Abstract
Scant attention has been paid to the potential role of gonadotropins in bone tissue homeostasis. The focus on estrogen and estrogen replacement therapy for osteoporosis as far back as the 1940’s may account for the paucity of gonadotropin studies in bone biology. It is conceivable that prevailing dogma may have subconsciously steered us away from addressing whether gonadotropins have a place in skeletal physiology. However an examination of bone tissue catabolism in ovariectomized (OVX) and luteinizing hormone-releasing hormone (LHRH) agonist (Zoladex®)-treated rats generated some interesting and conflicting data; Zoladex-treated rats, unlike the OVX group, failed to exhibit increased bone collagen catabolism despite clear evidence for estrogen deficiency. The findings, although controversial, supported the possibility that elevated gonadotropins in the OVX model were in some way accountable for increased bone catabolism. In response to these initial findings further studies were performed to determine if altered LH status may in some way impact on the skeleton To this end an investigation of bone mass and histomorphometry were conducted in LH receptor nullizygous mice and human chorionic gonadotropin (hCG) overexpressing mice. There were clear phenotypic differences; the LH receptor knockout mice displayed reduced bone mass whereas the hCG overexpressing animals had stark increases in bone mass. Much more recently the team of the Mount Sinai Bone Program have made a significant discovery that bone-resorbing osteoclasts express receptors for follicle-stimulating hormone (FSH) and that mice nullizygous for FSH receptor are resistant to bone loss despite severe estrogen deficiency. Details of these fascinating models will be presented together with additional findings that give credence for exploring gonadotropin action on the skeleton as we enter the twilight of this Decade of the Bone and Joint.
Estrogen’s claim on bone: an historical perspective
In 1941 Fuller Albright and colleagues were the first to communicate the association of osteoporosis (OP) with the menopause. At the time this important revelation was made it was generally accepted that the menopause was associated with ovarian failure and that estrogen levels declined with the menopause. Since ovarian failure seemed synonymous with increased bone loss it was postulated, as early as the 1940’s, that replacement estrogen might prevent OP. Certainly in the past two decades, estrogen, or hormone replacement therapy (HRT), has been an established prophylaxis for OP because it has successfully reduced the extent of bone loss and/or preserved bone mass postmenopause (CitationLindsay 2004; CitationStevenson 2005). Although it is generally agreed that estrogen plays a role in skeletal homeostasis the precise mechanism by which it exerts its sparing effect on bone is still not fully understood. To this end a concerted global effort has ensued to identify how estrogen contributes to bone metabolism and to ascertain if the effects of this steroid hormone are direct or otherwise.
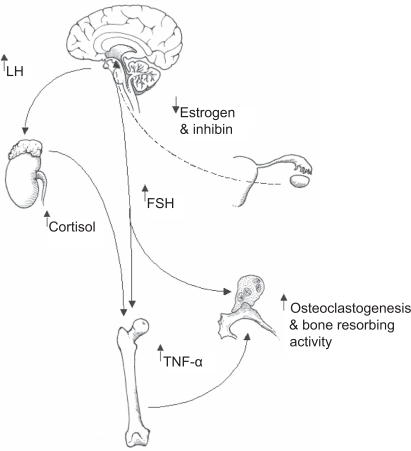
Figure 1 A summary of how follicle stimulating hormone (FSH) and luteinizing hormone (LH) might co-operate in promoting bone catabolism postmenopause. Elevated levels of LH are known to induce adrenal cortex hyperfunction and resultant increases in circulating cortisol. It is postulated that increased cortisol in response to raised LH postmenopause impacts upon bone forming osteoblasts by inhibiting bone matrix synthesis through dampening osteoblast activity. In the long term the effect of raised cortisol will contribute to osteopaenia and the subsequent development of osteoporosis. Increasing levels of FSH could target at least two different compartments of bone tissue; the bone resorbing osteoclast population and bone marrow macrophage precursors. With regard to the former, FSH has been shown to directly activate osteoclastic resorption. With reference to the bone marrow, resident macrophage precursors are now known to mobilise tumor necrosis factor alpha (TNF-α) in response to FSH. TNF-α is a key player in the demise of bone postmenopause through stimulating osteoclastic bone resorption. In this model elevated LH and FSH co-operate in fuelling net bone loss thereby increasing the risk of developing osteoporosis.
