vector-driven gene strategies (CitationLuisetti and Travis 1996; CitationStecenko and Brigham 2003). Furthermore, synthetic inhibitors of neutrophil elastase have been developed, including ONO-5046, FR901277, DX-890, and midesteine (CitationLuisetti et al 1996; CitationFujie et al 1999; CitationBarnes and Stockley 2005; CitationMolfino 2005). In animal models, FR901277 neutralized the action of elastase and other neutrophil serine proteinases such as cathepsin and proteinase 3, thus inhibiting acute inflammation and pulmonary emphysema (CitationFujie et al 1999). DX-890 is a recombinant protein derived from the human inter-α-trypsin inhibitor (CitationWark 2002), which after inhalation was well tolerated and detectable in its active form in BALF from healthy volunteers (CitationMolfino 2005). Midesteine is an orally active elastase inhibitor that, given for 4 weeks to patients with COPD, in a subgroup of treated subjects induced a post-treatment decrease of urine desmosine levels (CitationLuisetti et al 1996). Non-selective MMP inhibitors such as marimastat seem to induce relevant muscular-skeletal side-effects (CitationBelvisi and Bottomley 2003). This problem could perhaps be overcome by the development of specific inhibitors of MMP-9 (CitationMatter and Shudok 2004), which is the major elastolytic enzyme released by alveolar macrophages from COPD patients (CitationRussell et al 2002).
Antioxidants
Oxidative stress plays a key role in the development of COPD, in that the major cause of this disease is cigarette smoking, which represents a rich source of oxidant agents. Furthermore, other factors involved in COPD pathogenesis and progression, such as air pollutants, occupational dusts, and respiratory infections, also have the ability to produce oxidative stress. Indeed, smokers and patients with COPD are characterized by high concentrations of exhaled hydrogen peroxide (H2O2), which become even higher during disease exacerbations (CitationMacNee 2001). Moreover, increased levels of lipid peroxides, including 8-isoprostane and hydrocarbons such as ethane and pentane, are also detectable in the exhaled air condensate of patients with COPD (CitationHabib et al 1995). Lipid peroxidation products positively correlate with airway obstruction, thus suggesting that oxidative stress is closely associated with the progressive decline in lung function occurring in COPD (CitationBoots et al 2003). In addition, oxidative inactivation of the antiproteinase α 1-AT favors the increase in elastase burden which is responsible for the development of pulmonary emphysema. Oxidants largely contribute to the inflammatory and structural changes underlying COPD by inducing the production of several mediators and cytokines such as TNF-α and IL-8. In this regard, we have reported that H2O2 elicited a concentration-dependent increase in the amount of IL-8 released from bronchial epithelial cells, whose apoptotic death was also stimulated by oxidative stress (CitationPelaia et al 2004). H2O2 is also able to induce, in a time-dependent manner, the acetylation of histone H4 and the closely related synthesis of IL-8 by both bronchial and alveolar epithelial cells (CitationGilmour et al 2003; CitationTomita et al 2003). In other cell types such as alveolar macrophages, it has been shown that H2O2 and cigarette smoke can stimulate IL-8 secretion by inhibiting the activity of HDAC enzymes (CitationIto et al 2001). HDACs indeed repress gene transcription by deacetylating core histones, thus enhancing chromatin condensation and DNA supercoiling (CitationAyer 1999). In particular, oxidative stress might impair HDAC activity by enhancing, in the presence of high NO levels, the production of peroxynitrite and the subsequent nitration of tyrosine residues on HDAC or associated proteins. This mechanism could also help explain the low therapeutic efficacy, observed in COPD patients when compared with asthmatics, of inhaled glucocorticoids, whose anti-inflammatory actions largely depend on their ability to recruit and activate HDACs (CitationBarnes et al 2004).
