Abstract
Background
Isolated pulmonary hypertension secondary to systemic sclerosis is not uncommon. Our patient with systemic sclerosis presented with a very aggressive form of pulmonary hypertension due to a lethal combination of pulmonary veno-occlusive disease (PVOD) and pulmonary arterial thrombosis. This combined presentation has never before been reported in medical literature.
Case report
A 75-year-old woman with a 4-month history of atypical chest pains was admitted with a 3-week history of worsening symptoms of shortness of breath, reduced exercise tolerance, and bilateral pitting edema. On examination she had thickened skin in her hands, telangiectasia on her face, maculopapular rash in her legs, raised jugular venous pressure, and bilateral pitting edema. Her autoimmune profile revealed positive anticentromere antibodies, and her echocardiogram showed right ventricular systolic pressure of 91 mmHg. She also had renal impairment secondary to hypoperfusion. A diagnosis of isolated pulmonary hypertension secondary to limited systemic sclerosis was made. As she was clinically improving on slow diuretic infusion and awaiting transfer to a specialist center for management of pulmonary hypertension, our patient died due to cardiopulmonary arrest. Her postmortem revealed that she died of a combination of PVOD and pulmonary arteriopathy due to thrombosis.
Conclusion
This is clearly a unique case both in presentation and difficulty of management. Pulmonary vasodilators used in therapy of pulmonary arteriopathy can be detrimental in patients with PVOD. There is no definitive investigation, curative treatment, or management, that exists for a combination of PVOD and pulmonary arteriopathy due to thrombosis secondary to systemic sclerosis.
Introduction
Systemic sclerosis (SSC) is a chronic disorder of connective tissue with the features of inflammation and fibrosis, and produces degenerative changes in blood vessels, skin, joints, skeletal muscle, gastrointestinal (GI) tract, lungs, heart, and kidneys. Commonly noted features are the involvement of kidneys, causing renal crisis and gastrointestinal disease, but in contrast, another serious vascular problem, “intrinsic” pulmonary hypertension (PH), not secondary to pulmonary fibrosis, is found in 12% of patients with SSc, typically late in the course of the disease.Citation1 They are mainly associated with limited SSc and rarely with diffuse SSc. Our patient presented with a very aggressive form of PH secondary due to both pulmonary veno-occlusive disease (PVOD) and pulmonary arterial thrombosis.
Summary
A 75-year-old woman was admitted with a 3-week history of shortness of breath, bilateral pedal edema, reduced exercise tolerance of 10 to 15 yards, loss of appetite, and lethargy.
During the last 4 months before this current admission the patient had been under the care of cardiologists for investigation of atypical chest pains. She was fit and well before, and was not on any other regular medications. Symptoms started gradually, 4 months previously, with intermittent chest pain and exertional dyspnea after suffering from a flu-like illness. She was referred to the cardiologists. Further investigation with echocardiogram revealed mild to moderate pericardial effusion, normal left, and right, ventricular function. Right ventricular systolic pressure (RVSP) was mildly elevated at 28 mmHg at rest. She was diagnosed with viral pericarditis and discharged with nonsteroidal anti-inflammatory medications. Various laboratory tests were performed. She had mild renal impairment; normal liver function tests; normal thyroid function tests; slightly raised erythrocyte sedimentation rate at 28 mm/hr; slightly raised C-reactive protein; normal full blood count, and normal clotting screen. Her autoimmune profile for various immunological disorders was essentially normal apart from positive antinuclear human epidermoid carcinoma (HEp-2) antibodies. This isolated result was not given much importance at that time and the patient was not referred to the rheumatology department as she did not have any other significant symptoms or clinical features of SSc. On treatment with anti-inflammatories the patient was feeling a bit better and was discharged with regular outpatient follow-ups. The last 4 months she had been suffering from fluctuating symptoms of exertional dyspnea and leg swelling, and had been prescribed diuretics by her general practitioner, which subsequently caused deterioration in renal functions.
