Abstract
Gastrointestinal stromal tumor (GIST) is a rare primary neoplasm of the gastrointestinal tract, mesentery, or omentum. In the past, surgery has been the only effective treatment. The diagnosis and treatment of GIST has been revolutionized over the past decade, since expression of the receptor tyrosine kinase KIT was shown to occur on these tumors. Mutations in this proto-oncogene commonly cause constitutive activation of the KIT tyrosine kinase receptor, an important factor in the pathogenesis of the disease. The development of specific tyrosine kinase inhibitors, such as imatinib mesylate, has led to a breakthrough in the treatment of advanced GIST. Treatment with this drug has led to significant improvements in survival, with overall response rates in excess of 80%. Side effects are common, but usually manageable. The success of this drug has led to further trials investigating its use in the pre- and postoperative situation. This review summarizes the current knowledge of GIST and imatinib treatment and possible future developments.
Introduction
Gastrointestinal stromal tumor (GIST) is a rare tumor, accounting for less than 1% of primary gastrointestinal (GI) neoplasms. It is, however, the commonest non-epithelial tumor of the gastrointestinal tract. The median age of diagnosis is approximately 60 years, with the annual incidence estimated at 10–20 cases per million (CitationNilsson et al 2005). It is very rare in children and affects males and females equally. GIST is mainly a disease of the GI tract, mesentery, and omentum. Most commonly, it originates in the stomach (60%), followed by the small intestine (30%), the colon and rectum (5%), and the oesophagus (5%) (CitationVan der Zwan and DeMatteo 2005). Many are found incidentally at surgery or autopsy. GIST can be classified into different risk groups. At presentation, only 44% are overtly malignant or high risk, while 32% are of low or very low risk (CitationNilsson et al 2005). Although the exact pathogenesis is not fully known, it is thought to originate from the same lineage as the interstitial cells of Cajal. These are pacemaker cells of the GI tract, which are phenotypically similar to GIST cells (CitationKindblom et al 1998).
Most GISTs are spindle cell tumors, which were previously classed as either leiomyoma or leiomyosarcoma. Following the introduction of immunohistochemistry in the 1980s, Mazur and Clark coined the term GIST (CitationMazur and Clark 1983), but it was not until the 1990s that this entity was widely recognized. Most of these stromal tumors stained positively for CD34 (CitationMiettinen et al 1995). In 1998, it was discovered that these tumors had gain of function mutations in the KIT proto-oncogene (CitationHirota et al 1998). The KIT protein is a transmembrane receptor for stem cell factor. The intracytoplasmic portion of this receptor functions as a tyrosine kinase. The availability of the immunohistochemical marker, CD117, to the KIT protein, has revolutionized the diagnosis of GIST, by identifying a treatment target. Approximately 95% of GISTs stain positive for CD117, making it a very useful marker for diagnosis (CitationMiettinen and Lasota 2001). This has led to the development of the targeted therapy imatinib mesylate (STI-571; Glivec®, Novartis, Basel, Switzerland). This drug inhibits several tyrosine kinase receptors with varying affinity, including KIT, the BCR-ABL fusion protein, and the platelet derived growth factor receptor (PDGFR) (CitationHeinrich et al 2000, CitationDe Giorgi and Verweij 2005).
Presentation
Because GIST was only recently recognized as a separate clinicopathological entity, the literature prior to 2000 did not give an accurate account of the clinical behavior of GIST.
GIST can present in many ways. Thirty percent are diagnosed incidentally on a pathological or autopsy resection specimen (CitationNilsson et al 2005). Small tumors may be asymptomatic and GISTs can grow to a large size before producing any symptoms. This may be because GISTs grow by displacing adjacent structures rather than invading them. Presenting symptoms can therefore include non-specific GI symptoms such as nausea, vomiting, dyspepsia, abdominal pain, distension, or change in bowel behavior. Less commonly, there may be symptoms of obstruction, bleeding, or rupture into the peritoneal cavity.
Despite radical resection with clear margins, 40%–80% recur within the abdominal cavity. However, the majority of recurrences are solitary and thus may be resectable. The most common sites of metastases are the peritoneum and liver, whereas lymph node metastases are relatively rare. In contrast to leiomyosarcomas, pulmonary and bone metastases occur late and are uncommon.