Therefore, antioxidants may exert potentially beneficial effects in COPD. In fact, in rats exposed to tobacco smoke, the intra-tracheal administration of a catalytic antioxidant (AEOL 10150) elicited a significant decrease in BAL neutrophils and macrophages, detected at 2 days and 8 weeks, respectively (CitationCrapo 2003). Anti-oxidant defences can be potentiated by the cysteine-donor N-acetyl cysteine (NAC), able to stimulate the production of the endogenous tripeptide glutathione (GSH), which provides an effective intra- and extracellular shield against oxidative stress. However, a large trial carried out on 523 patients with COPD, randomly receiving oral NAC (600 mg daily) or placebo for 3 years, has very recently demonstrated that NAC did not produce any change neither in the annual decline in FEV1 nor in the number of exacerbations per year (CitationDecramer et al 2005). Nacystelyn is a NAC-related thiol antioxidant that has been shown to be effective in vitro, as well as in in vivo animal models (CitationAntonicelli et al 2004); it also has the advantage of being suitable for aerosolized administration. Furthermore, the development of more powerful antioxidants including stable GSH compounds, analogs of superoxide dismutase, and selenium-based drugs is underway (CitationBarnes and Stockley 2005). In this regard, the potential therapeutic utility for COPD of BXT-51072, the lead compound in a series of small-molecule GSH peroxidase mimics, is interesting. BXT-51072 is capable of increasing the rate of peroxide metabolism, as well as of inhibiting both inflammation and oxidative damage (CitationMolfino 2005).
Conclusions
Because COPD is not currently well controlled by available drugs, which are unable to resolve inflammation and to prevent lung tissue destruction and the associated progressive decline in pulmonary function, novel and more effective therapies are badly required. This need is made even more urgent by the known difficulties, due to both addiction and psychological reasons, encountered by patients and healthy subjects trying to quit smoking. Therefore, the common goal of the several different experimental approaches in development for COPD treatment should be a more successful targeting of disease causative mechanisms. In this regard, there is no doubt that during the past few years significant progress has been made towards a better understanding of COPD pathophysiology. As a result, such new advances have led to a significant expansion of the pharmacological options now under evaluation. Within this renewed therapeutic context, many strategies appear to be quite promising for the near future, including selective PDE inhibition, interferences with the cytokine/chemokine network, and blockade of MAPK signal transduction pathways. However, knowledge of the basic cellular and molecular events underlying COPD pathogenesis still needs to be further improved, possibly through the identification of the genetic factors involved, and also via the application of proteomic techniques aimed at characterizing the specific patterns of protein expression related to the various COPD phenotypes. The potential future achievements will probably open the way for exciting perspectives, thus theoretically leading to the design of compounds capable of either directly modulating relevant genes or, alternatively, targeting their protein products.
Finally, a further contribution to the progress of COPD treatment may arise from the so-called regenerative medicine, aimed at restoring the integrity of lung tissue structure, destroyed in patients with emphysema. In particular, interesting data have been provided by experiments showing the activation of growth and repair processes in rat alveoli, induced by retinoic acid (CitationBelloni et al 2000). In this regard, more beneficial results may perhaps be pursued by biological strategies based on the use of stem cells (CitationOtto 2002). Therefore, our hope is that the combination of all these new approaches will eventually enable, in the next decades, a definitive cure for such a widespread and severe respiratory disease.