For a considerable time the effects of estrogen on bone were thought to be indirect since receptors for estrogen had not been isolated from bone tissue. The identification of estrogen receptors in human primary osteoblasts and osteoblast-like osteosarcoma cells twenty years ago provided the first indications that a direct effect of estrogen upon bone tissue might be involved (CitationEriksen et al 1988; CitationKomm et al 1988). Gideon CitationRodan (1991) proposed that estrogen might exert an anabolic action in bone either directly or via the generation of local growth factors. Early evidence in support of an anabolic action of estrogen in bone came from studies using rat osteoblasts (CitationErnst et al 1989; CitationGray 1989) in which type I collagen mRNA expression increased in response to estrogen. However estrogen administration to rats does not stimulate bone formation (CitationWesterlind et al 1993). Also, research conducted in Rigg’s group found no direct effect of estrogen on human osteoblast growth and maturation (CitationKeeting et al 1991). Similarly estrogen was without effect on type I collagen protein synthesis in human fetal (CitationRobinson et al 1997) and adult (CitationMahonen et al 1998) osteoblasts. Although attractive, the possibility that estrogen might elicit a direct anabolic action in mammalian bone is certainly not compelling; estrogen actually inhibits pre-osteoblast proliferation and decreases osteoblast activity (CitationTurner et al 1994) and only modest increases in bone mass have been obtained for mice treated with suprapharmacological doses of the steroid (CitationSamuels et al 1999). Furthermore a dose of 1500 μg estrogen/kg murine body weight was unable to induce endocortical bone formation (CitationBain et al 1993). Given that 5 μg estrogen/kg is sufficient to saturate the estrogen receptor ligand-binding sites it is highly unlikely that the effect of estrogen on any bone formation is via a “conventional pathway” (CitationTurner 1999). Turner comments that the increased bone mass (more specifically, osteosclerosis) reported by CitationSamuels and colleagues (1999) is attributed to bone marrow toxicity due to the very high circulating estrogen titre. Suffice it to say estrogen does not appear to be a particularly potent factor in the regulation of bone mass; mice nullizygous for either estrogen receptor alpha or beta have normal bone mass, furthermore only modest osteopenia is observed in mice lacking both receptors (CitationLindberg et al 2001; CitationMcCauley et al 2002; CitationWindahl et al 2002).
One other possible influence of estrogen on the skeleton is to inhibit the process of bone resorption by osteoclasts. With over half a century of basic and clinical research conducted after Albright and colleagues made the important connection between estrogen deficiency and OP, CitationPacifici (1998, 2008) distilled the biological basis of postmenopausal OP down to increased bone-resorbing cytokine production. The miscreants responsible are chiefly interleukin-1 (IL-1) and tumor necrosis factor-alpha (TNF-α) and their significance to the pathogenesis of postmenopausal OP will become clearer in due course. Most recently CitationNakamura and colleagues (2007) have provided evidence that estrogen withdrawal may prolong the life span of osteoclasts which in turn might be to the detriment of bone during periods of prolonged estrogen deficiency. In light of the proven efficacy of HRT in preserving bone mass postmenopause it is likely that estrogen dampens the process of bone resorption rather than promoting matrix synthesis.
Menopausal endocrinology is not a simple case of estrogen decline. At the time of Albright’s publication in 1941, far less was known about menopausal endocrinology. Although extragonadal factors (primarily from the pituitary) were believed to influence gonadal function Herbert McLean Evans and colleagues only coined the terms follicle-stimulating hormone (FSH) and luteinizing hormone (LH) four years prior to Albright’s publication (CitationMedvei 1982). Furthermore the association between LH/FSH and estrogen within the ovarian – pituitary axis was not understood and this continued for many years to come. Indeed the dynamic link between estrogen and gonadotropins even escaped Pincus and Kirsch in 1965 (CitationMedvei 1982). It is widely recognized that even modest estrogen withdrawal culminates in a corresponding rise in both LH and FSH from the anterior pituitary. It is particularly noteworthy why investigations into LH/FSH on bone metabolism have not, until quite recently, been considered despite the inverse and inextricable link between circulating estrogen and LH/FSH. It is a genuine possibility that raised gonadotropin levels might participate in bone resorption and contribute to the pathogenesis of postmenopausal OP. A description of how they came to be identified in the context of bone biology and how they might impact upon the skeleton are presented below.