The last 3 weeks she presented with worsening symptoms of shortness of breath on exertion and then rest, reduced exercise tolerance, atypical chest pains, nonproductive cough, muscle and joint aches, lethargy, and severe Raynaud’s phenomenon in her hands and feet. The patient had no significant history of dysphagia or joint swelling. On general examination the patient had thickened skin on her palms and fingers, some telangiectasia of skin on the perioral and maxillary regions of the face, and urticarial maculopapular rash on her feet and around the digits, with some purpura. No digital ulcers or gangrene were noted. On systemic examination patient was hemodynamically stable, but had marked jugular venous pulsations and pitting edema up to her knees. Her renal function tests revealed her creatinine, at 420 μmol/L, was significantly higher than her normal level of 120 μmol/L. Her urine analysis revealed mild proteinuria, traces of leucocytes and ketones. No evidence of nitrates, blood, or casts were noted. The patient was reviewed by renal physicians, and a provisional diagnosis of renal impairment secondary to hypoperfusion was made. The patient was started on intravenous diuretic infusion over 48 hours, and all other nephrotoxic medications were withheld. Her autoimmune profile can be found in . The patient was reviewed by rheumatologists, the rash was biopsied, and its histology examination revealed mild inflammation of the skin, and the cutaneous vasculitis pattern of lymphocytes infiltrates within the partially damaged subcutaneous tissue structures was noted. The results were inconclusive as they did not point to the cause of the vasculitis. The patient had no clinical features of other rheumatological diseases associated with cutaneous vasculitis. Repeat echocardiogram and Doppler studies revealed minimal pleural function, normal left ventricular (LV) function and hypertrophied right ventricle (RV) with moderate RV failure and RVSP of 91 mmHg with dilated pulmonary arteries. Right heart catheter study was not performed because of lack of facilities in our district general hospital. Computed tomography (CT) pulmonary angiogram was performed, which revealed no evidence of clots in the main branches of the pulmonary artery with dilated pulmonary artery vasculature. Lung parenchyma showed no evidence of any interstitial lung disease or emphysematous changes (). The patient was discussed in the multidisciplinary team meeting which consisted of rheumatologists, chest physicians, renal physicians, and the radiologists. A diagnosis of pulmonary hypertensive disease secondary to limited SSc was made on the following features of Raynaud’s phenomenon: skin thickening in her palm and finger regions, telangiectasia, worsening dyspnea, positive anticentromere antibodies, dilated pulmonary arterial vasculature, and raised RVSP. The patient was urgently referred to a specialist center for further investigations and management of PH. The patient was only on diuretics and a small dose of calcium channel blockers. The patient was not put on any specific pulmonary vasodilators at that time as she was symptomatically and hemodynamically better, and was also awaiting further interventional studies in the specialist center. Her renal functions improved with the creatinine of 158 μmol/L, with a cautious infusion of diuretics pretransfer. Renal impairment improved without usage of any immunosuppressive agents. So, renal crisis secondary to SSc was ruled out, and renal biopsy was not performed.
Figure 1 Computed tomography thorax reveals marked enlargement of pulmonary artery (arrow).
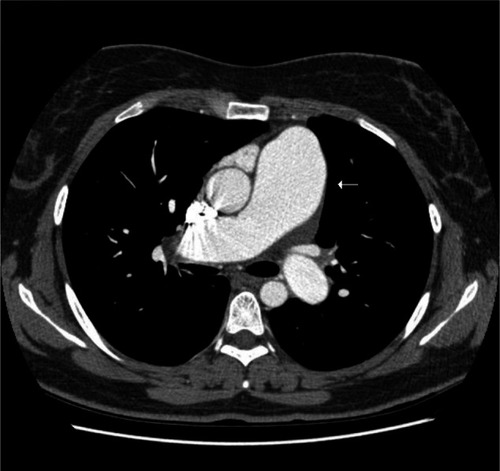
Table 1 Patient’s autoimmune profile
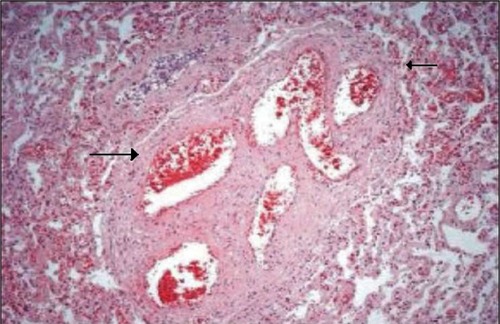
The night before the transfer to the specialist center, she unfortunately had a cardiopulmonary arrest with pulse-less electrical activity. The patient’s chest was clear, with no evidence of pulmonary edema. Her postmortem revealed the cause of her death was due to PVOD combined with pulmonary arteriopathy due to thrombosis. The histopathological images of the above are noted in and , respectively. The histopathology of pulmonary venous vasculature revealed () multiple fibrotic interlobular septae containing venules whose lumen was occluded by dense, collagen-rich fibrous tissue suggestive of PVOD. The specimen also demonstrated (), multiple lumens of small arterioles with recanalized thrombus. No evidence of plexiform lesions was found. No obvious blood clots were found in the main branches of the pulmonary arteries or the two subdivisions of the pulmonary arteries, which goes against the massive pulmonary emboli causing the cardiopulmonary arrest.
Figure 2 Pulmonary veno-occlusive disease reveals multiple septae with fibrinous exudates.