References
- AdamsRHPorrasAAlonsoG2000Essential role of p38α MAP kinase in placental but not embryonic cardiovascular developmentMol Cell61091610949032
- AlabasterVA1997Discovery and development of selective M3 antagonists for clinical useLife Sci601053609121347
- AntonicelliFBrownDParmentierM2004Regulation of LPS-mediated inflammation in vivo and in vitro by the thiol antioxidant nacystelynAm J Physiol Lung Cell Mol Physiol286L13192715136298
- AsadullahKSterryWVolkHD2003Interleukin-10 therapy – review of a new approachPharmacol Rev552416912773629
- AyalaJMGoyalSLivertonNJ2000Serum-induced monocyte differentiation and monocyte chemotaxis are regulated by the p38 MAP kinase signal transduction pathwayJ Leukoc Biol678697510857861
- AyerDE1999Histone deacetylases: transcriptional repression with SINers and NuRDsTrends Cell Biol9193810322454
- BaldwinAS1996The NF-κ B and Iκ B proteins: new discoveries and insightsAnnu Rev Immunol14649818717528
- BarnesPJ2003Theophylline: new perspectives on an old drugAm J Respir Crit Care Med167813812623857
- BarnesPJAdcockIMItoK2005Histone acetylation and deacetylation: importance in inflammatory lung diseasesEur Respir J255526315738302
- BarnesPJHanselTT2004Prospects for new drugs for chronic obstructive pulmonary diseaseLancet3649859615364192
- BarnesPJItoKAdcockIM2004Corticosteroid resistance in chronic obstructive pulmonary disease: inactivation of histone deacetylaseLancet363731315001333
- BarnesPJShapiroSDPauwelsRA2003Chronic obstructive pulmonary disease: molecular and cellular mechanismsEur Respir J226728814582923
- BarnesPJStockleyRA2005COPD: current therapeutic interventions and future approachesEur Respir J25108410615929966
- BeehKMKornmannOBuhlR2003Neutrophil chemotactic activity of sputum from patients with COPD: role of interleukin 8 and leukotriene B4Chest1231240712684317
- BelloniPNGarvinLMaoCP2000Effects of all-trans-retinoic acid in promoting alveolar repairChest117S235S41
- BelvisiMGBottomleyKM2003The role of matrix metalloproteinases (MMPs) in the pathophysiology of chronic obstructive pulmonary disease (COPD): a therapeutic role for inhibitors of MMPs?Inflamm Res529510012755372
- BirkeFWMeadeCJAnderskewitzR2001In vitro and in vivo pharmacological characterization of BIIL 284, a novel and potent leukotriene B(4) receptor antagonistJ Pharmacol Exp Ther2974586611259574
- BootsAWHaenenGRBastA2003Oxidant metabolism in chronic obstructive pulmonary diseaseEur Respir J46s1427
- BucchioniEKharitonovSAAllegraL2003High levels of interleukin-6 in the exhaled breath condensate of patients with COPDRespir Med97129930214682411
- CaramoriGRomagnoliMCasolariP2003Nuclear localization of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbationsThorax583485112668802
- CarpenterDCRumseyWLBusch-PetersenJ2004The selective CXCR2 receptor antagonist SB-656933 inhibits CXCL1-induced neutrophil CD11b expression in human whole bloodEur Respir J24Suppl 48s218
- CasaburiRMahlerDAJonesPW2002A long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary diseaseEur Respir J192172411866001
- CazzolaMDonnerCF2000Long-acting β 2 agonists in the management of stable chronic obstructive pulmonary diseaseDrugs603072010983735
- CazzolaMMateraMGLotvallJ2005Ultra long-acting β 2-agonists in development for asthma and chronic obstructive pulmonary diseaseExpert Opin Invest Drugs1477583
- CelliBRMacNeeWATS/ERS Task Force2004Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paperEur Respir J239324615219010
- ChurgADaiJTaiH2002Tumour necrosis factor-α is central to acute cigarette smoke-induced inflammation and connective tissue breakdownAm J Respir Crit Care Med1668495412231496