Rare familial cases of GIST with a mutated KIT have been recognized. This may be as part of the Carney triad of gastric GIST, functioning extra-adrenal paraganglioma and pulmonary chondroma. This mainly affects young women and was first described in by Carney in 1977 (CitationCarney et al 1977). Although it is thought to be hereditary (young age and multiple specific tumors), no genetic abnormality has been identified. GIST has also been reported in association with neurofibromatosis type 1 (Von Reckinghausen’s disease) (CitationIshida et al 1996).
GIST can range in size from less than 1 cm to over 30 cm in diameter. Various investigations may be used in the diagnosis of GIST. Gastric tumors are often detected by endoscopy. Macroscopically, primary GIST is usually a well-circumscribed submucosal vascular tumor, protruding into the lumen. Computed tomography (CT) is useful to assess the extent of the primary disease and to assess for the presence of metastatic disease. Magnetic resonance imaging (MRI) may provide further soft tissue delineation. Positron emission tomography (PET) with the tracer 18-fluorodeoxyglucose (18-FDG) demonstrates intense uptake, but may not distinguish GIST from other malignancies. Its main use is in demonstrating the presence or absence of metastases.
Smaller tumors are usually treated by excision biopsy. For larger lesions, the use of preoperative biopsy is controversial. This is because there is a risk of tumor rupture, hemorrhage, and perforation of viscera (CitationJoensuu et al 2002). The diagnostic yield from endoscopic biopsy is approximately 50%.
Diagnosis
Macroscopically, GIST has the appearance of a friable unencapsulated mass arising from the muscle layer, rather than the epithelium. Central necrosis is often present in larger lesions (CitationD’Amato et al 2005). Microsopically, most GISTs consist of a uniform population of spindle cells (70%). Some are characterized as epithelioid type (20%), which typically arise in the stomach (CitationMiettinen et al 2002). The remaining 10% consist of a mixture of these two morphologies. The spindle cells are arranged in short fasicles or whorls, with prominent nuclear palisading. Either type may contain curvilinear collections of extracellular collagen called skeinoid fibres (CitationCorless et al 2004). The eosinophilic subtype consists of rounded cells with eosinophilic or clear cytoplasm (CitationVan der Zwan and DeMatteo 2005).
Ninety-five percent of GISTs stain positively for the CD117 antigen, which is an epitope for the KIT receptor tyrosine kinase. KIT is usually widespread and gives cytoplasmic staining and can show a characteristic dot-like “golgi” pattern (CitationFletcher et al 2002). Other markers which may be positive include BCL-2 (80%), CD34 (70%), muscle specific actin (50%), smooth muscle actin (35%), s-100 (10%), and desmin (5%) (CitationCorless et al 2004). KIT positivity is important as it is one of the targets of the tyrosine kinase inhibitor imatinib mesylate. KIT overexpression is common in a variety of tumor types including GISTs, seminomas, adenoid cystic carcinomas, malignant melanomas, and lung carcinomas (non-small cell and small cell types) (CitationTakahashi et al 1995; CitationWent et al 2004). But it is worth noting that positive staining for KIT does not necessarily indicate KIT activation or KIT mutation (CitationJoensuu et al 2002). KIT mutations, however, are present in 95% of GISTs. Other research has shown activating KIT muations in seminomas, chronic myeloproliferative disorder, acute leukemia, mast cell neoplasia, and sinonasal NK/T-cell lymphoma (CitationMiettinen et al 2002). Platelet-derived growth factor receptor alpha (PDGFRα) mutations are found in 5%–10% GISTs (CitationSihto et al 2005).
Many efforts have been made to produce a reliable prognostic classification () (CitationFletcher et al 2002). Mitotic count and tumor size have been shown to be very important. A large study examined 1765 tumors for prognostic markers (CitationMiettinen et al 2005). Tumours less than 10 cm with less than 5 mitoses per 50 high powered fields (HPFs) had only a 2%–3% risk of metastases. Conversely, the metastatic rate for tumors greater than 10 cm, with greater than 5 mitoses per 50 HPFs, was as high as 86%. Non-gastric primary tumor location and male gender may also be independent adverse prognostic factors (CitationRutkowski et al 2007).
Table 1 Proposed approach for defining risk of aggressive behavior in GISTs
Evidence is now emerging that mutational analysis can help to predict prognosis. A recent study showed improved survival in those with KIT exon 11 point mutation compared to deletions in gastric GISTs. The same was true with comparison with point mutations from other locations (CitationSteigen et al 2007). This is discussed further below.