Zoladex versus ovariectomy in the rat; early indications for a role of gonadotropins in stimulating bone loss
The biochemical composition and metabolism of bone collagen in OP formed the PhD thesis of the author. Part of the PhD studies involved the application of the ovariectomized (OVX) rat model, a widely used and accepted experimental tool to study bone in response to estrogen withdrawal (CitationSaville 1969; CitationMcOsker and Li 1991; CitationWronski and Yen 1991). In addition to the OVX model, I was keen to explore alternatives to OVX, primarily from a welfare point of view. One particularly attractive alternative to OVX was the application of a depot formulation of the luteinizing hormone-releasing hormone (LHRH) agonist goserelin (Zoladex) that had been shown to last at least one month (CitationWard et al 1989). Although the LHRH agonist buserelin had been used previously in rats (CitationGoulding and Gold 1989), this required daily injections and therefore constant animal handling, which was something I wanted to avoid.
Total hydroxyproline (Hypro) excretion relative to creatinine clearance was used to monitor collagen catabolism in the OVX and LHRH models on a weekly basis. As expected Hypro excretion rose markedly within the first week of OVX and remained elevated for up to 3 weeks after which it steadily fell to control levels by the sixth week. In marked contrast Zoladex administration led to reduced Hypro excretion which returned to control levels after one month. A second depot likewise resulted in a significant reduction in Hypro excretion, the levels once again rising to controls by week 8. A clear polarization in Hypro excretion was evident; OVX was producing the expected rise in collagen catabolism whereas estrogen deficiency triggered by Zoladex was actually decreasing it. Although the use of Zoladex was employed as an alternative to OVX for the induction of estrogen deficiency, it was particularly interesting to note a very different response of the model to agonist treatment in terms of bone catabolism. CitationGoulding and Gold (1989) had stated that “the degree of osteopenia elicited by OVX and buserelin treatment was similar” and that “administration of buserelin provides a new way of inducing estrogen-deficiency osteopenia in the rat without removing the ovaries”. However, their own data do not really give credence to these statements. For example all the urine Hypro analyses for the agonist-treated rats gave lower values than the OVX group. In addition for half of the time points the urine Hypro concentration between controls and agonist treated animals were similar. What is also important to note is that although Goulding and Gold pitched their article advocating buserelin as a surrogate for OVX the authors postulate that the differences identified for the two models may account for some steroidal protection in agonist treated rats. It is also possible that what Goulding and Gold had actually found was evidence of gonadotropins contributing to bone turnover but had overlooked their potential significance. Similarly CitationTobias and colleagues (1994) noted key differences between OVX and buserelin treatment on indices of trabecular bone turnover in rats; whereas 90 days post OVX produced up to 80% tibial cancellous bone loss, buserelin resulted in approximately 40% loss. In addition parameters reflecting changes in bone formation rate differed for the two models since buserelin treatment was not accompanied by increases in bone formation. Like Goulding and Gold, Tobias and colleagues suggested that the tool itself was to blame rather than providing a suggestion that gonadotropins in some way might be accountable. Importantly CitationYeh and colleagues (1996) subsequently revealed that an intact pituitary was necessary to trigger bone loss in response to estrogen withdrawal; whereas OVX resulted in marked bone loss, hypophysectomy in combination with OVX attenuated the osteopenic response. In light of the data that I had observed between Zoladex and OVX, I stated in my doctoral thesis that “These observations may support an important function for the gonadotropins in modulating the process of bone resorption.” These data have since been published (CitationMansell et al 2007), but for a few years after obtaining my PhD, bone biologists were reluctant to believe that gonadotropins had any role to play in skeletal metabolism following reports that LHRH agonists could cause osteopenia.
To further investigate the potential role of gonadotropins in skeletal development and morphology static histomorphometry and bone mineral density (BMD) were determined in mice nullizygous for the LH receptor (LuRKO) or mice overexpressing hCG. (CitationYarram et al 2003). Both groups exhibited skeletal phenotypes; male LuRKO mice had reduced bone mass for both femur and tibia whereas female hCG overexpressing mice had greatly elevated BMD at these sites. With regards to the latter model the circulating estrogen concentration was approximately double that of the wild type mice and would not have been sufficient to cause the large increase (30%) in murine BMD given that only suprapharmacological levels of this steroid yield less than comparable BMD in mice (CitationSamuels et al 1999). Importantly the increase in BMD reported for female hCG overexpressing mice could be prevented by OVX, thereby supporting a role of the ovary in generating the skeletal phenotype of these mice. One possible explanation however might be raised testosterone from hCG receptive thecal cells (CitationKumar 2005). Although the level of testosterone in the hCG overexpressing mice was elevated (∼6-fold) the potential for this steroid to explain the bone phenotype requires corroboration.