- ComptonCHGubbJNiemanR2001Cilomilast, a selective phosphodiesterase-4 inhibitor for treatment of patients with chronic obstructive pulmonary disease: a randomised, dose-ranging studyLancet3582657011498212
- CrapoJD2003Oxidative stress as an initiator of cytokine release and cell damageEur Respir J22Suppl 44s46
- CrooksSWBayleyDLHillSL2000Bronchial inflammation in acute bacterial exacerbations of chronic bronchitis: the role of leukotriene B4Eur Respir J152748010706491
- DavenpeckKLBerensKLDixonRA2000Inhibition of adhesion of human neutrophils and eosinophils to P-selectin by the sialyl Lewis antagonist TBC1269: preferential activity against neutrophil adhesion in vitroJ Allergy Clin Immunol1057697510756228
- de BoerWI2003Potential new drugs for therapy of chronic obstructive pulmonary diseaseExpert Opin Invest Drugs12106786
- de BoerWI2005Perspectives for cytokine antagonist therapy in COPDDrug Discov Today109310615718158
- de BoerWISontJKvan SchadewijkA2000Monocyte chemoattractant protein 1, interleukin 8, and chronic airways inflammation in COPDJ Pathol1906192610727989
- DecramerMRutten-van MolkenMDekhuijzenPN2005Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (bronchitis randomized on NAC cost-utility study, BRONCUS): a randomized placebo-controlled trialLancet36515526015866309
- DentG2002CiclesonideCurr Opin Investig Drugs37883
- DignettiPLevaMMincariniM2002Cytokine concentrations in induced sputum of mild COPD patients: correlation to clinical response to inhaled corticosteroidsEur Respir J20Suppl 38s309
- Di StefanoACaramoriGOatesT2002Increased expression of NF-kB in bronchial biopsies from smokers and patients with COPDEur Respir J205566312358328
- Di StefanoACaramoriGCapelliA2004STAT4 activation in smokers and patients with chronic obstructive pulmonary diseaseEur Respir J24788515293608
- DriesslerFVenstromKSabatR2004Molecular mechanism of interleukin-10-mediated inhibition of NF-kB activity: a role for p50Clin Exp Immunol135647314678266
- DumasJSibleyRRiedlB2000Discovery of a new class of p38 kinase inhibitorsBioorg Med Chem Lett1020475010999467
- EnglishJMCobbMH2002Pharmacological inhibitors of MAPK pathwaysTrends Pharmacol Sci2340511804650
- FujieKShinguhYYamazakiA1999Inhibition of elastase-induced acute inflammation and pulmonary emphysema in hamsters by neutrophil elastase inhibitor FR901277Inflamm Res48160710219659
- FukeSBetsuyakuTNasuharaY2004Chemokines in bronchiolar epithelium in the development of chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol314051215220136
- GallagherTFFier-ThompsonSMGarigipatiRS19952,4,5-Triarylimidazole inhibitors of IL-1 biosynthesisBioorg Med Chem Lett511716
- GambleEGrootendorstDCBrightlingCE2003Antiinflammatory effects of the phosphodiesterase-4 inhibitor cilomilast (Ariflo) in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1689768212816740
- GilmourPSRahmanIDonaldsonK2003Histone acetylation regulates epithelial IL-8 release mediated by oxidative stress from environmental particlesAm J Physiol Lung Cell Mol Physiol284L5334012573991
- GompertzSStockleyRA2002A randomized, placebo-controlled trial of a leukotriene synthesis inhibitor in patients with COPDChest1222899412114372
- GoodarziKGoodarziMTagerAM2003Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissuesNat Immunol49657312949533
- GottliebDJStonePJSparrowD1996Urinary desmosine excretion in smokers with and without rapid decline of lung function: the Normative Aging StudyAm J Respir Crit Care Med154129058912738
- GronkeLBeehKM2002LTB019A, a leukotriene B(4) receptor antagonist, has no effect on the levels of neutrophils, MPO, IL-8 and TNF-α in induced sputum of patients with COPDAm J Respir Crit Care Med165A598