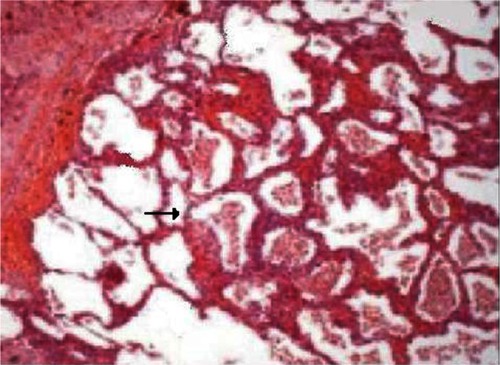
Figure 3 Pulmonary arteriopathy with thrombotic lesions arterioles with recanalized thrombi (arrows).
Discussion
This is an unusual case of PVOD and pulmonary arterial thrombosis secondary to SSc, without any lung parenchyma disease present, resulting in cardiopulmonary arrest and sudden death. No case has been reported with similar presentation and pathology in medical literature.
Limited SSc is characterized by sclerotic changes to hands, face, feet, or forearms, in addition to two or more of the following features, for example: atrophic changes of the ala nasi and lips; facial amimia; telangiectasia of the skin; late involvement of the lungs; late development of PH; anticentromere antibodies; dilated capillary loops in nail folds, and cutaneous calcification. Our patient fit well into the spectrum of limited SSc.
Isolated PH in SSc is one of the atypical complications of SSc. In the proceedings of the Fourth World Symposium on PH at Dana Point meeting, the new hemodynamic definition of a partial pressure of $25 mmHg was suggestive of PH.Citation2 This definition covers all forms of PH. Even though our patient did not have the right heart catheter study, the diagnosis of PH was made based on the symptom complex of PH, CT scan images, echocardiogram, and Doppler studies of the heart. Isolated PH in SSc has a high mortality compared with nonPH patients.
Pathophysiology of isolated PH secondary to SSc is complex. It is a type of vasculopathy in the lungs which is very aggressive and destructive in nature, with abrupt onset and lethal outcome.
Endothelial cell injury is the important process in the pathogenesis of SSc associated with pulmonary vascular disease. Endothelial dysfunction in the vessels starts a series of chemical responses with uncontrolled and imbalanced production of vasoactive mediators, vascular remodelling, and in situ thrombus formation, resulting in progressive luminal narrowing, which is further enhanced by sympathetic nervous system activity, as well as serotonin uptake in vessels.Citation3 The vascular resistance gradually builds up and elevated and sustained PH and RV overload, and hypertrophy, results in right heart failure.Citation4
There are five major histopathological features of PH noted in SSc patients. The majority of them belong to the group involving the pulmonary arteries and arterioles. They include pulmonary arteriopathy with plexiform lesions, pulmonary arteriopathy with thrombotic lesions, and arteriopathy without these lesions. A few of them belong to the group of either veno-occlusive disease or chronic pulmonary venous hypertension.Citation5 Our patient had both pulmonary arterial and venous disease causing worsening PH, deterioration in her clinical condition, and sudden death.
Pulmonary arteriopathy with thrombotic lesions is characterized by presence of thrombi in arterioles with evidence of recanalization. It is usually a histopathological diagnosis and very difficult to interpret from a routine CT scan image. CT scans used in diagnosis of PVOD and arteriopathy with thrombotic lesions are not very sensitive. CT scan of these patients reveals thickened septae, enlarged pulmonary arteries, and occasional pleural effusion. In our patient there were no obvious septae, pleural effusions were found, while enlarged pulmonary arteries with no interstitial lung disease were noted. Cutaneous vasculitis in SSc is not uncommon.Citation6 In our patient there was no clinical evidence of other rheumatological diseases which are normally associated with cutaneous vasculitis.
Immunology plays an important role in the diagnosis and prognosis of patients with SSc, especially that associated with anti-B23 autoantibodies, which are strongly associated with isolated PH related to SSc. Nearly 60% of these patients have these antibodies, as opposed to only 24% of nonPH SSc patients. B23 seropositivity is also associated with anti-U1RNP antibodies, and with decreased lung capacity.Citation7 It is also noted that anti-B23 autoantibodies are useful in detection of moderate-to-severe pulmonary arterial hypertension, suggesting that anti-B23 autoantibodies can be used for the subsequent development of PH in limited SSc. Recently, plasma concentrations of plasmin-a2-plasmin inhibitor complex (PIC) were found to be very high in SSc patients compared with normal control subjects.Citation8 The incidence of PH was higher in patients with elevated plasma PIC levels than in those with normal PIC levels. Plasma concentrations of PIC were also correlated with RV systolic pressures by echocardiogram. Currently, both B23 and PIC are investigational and are not available for clinical applications.