As stated above, the research gleaned from the application of LHRH agonists to treat prostate cancer and endometriosis has raised concerns regarding their impact on the skeleton. Importantly the research gathered from the use of these agonists indicates that gonadotropins do not participate in bone tissue metabolism. However, the magnitude of bone loss appears to be small and “unlikely to be of any clinical relevance” (CitationFogelman 1992). Although there are studies identifying bone loss in response to LHRH agonist use there are also reports in which the risk of developing OP is no different to age-atched individuals in the community (eg, CitationPeters et al 2001). Given that FSH/LH are greatly elevated at menopausal onset and that LHRH agonist administration does not mirror the skeletal changes observed for OVX it is possible that gonadotropins in some way contribute to bone catabolism.
Adrenal hyperplasia in response to LH: Could LH promote bone loss via cortisol upregulation?
A stark characteristic of the OVX rat/mouse is obesity; despite pair feeding with sham-operated controls, OVX animals always gain weight because of increases in intraabdominal fat. The increased fat and its distribution following OVX is likely linked to a Cushingoid state attributed to elevated glucocorticoids. Since the discovery of LH receptors in the adrenal gland and adrenal hyperfunction post OVX and the menopause (CitationKero et al 2000; CitationCarlson 2007), it is tempting to hypothesize that raised LH post-OVX might drive bone catabolism via increased cortisol production. Excessive cortisol, as found for patients with Cushing’s syndrome, increases the risk of developing OP because glucocorticoids diminish bone-forming osteoblast numbers (CitationManolagas 2000; CitationMancini et al 2004).
Bone-resorbing osteoclasts are targets for FSH: the Mount Sinai Bone Program revelation
The perimenopausal period is associated with the most rapid phase of bone loss (CitationRandolph et al 2004; CitationSowers et al 2006) even in the face of higher-than-premenopausal estrogen levels but very high circulating FSH (CitationPrior 1998). CitationDevleta and colleagues (2004) have recently provided a compelling association between raised FSH and osteopenia in ammenorrheic women. Similarly CitationKawai and colleagues (2004) correlate reduced FSH with bone accrual in response to estrogen. Collectively these findings inspired the Mount Sinai Bone Program to investigate whether bone could be a target for FSH.
A combination of reverse transcription polymerase chain reaction (RT-PCR), flow cytometry, Western blotting, and immunocytochemistry revealed that human and murine osteoclasts, and their precursors, expressed FSH receptors (FSHR). Osteoclastogenesis occurred when both human and murine bone marrow precursors were co-treated with FSH and receptor activator for nuclear factor kappa B (NF-κB)-ligand (RANK-L), a factor central for osteoclast formation. Furthermore FSH stimulated bone resorption and triggered phosphorylation of Erk, Akt, and Iκ-Bα, effectors of the proresorptive actions of RANK-L. There are now good data supporting the expression of Gi2α-coupled FSHR on osteoclasts and their precursors (CitationSun et al 2006).
In vivo, mice haploinsufficient for FSHβ have raised bone mass precipitated by reduced osteoclast activity. Severe estrogen deficiency occurs in response to FSHβ or FSHR ablation yet mice nullizygous for either have preserved bone mass despite being estrogen-deficient. These striking findings indicate that FSH directly participates in the regulation of bone mass (CitationSun et al 2006; CitationZaidi et al 2007). Recall that a major local stimulus for driving bone resorption postmenopause is TNF-α and there are compelling studies putting TNF-α as a key mediator of bone loss in response to estrogen withdrawal (CitationPacifici 1998, 2008); mice nullizygous for TNF-α do not lose bone in response to OVX (CitationRoggia et al 2001) and insensitivity to TNF-α protects against estrogen deficient osteopaenia (CitationAmmann et al 1997; CitationKimble et al 1997; CitationCharatcharoenwitthaya et al 2007). It is now known that this cytokine is mobilized by bone marrow precursors in response to FSH (CitationIqbal et al 2006).