- GuglielmottiAD’OnofrioEColettaI2002Amelioration of rat adjuvant arthritis by therapeutic treatment with bindarit, an inhibitor of MCP-1 and TNF α productionInflamm Res51252812056513
- HabibMPClementsNCGarewalHS1995Cigarette smoking and ethane exhalation in humansAm J Respir Crit Care Med1511368727735586
- HagimotoNKuwanoKInoshimaI2002TGF-α 1 as an enhancer of Fas-mediated apoptosis of lung epithelial cellsJ Immunol1686470812055267
- HandleyDAAndersonAJKoeslerJ2000New millennium bronchodilators for asthma: single-isomer beta agonistsCurr Opin Pulm Med643910608425
- HibiTInoueNOgataH2003Introduction and overview: recent advances in the immunotherapy of inflammatory bowel diseaseJ Gastroenterol38Suppl 15364212698869
- HildebrandtGCCorrionLAOlkiewiczKM2004Blockade of CXCR3 receptor-ligand interactions reduces leukocyte recruitment to the lung and the severity of experimental idiopathic pneumonia syndromeJ Immunol1732050915265940
- HillATBayleyDStockleyRA1999The interrelationship of sputum inflammatory markers in patients with chronic bronchitisAm J Respir Crit Care Med160893810471615
- HoggJCChuFUtokaparchS2004The nature of small airway obstruction in chronic obstructive pulmonary diseaseN Engl J Med35026455315215480
- ItoKLimSCaramoriG2001Cigarette smoking reduces histone deacetylase 2 expression, and inhibits glucocorticoid actions in alveolar macrophagesFASEB J151110211292684
- ItoKItoMElliottWM2005Decreased histone deacetylase activity in chronic obstructive pulmonary diseaseN Engl J Med35219677615888697
- JoosGFPauwelsRA2001Tachykinin receptor antagonists: potential in airways diseasesCurr Opin Pharmacol12354111712745
- KeatingsVMCaveSJHenryMJ2000A polymorphism in the tumour necrosis factor-α gene promoter region may predispose to a poor prognosis in COPDChest118971511035665
- KeatingsVMCollinsPDScottDM1996Differences in interleukin-8 and tumour necrosis factor-α in induced sputum from patients with chronic obstructive pulmonary disease or asthmaAm J Respir Crit Care Med15353048564092
- KikkawaHKannoKIkezawaK1994TA-2005, a novel, long-acting, and selective β 2-adrenoceptor agonist: characterization of its in vivo bronchodilating action in guinea pigs and cats in comparison with other β 2-agonistsBiol Pharm Bull171047527820105
- KitagawaKWadaTFuruichiK2004Blockade of CCR2 ameliorates progressive fibrosis in kidneysAm J Pathol1652374615215179
- KussHHoefgenNJohanssenS2003In vivo efficacy in airway disease models of N-(3,5-dichloropyrid-4-yl)-[1-(4-fluorobenzyl)-5-hydroxy-indole-3-yl]-glyoxylic acid amide (AWD 12-281), a selective phosphodiesterase inhibitor for inhaled administrationJ Pharmacol Exp Ther3073738512944497
- LacosteJYBousquetJChanezP1993Eosinophilic and neutrophilic inflammation in asthma, chronic bronchitis and chronic obstructive pulmonary diseaseJ Allergy Clin Immunol92537488409114
- LamontagneSMeadowsELukP2001Localization of phosphodiesterase-4 isoforms in the medulla and nodose ganglion of the squirrel monkeyBrain Res920849611716814
- LeeJCBadgerAMGriswoldDE1993Bicyclic imidazoles as a novel class of cytokine biosynthesis inhibitorsAnn NY Acad Sci696149708109825
- LeeJCKumarSGriswoldDE2000Inhibition of p38 MAP kinase as a therapeutic strategyImmunopharmacology4718520110878289
- LehnerMDSchwoebelFKotlyarovA2002Mitogen-activated protein kinase-activated protein kinase 2-deficient mice show increased susceptibility to Listeria monocytogenes infectionJ Immunol16846677311971016
- LimSRocheNOliverBG2000Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10Am J Respir Crit Care Med16213556011029344