Management of the PH involving vasodilators, immunosuppressive agents, or glucocorticoids are ineffective and can even be detrimental. Currently, the goals of therapy are to increase exercise tolerance, improve quality of life, and prolong survival.Citation9 Optimal management requires accurate evaluation and staging of pulmonary arterial hypertension, careful serial monitoring, and supportive treatment using a combination of anticoagulants, inotropic agents, diuretics, and cardiopulmonary rehabilitation. Supplemental oxygen is appropriate in patients with resting or exertional hypoxemia. Calcium channel blockers are found to be not very responsive and also have a negative impact on LV function, causing hypotension and syncope. They should be used with great caution in SSC patients with advanced PH. Synthetic prostacyclin analogue that produces potent vasodilatory, antithrombotic, and inotropic effects, has been shown to cause long-term hemodynamic improvement.Citation9,Citation10 But these agents tend to cause deterioration in PVOD. So there are no clear guidelines for treatment of patients with both PVOD and pulmonary arteriopathy with thrombosis. Anticoagulation in isolated PH secondary to SSc has always been an uncertain topic.
Thrombotic arteriopathy has been implicated in the pathophysiology of pulmonary arterial hypertension. Overall, the literature supports a treatment effect of warfarin on survival of isolated PH in SSc. These results suggest that interruption of ongoing thrombosis with effective systemic anticoagulation therapy is associated with a better prognosis, especially for patients where the disease is not responsive to vasodilators. It is also important to recognize that anticoagulation with warfarin is associated with risk of intracranial and gastrointestinal bleeding.Citation11 It is important to note that our patient was not prescribed warfarin as no clear consensus was reached among the various physicians who dealt with our patient. This was because there were no clear guidelines in our hospital trust, or at the national level.
Conclusion
Prognosis of isolated PH due to SSc is worse than primary PH and secondary PH, due to interstitial lung disease and obstructive airways disease. There are also differences with respect to survival when the patients were classified based on their type of pathology. Patients having arteriopathy with thrombotic lesions have the best survival compared with those having arteriopathy with plexiform lesions and PVOD. These differences in survival between thrombotic and plexiform arteriopathies and between the thrombotic arteriopathy and veno-occlusive disease are statistically significant.Citation5 The differences are most apparent when survival during the first 6 months was compared between the three groups in the first 6 months: 71% of patients having PVOD, and 52% of those having plexiform arteriopathy were dead, compared with 30% mortality in patients with thrombotic arteriopathy. It is clearly evident that the combination of thrombotic arteriopathy and venoocclusive disease would be really very destructive in our patient’s condition. There exists no definitive investigation, curative treatment, or management for isolated PH secondary to SSc.
Disclosure
The author reports no conflict of interest with this work.
References
- VealeDJCollidgeTABelchJJFIncreased prevalence of symptomatic macrovascular disease in systemic sclerosisAnn Rheum Dis1995548538557492228
- HooperMNew definition of pulmonary hypertensionEur Respir J20093479079119797667
- ReichenbergerFSchauerJKellnerKDifferent expression of endothelin in the bronchoalveolar lavage in patients with pulmonary diseasesLung2001179316317411891606
- AbrahamDJVancheeswaranRDashwoodMRIncreased levels of endothelin-1 and differential endothelin type A and B receptor expression in scleroderma-associated fibrotic lung diseaseAm J Pathol199715138318419284832
- PietraGGMetteSAPalevskyHIHistopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung and Blood Institute, Primary Pulmonary Hypertension RegistryCirculation198980119812062805258
- HerrickALOogarahPKFreemontAJMarcusonRHaeneyMJaysonMIVVasculitis in patients with systemic sclerosis severe digital ischaemla requiring amputationAnn Rheum Dis1994533233268017986
- OkanoYSteenVDMedsgerTAJrAutoantibody to U3 nucleolar ribonucleoprotein (fibrillarin) in patients with systemic sclerosisArthritis Rheum1992351951001731817
- TormeyVJBunnCCDentonCPAnti-fibrillarin antibodies in systemic sclerosisRheumatology200140101157116211600746
- GaliNKimNHPulmonary microvascular disease in chronic thromboembolic pulmonary hypertensionProc Am Thorac Soc20063571616963536
- RubinLJBadeschDBEvaluation and management of the patient with pulmonary arterial hypertensionAnn Intern Med200514328229216103472
- DuchiniASessomsSLGastrointestinal hemorrhage in patients with systemic sclerosis and CREST syndromeAm J Gastroenterol199893145314569732924