The low-density lipoprotein receptor-related protein 5 (LRP5) gene is linked to circulating FSH in normal postmenopausal women
Low-density lipoprotein receptor-related protein 5 (LRP5) is a co-receptor for Wnt/β–catenin signalling (CitationHe et al 2004). It is now known that LRP5 is required for osteoblast proliferation (CitationGong et al 2001; CitationKato et al 2002) and possibly the prevention of osteoblast apoptosis (CitationBabij et al 2003). In a recent review of the literature (CitationMizuguchi et al 2004) it is becoming clear that LRP5/Wnt signaling is central for postnatal bone development and adult bone accrual making LRP5 an exciting candidate for further study in the context of musculoskeletal diseases, including osteoporosis and osteoarthritis.
Loss-of-function mutations in LRP5 result in osteoporosis-pseudoglioma syndrome (OPPG), an autosomal recessive disorder (CitationGong et al 2001). Conversely a gain-in-function mutation, for example a G171V point mutation, results in elevated bone mass, enhanced alkaline phosphatase activity and raised active osteoblast number (CitationBabij et al 2003). Furthermore mutation analysis of families and patients has revealed at least 19 LRP5 sequence variants of which 6 are thought to result in a high bone mass phenotype (CitationVan Wesenbeeck et al 2003).
It has recently been reported that LRP5 is associated with circulating FSH in normal postmenopausal women (CitationZofkova et al 2007). A compelling association between serum FSH levels and a C/T (c.4037:A1330V) polymorphism in the LRP5 gene was identified whereas no relationship was found for LH, estrogen testosterone or their precursors. The link between FSH and LRP5 strengthens the postulation that LRP5 is implicated in the changes accompanying the menopause. In addition their relationship supports the concept of co-evolutionary mechanisms linking calcium homeostasis with reproductive biology.
Given the compelling association between the regulation of bone mass with LRP5 and the association between this co-receptor and circulating FSH, the dynamic relationship between LRP5 and FSH certainly warrants closer investigation into bone metabolism postmenopause.
Concluding remarks
The revelation made by CitationSun and colleagues (2006) that FSH is the chief miscreant for postmenopausal OP has sparked considerable debate amongst leading researchers in the field (CitationBaron 2006; CitationMartin and Gaddy 2006; CitationSeibel et al 2006; CitationPrior 2007); CitationSeibel and colleagues (2006) postulate that the osteopaenic resistance in FSHR/ FSHβ nullizygous mice is attributed to raised testosterone in the face of elevated LH. Furthermore there are no reports of lower rates of bone loss for individuals with pituitary insufficiency compared to those with raised gonadotropins in response to natural or surgical menopause. In this regard CitationSeibel and colleagues suggest that differences in the levels of sex steroids explain the skeletal phenotype reported by Sun and colleagues (2006). However it is important to recognize those clinical and natural endocrine events in which the estrogen-deficiency-osteoporosis model does not hold; importantly the rate of bone loss is greater during the perimenopause than during the first years postmenopause when estrogen levels are lower. Also, both estrogen and progesterone are required to negate premenopausal bone losses. Whilst Jerilynn CitationPrior (2007) acknowledges that the estrogen-deficiency-osteoporosis model “faces considerable challenge” all are in agreement that much more research into the potential role of gonadotropins in skeletal physiology will have to be undertaken to displace it. Although we are far from a paradigm shift it is highly likely that our understanding of the skeletal response to estrogen deficiency will widen and with it changes in our approach to treating and preventing postmenopausal OP.
Acknowledgements
The author is indebted to Dr Alice May Roberts, University of Bristol, for the artwork presented in this review. The authors report no conflicts of interest in this work.