- LuisettiMSturaniCSellaD1996MR889, a neutrophil elastase inhibitor, in patients with chronic obstructive pulmonary disease: a double-blind, randomized, placebo-controlled clinical trialEur Respir J9148268836663
- LuisettiMTravisJ1996Bioengineering:/1-proteinase inhibitor site-specific mutagenesis. The prospect for improving the inhibitorChest110Suppl 6S27883
- MahlerDAHuangSTabriziM2004Efficacy and safety of a monoclonal antibody recognizing IL-8 in COPD; a pilot studyChest1269263415364775
- MartinTRPistoreseBPChiEY1989Effects of leukotriene B4 in the human lung: recruitment of neutrophils into the alveolar spaces without a change in protein permeabilityJ Clin Invest841609192553777
- MaselliRPelaiaGSinghKK2006Oxidative stress and respiratory diseaseOxidative stress, disease and cancerLondonImperial College Press67385
- MacNeeW2001Oxidative stress and lung inflammation in airways diseasesEur J Pharm429195207
- MateraMGCazzolaM2004New anti-inflammatory approaches in COPDDrug Discov Today133543
- MatterHSchudokM2004Recent advances in the design of matrix metalloprotease inhibitorsCurr Opin Drug Discov Devel751335
- McIntyreKWShusterDJGilloolyKM2003A highly selective inhibitor of 1β kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in miceArthritis Rheum482652913130486
- MolfinoNA2004Genetics of COPDChest12519294015136409
- MolfinoNA2005Drugs in clinical development for chronic obstructive pulmonary diseaseRespiration721051215753645
- MontuschiPKharitonovSACiabattoniG2003Exhaled leukotrienes and prostaglandins in COPDThorax58585812832671
- MudgettJSDingJGuh-SieselL2000Essential role for p38 α mitogen-activated protein kinase in placental angiogenesisProc Natl Acad Sci U S A9710454910973481
- NelsonDRTuZSoldevila-PicoC2003Long-term interleukin 10 therapy in chronic hepatitis patients has a proviral and anti-inflammatory effectHepatology388596814512873
- NickJAYoungSKBrownKK2000Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammationJ Immunol1642151910657669
- NishimotoNKishimotoT2004Inhibition of IL-6 for the treatment of inflammatory diseasesCurr Opin Pharmacol43869115251133
- NogueraABusquetsXSauledaJ1998Expression of adhesion molecules and G proteins in circulating neutrophils in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med158166489817722
- OlsenNJSteinCM2004New drugs for rheumatoid arthritisN Engl J Med35021677915152062
- OttoWR2002Lung epithelial stem cellsJ Pathol197527+3512115868
- PaikrtDvan der PollTLeviM1997Interleukin-10 inhibits activation of coagulation and fibrinolysis during human endotoxemiaBlood89270159108387
- PelaiaGCudaGVatrellaA2003Effects of transforming growth factor-/and budesonide on mitogen-activated protein kinase activation and apoptosis in airway epithelial cellsAm J Respir Cell Mol Biol29121812600835
- PelaiaGCudaGVatrellaA2004Effects of hydrogen peroxide on MAPK activation, IL-8 production and cell viability in primary cultures of human bronchial epithelial cellsJ Cell Bioc9314252
- PelaiaGCudaGVatrellaA2005Mitogen-activated protein kinases and asthmaJ Cell Physiol2026425315316926
- ReidP2002RoflumilastCurr Opin Investig Drugs3116570
- RomanoSJSleeDH2001Targeting selectins for the treatment of respiratory diseasesCurr Opin Investig Drugs290713
- RussellRECulpittSVDeMatosC2002Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol26602911970913
- SaettaMBaraldoSCorbinoL1999CD8+ cells in the lungs of smokers with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med160711710430750
- SaettaMMarianiMPanina-BordignonP2002Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1651404912016104