Pathogenesis
GIST cells are phenotypically very similar to the KIT positive interstitial cells of Cajal of the GI tract. They are pacemaker cells which regulate motility and their development depends on cellular signalling regulated by the KIT protein (CitationMiettinen et al 2002). The KIT proto-oncogene on chromosome 4 (4q11–q12) encodes for the KIT protein, which is also expressed on mast cells, germ cells, and hemopoietic cells. Stem cell factor is the ligand for this receptor and binding activates cell growth, differentiation, and survival. In GIST, ligand independent activation occurs due to the presence of an activating mutation, which was first described by Hirota and colleagues. They showed that GIST cells were able to grow without their ligand in vitro and in nude mice (CitationHirota et al 1998). Since then, many more gain-of-function mutations have been identified (CitationSanborn and Blanke 2005). It has been shown that the site of mutation has a prognostic importance. The most frequent are exon 11 (65%–70%), 9 (10%–20%), 13 (1%–2%) and 17 (<1%) (). Mutations of the juxtamembrane domain (exon 11) of the KIT receptor are the most frequently seen, where small in-frame deletions and insertions or point mutations are responsible for ligand-independent receptor dimerization. These mutations are most commonly found in GIST of the stomach. Deletions and insertions tend to affect the first part of the exon (especially codons 557 to 559), whereas point muations are limited to just 4 codons (557, 559, 560, and 576) (CitationCorless et al 2004). Exon 9 mutations are located in the extracellular domain of the KIT receptor, whereas exons 13 and 17 encode for the intracellular part of the receptor (kinase 1 domain and activation loop respectively).
Figure 1 KIT receptor and location and frequency of mutations.
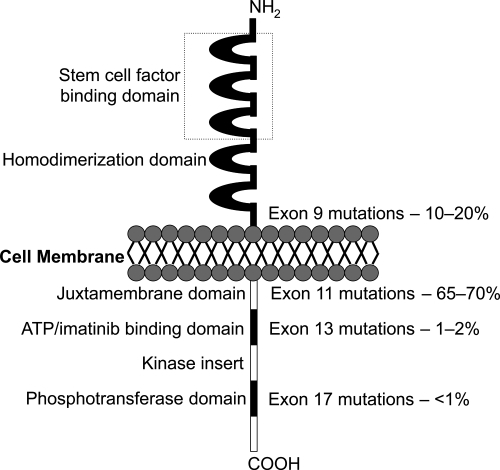
Some studies have suggested a more aggressive clinical behaviour in tumors with exon 11 mutations (CitationErnst et al 1998; CitationLasota et al 1999; CitationTaniguchi et al 1999; CitationLi et al 2000). Others have suggested that KIT mutations were not identified more commonly in higher grade GISTs (CitationRubin et al 2001) and that mutations of exon 11 were common in histologically benign GISTs. So there is continuing controversy as to whether exon 11 mutations confer aggressive behavior or not. As imatinib targets the ATP (adenosine triphosphate) binding site of the KIT receptor (encoded by exon 11), tumors with exon 11 mutations treated with imatinib have a significantly better partial response rate, event free and overall survival compared to exon 9 or no detectable mutation. A partial response rate of up to 83% has been achieved, showing how sensitive these tumors can be (CitationHeinrich et al 2003a; CitationCorless et al 2004; CitationDebiec-Rychter et al 2006).
Not all GISTs demonstrate KIT mutation. An important study of 40 KIT-negative GISTs demonstrated PDGFRα activating mutations in 14 (35%) (CitationHeinrich et al 2003b). The authors also showed that the mechanisms of cytogenetic progression and signal transduction were similar to those seen in KIT positive GISTs. These two mutational events are mutually exclusive of each other. Other studies have confirmed similar findings (CitationHirota et al 2003; CitationWardelmann et al 2004). Activating mutations of PDGFRα do not appear in other tumor types (CitationBurger et al 2005; CitationSihto et al 2005).
KIT or PDGFRα mutations occur early in the pathogenesis of GIST, as they are found even in very small tumors. Progression of GIST has been shown to be related to various chromosomal aberrations (CitationGunawan et al 2002). The most common chromosomal aberration seen in malignant GIST are losses of 14q32 and 22q11.The loss of the long arm of chromosome 22 is particularly associated with progression to a borderline or malignant lesion (CitationDebiec-Rychter et al 2001).