References
- AlbrightFSmithPHRichardsonAM1941Postmenopausal osteoporosis-its clinical featuresJAMA116246574
- AmmannPRizzoliRBonjourJP1997Transgenic mice expressing soluble tumor necrosis factor-receptor are protected against bone loss caused by estrogen deficiencyJ Clin Invest9916997039120014
- BabijPZhaoWSmallC2003High bone mass in mice expressing a mutant LRP5 geneJ Bone Miner Res69607412817748
- BainSDBaileyMCCelinoDL1993High-dose estrogen inhibits bone resorption and stimulates bone formation in the ovariectomized mouseJ Bone Miner Res8435428475793
- BaronR2006FSH versus estrogen: who’s guilty of breaking bones?Cell Metab3302516679287
- CarlsonHE2007Human adrenal cortex hyperfunction due to LH/hCGMol Cell Endocrinol269465017363138
- CharatcharoenwitthayaNKhoslaSAtkinsnEJ2007Effect of blockade of tumor necrosis factor-alpha and interleukin-1 action on bone resorption in early postmenopausal womenJ Bone Miner Res22724917295604
- DevletaBAdemBSenadaS2004Hypergonadotropic amenorrhea and bone density: new approach to an old problemJ Bone Miner Res223604
- EriksenEFColwardDSBeryNJ1988Evidence of oestrogen receptors in normal human osteoblast-like cellsScience241843388021
- ErnstMHealthJKRodanGA1989Estradiol effects on proliferation, messenger ribonucleic acid for collagen and insulin-like growth factor I, and parathyroid hormone-stimulated adenylate cyclase activity in osteoblastic cells from calvariae and long bonesEndocrinology125825332752978
- FogelmanMD1992Gonadotropin-releasing hormone agonists and the skeletonFertil Steril57715241555680
- GongYSleeRBFukaiN2001LDL receptor-related protein 5 (LRP5) affects bone accrual and eye developmentCell1075132311719191
- GouldingAGoldE1989A new way to induce oestrogen deficient osteopaenia in the rat: comparison of the effects of surgical ovariectomy and administration of the LHRH agonist buserelin on bone resorption and compositionJ Endocrinol12129382502594
- GrayTK1989Oestrogens and the skeleton: cellular and molecular mechanismsJ Steroid Biochem3428572626018
- HeXSemenovMTamaiK2004LDL receptor related protein 5 and 6 in Wnt/beta-catenin signalling: arrows point the wayDevelopment13116637715084453
- IqbalJLSunLKumarTR2006Follicle stimulating hormone stimulates TNF production from immune cells to enhance osteoblast and osteoclast formationProc Natl Acad Sci U S A103149253017003115
- KatoMPatelMSLevasseurR2002Cbfa-1 independent decrease in osteoblast proliferation,osteopaenia, and persistent embryonic eye vascularisation in mice deficient in LRP5, a Wnt coreceptorJ Cell Biol1573031411956231
- KawaiHFuruhashiMSuganumaN2004serum follicle stimulating hormone level is a predictor of bone mineral density in patients with hormone replacement therapyArch Gynecol Obstet269192513680264
- KeetingPEScottREColwardDS1991Lack of a direct effect of oestrogen on proliferation and differentiation of normal human osteoblast-like cellsJ Bone Miner Res62973042035356
- KeroJPoutanenMZhangF-P2000Elevated luteinizing hormone induces expression of its receptor and promotes steroidogenesis in the adrenal cortexJ Clin Invest1056334110712435
- KimbleRBainSPacificiR1997The functional block of TNF but nor of IL-6 prevents bone loss in ovariectomized miceJ Bone Miner Res12935419169353
- KommBSTerpeningCMBinzDJ1988Estrogen binding, receptor mRNA and biologic response in osteoblast-like osteosarcoma cellsScience2418143164526
- KumarTR2005What have we learned about gonadotropin function from gonadotropin subunit and receptor knockout miceReproduction13029330216123236
- LindbergMKAlataloSLHalleenJM2001Estrogen receptor specificity in the regulation of the skeleton in female miceJ Endocrinol1712293611691642
- LindsayR2004Hormones and bone health in postmenopausal womenEndocrine242233015542889
- MahonenAJukkolaARisteliL1998Type I procollagen synthesis is regulated by steroids and related hormones in human osteosarcoma cellsJ Cell Biochem68151639443071
- ManciniTDogaMMazziottiG2004Cushing’s syndrome and bonePituitary7243616132204
- ManolagasSC2000Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosisEndocr Rev211153710782361
- MansellJPBaileyAJYarramSJ2007Could bone tissue be a target for luteinizing hormone/chorionic gonadotropin?Mol Cell Endocrinol2699910617368927
- MartinJTGaddyD2006Bone loss goes beyond estrogenNature Med12612316761003