- SalmonMCarpenterDCDehaasCJ2004Inhibition of LPS-induced neutrophil recruitment in the lungs correlates with modulation of neutrophil CD11b expression using the selective CXCR2 receptor antagonist SB-656933Eur Respir J24Suppl 48218s
- SandhausRA2004α 1-Antitrypsin deficiency. 6: new and emerging treatments for α 1-antitrypsin deficiencyThorax59904915454659
- SasakiTIrie-SasakiJJonesRJ2000Function of PI3Kg in thymocyte development, T cell activation, and neutrophil migrationScience2871040610669416
- SchelfhoutVJJoosGFFerrerP2003Activity of LAS34273, a new long-acting anticholinergic antagonist, in COPD patientsAm J Respir Crit Care Med167A319
- ShaoMXNakanagaTNadelJA2004Cigarette smoke induces MUC5AC mucin overproduction via tumour necrosis factor-α-converting enzyme in human airway epithelial (NCI-H292) cellsAm J Physiol287L4207
- ShapiroSD1999The macrophage in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med160S293210556166
- ShapiroSDIngenitoEP2005The pathogenesis of chronic obstructive pulmonary disease: advances in the past 100 yearsAm J Respir Cell Mol Biol323677215837726
- SironiMGuglielmottiAPolentaruttiN1999A small synthetic molecule capable of preferentially inhibiting the production of the CC chemokine monocyte chemotactic protein-1Eur Cytokine Netw104374210477401
- SmithSJCieslinskiLBNewtonR2004Discovery of BRL 50481, a selective inhibitor of phosphodiesterase 7: in vitro studies in human monocytes, macrophages and CD8+ T-lymphocytesMol Pharmacol6616798915371556
- SoderlingSHBeavoJA2000Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functionsCurr Opin Cell Biol12174910712916
- StecenkoAABrighamKL2003Gene therapy progress and prospects: α 1-antitrypsinGene Ther1095912571637
- SturtonGFitzgeraldM2002Phosphodiesterase 4 inhibitors for the treatment of COPDChest121Suppl 1S1926
- TakanashiSHasegawaYKanehiraY1999Interleukin-10 level in sputum is reduced in bronchial asthma, COPD and in smokersEur Respir J143091410515406
- TakizawaHTanakaMTakamiK2001Increased expression of transforming growth factor- α 1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD)Am J Respir Crit Care Med16314768311371421
- TamuraDYMooreEEJohnsonJL1998p38 mitogen-activated protein kinase inhibition attenuates intercellular adhesion molecule-1 up-regulation on human pulmonary microvascular endothelial cellsSurgery12440379706165
- TomitaKBarnesPJAdcockIM2003The effect of oxidative stress on histone acetylation and IL-8 releaseBiochem Biophys Res Commun301572712565901
- TravesSLCulpittSRussellREK2002Elevated levels of the chemokines GRO-a and MCP-1 in sputum samples from COPD patientsThorax57590512096201
- UnderwoodDCOsbornRRBochnowiczS2000SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lungAm J Physiol Lung Cell Mol Physiol279L89590211053025
- van der VaartHKoeterGHPostmaDS2005First study of infliximab treatment in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med172465915937294
- WardSSotsiosYDowdenJ2003Therapeutic potential of phosphoinositide 3-kinase inhibitorsChem Biol102071312670534
- WangZCanagarajahBJBoehmJC1998Structural basis of inhibitor selectivity in MAP kinasesStructure61117289753691
- WarkPA2002DX-890 (Dyax)IDrugs5586912802707
- WedzichaJASeemungalTAMacCallumPK2000Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levelsThromb Haemost84210510959691
- WinzenRKrachtMRitterB1999The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanismEMBO J1849698010487749
- YakymovychIEngstromUGrimsbyS2002Inhibition of transforming growth factor-α signaling by low molecular weight compounds interfering with ATP-α or substrate-binding sites of the TGF-α type I receptor kinaseBiochemistry4111000712206672