- McCauleyLKTözümTFRosolTJ2002Estrogen receptors in skeletal metabolism: lessons from genetically modified models of receptor functionCrit Rev Eukaryot Gene Expr128910012434924
- McOskerJELiXJ1991Use of rectilinear single photon absorptiometry to evaluate bone mass changes in rats. Scanning Microscopy International, ChicagoCell Mater Suppl193104
- MedveiVC1982A history of endocrinologyNorwell, MAKluwer Academic Publishers
- MizuguchiTFurutaIWatanabeY2004LRP5, low density-lipoprotein-receptor-related protein 5, is a determinant for bone mineral densityJ Hum Genet4980614727154
- NakamuraTImaiYMatsumotoT2007Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclastsCell1308112317803905
- PacificiR1998Editorial: cytokines, estrogen, and postmenopausal osteoporosis – the second decadeEndocrinology1392659619607769
- PacificiR2000Estrogen deficiency, T cells and bone lossCell Immunol2521–2688017888417
- PetersJLFairneyAKydP2001Bone loss associated with the use of LHRH agonists in prostate cancerProstate Cancer Prostatic Dis4161612497035
- PriorJC1998Perimenopause: the complex endocrinology of the menopausal transitionEndocrinol Rev19397428
- PriorJC2007FSH and bone-important physiology or not?Trends Mol Med131317141571
- RandolphJFSowersMBondarenkoIV2004Change in estradiol and follicle stimulating hormone across the early menopausal transition: effects of ethnicity and ageJ Clin Endocrinol Metab8915556115070912
- RobinsonJAHarrisSARiggsBL1997Estrogen regulation of human osteoblastic cell proliferation and differentiationEndocrinology1382919279202236
- RodanGA1991Mechanical loading, oestrogen deficiency, and the coupling of bone formation to bone resorptionJ Bone Miner Res652791887815
- RoggiaCGaoYGenciS2001Up-regulation of TNF-producing T cells in the bone marrow: a key mechanism by which estrogen deficiency induces bone loss in vivoProc Natl Acad Sci U S A9813960511717453
- SamuelsAPerryMJTobiasJH1999High-dose estrogen induces de novo medullary bone formation in female miceJ Bone Miner Res14178869933470
- SavillePD1969Changes in skeletal mass and fragility with castration in the rat: a model of osteoporosisJ Am Geriatrics Soc1715564
- SeibelMJDunstanCRZhouH2006Sex steroids, not FSH, influence bone massCell127107917174881
- SowersMRJannauschMMcConnellD2006Hormone predictors of bone mineral density changes during the menopausal transitionJ Clin Endocrinol Metab911261716403818
- StevensonJC2005Justification for the use of HRT in the long-term prevention of osteoporosisMaturitas511132615917151
- SunLPengYSharrowAC2006FSH directly regulates bone massCell1252476016630814
- TobiasJHChambersTJGallagherA1994Effect of administration and subsequent cessation of buserelin on cancellous bone of female ratsJ Bone Miner Res91919257872057
- TurnerRTRiggsBLSpelsbergTC1994Skeletal effects of estrogenEndocrinol Rev15275300
- TurnerRT1999Mice, estrogen, and postmenopausal osteoporosisJ Bone Miner Res14187919933471
- Van WesenbeeckLCleirenEGramJ2003Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone densityAm J Hum Genet727637112579474
- WardJAFurrBJValcacciaB1989Prolonged suppression of rat testis function by a depot formulation of Zoladex, a GnRH agonistJ Androl10478862533593
- WesterlindKCWakleyGKEvansGL1993Estrogen does not increase bone formation in growing ratsEndocrinology1332924348243320
- WindahlSHAnderssonGGustafssonJA2002Elucidation of estrogen receptor function in bone with the use of mouse modelsTrends Endocrinol Metab1319520012185665
- WronskiTJYenCF1991The ovariectomised rat as an animal model for postmenopausal bone loss. Scanning Microscopy International, ChicagoCells Mater Suppl16974
- YarramSJPerryMJChristopherTJ2003Luteinizing hormone receptor knockout (LuRKO) mice and transgenic human chorionic gonadotropin (hCG) overexpressing mice (hCGαβ+) have bone phenotypesEndocrinology14435556412865338
- YehJKChenMMAloiaJF1996Ovariectomy-induced high turnover in cortical bone is dependent on pituitary hormone in ratsBone18443508739902
- ZaidiMBlairHCIqbalJ2007Proresorptive actions of FSH and bone lossAnn NY Acad Sci11163768218083939
- ZofkovaIHillMZajickovaK2007Association of C/T polymorphism in the LRP5 gene with circulating follicle stimulating hormone in Caucasian postmenopausal womenPhysiol Res56735917087607