Surgery
Surgery remains the standard initial management for all localized GISTs. The tumor should be removed en bloc, with a clear margin. The pseudocapsule should be removed and not penetrated. Therefore, a wedge resection (stomach) or segmental resection (intestine) is required. If neighboring structures are involved, en-bloc resection should still be contemplated. Studies have evaluated the outcomes of surgery, comparing complete versus incomplete resections. In a series of 200 GISTs, median survival was 66 months for complete resection compared with 22 months for incomplete resection or unresectable disease (CitationDeMatteo et al 2000). The complete resection rate was approximately 85%. The optimum surgical margin has not been clarified. Lymph node dissection or biopsy is not recommended mainly due the pattern of spread of GISTs: lymph node metastases are rare.
More recently, there has been a move to laparoscopic surgery, particularly for gastric GISTs. One series of 50 consecutive patients showed this approach was associated with low morbidity and short hospitalization. All resections had clear margins and the long term disease free survival was 92% (CitationNovitsky et al 2006). In a second series of 22 clinically suspected GISTs, similar findings were shown, with only one case of recurrence (CitationBerindoague et al 2006).
Spontaneous tumor rupture, or rupture during surgery, increases the risk of peritoneal recurrence and is an adverse prognostic factor. Overall 5-year survival rates for primary resected disease are in the order of 50%–55%. One series has reported recurrence rates of up to 90% after surgical resection (CitationNg et al 1992). Most recurrences occurred within the first 2 years of resection. Prognostic factors for recurrence are shown in .
Table 2 Prognostic factors for recurrence
There is no standard follow up regimen for resected GIST. As recurrent disease can be treated by surgery or imatinib, active follow up is warranted. Suggested follow up protocols have been developed according to risk groups. For example, a high risk patient should have a CT scan every 3–4 months for 3 years, then every 6 months to 5 years. For low risk, a CT scan every 6 months for 5 years is acceptable (CitationBlay et al 2005).
The commonest sites of metastases are the liver and intraperitoneum. There is some evidence that metastatectomy can improve survival in selected patients (CitationChen et al 1998). Those that benefit most have a disease free interval of greater than 12 months, well differentiated histology and an isolated liver metastasis. Surgery can be used for palliation, but with the advent of imatinib, it has largely been superseded.
Chemotherapy
Interpretation of results from previous studies is difficult as it is likely that many tumors classified as leiomyosarcomas were actually GIST. Given this reservation, the efficacy of chemotherapy is low, with response rates less than 10% (CitationDematteo et al 2002). Doxorubicin and ifosfamide have limited activity in GIST compared to other soft tissue sarcomas. One study of 26 patients with GIST showed 38.4% to express P-glycoprotein and 35.4% expressed MRP1 (multi drug resistance protein). These levels were significantly higher than for soft tissue leiomyosarcoma. This may explain the poor response of GIST to standard chemotherapy (CitationPlaat et al 2000).
Radiotherapy
Radiation therapy has little role to play in the management of GIST. Most tumors are not amenable to treatment because of their location and close proximity to vital organs. GISTs are thought to be relatively radio-resistant. Nevertheless, radiotherapy can be successfully used in patients with advanced disease to control bleeding or other troublesome symptoms.
Hepatic artery embolization
This technique may provide palliation in patients with GIST metastatic to the liver. Due to the vascular nature of GIST, occluding the supplying artery may be effective. There has been more interest in chemoembolization, which allows increased local drug delivery, but reduced systemic effects due to high first pass metabolism in the liver. Two small studies have shown limited efficacy with responses lasting 8–12 months (CitationMavligit et al 1995; CitationRajan et al 2001).
Development of imatinib mesylate
Imatinib was developed as a tyrosine kinase receptor inhibitor. In 1989, a family of compounds called tyrphostins were shown to have specificity for the epidermal growth factor receptor. Further developments led to the identification of 2-phenylaminopyrimidine compounds as having the most promising inhibitory activity against receptor tyrosine kinases. This led to the development of imatinib, which was shown to inhibit the intracellular kinases ABL and BCR-ABL fusion protein in chronic myeloid leukemia (CML) cells (CitationDruker et al 1996), but was subsequently found to have comparable activity against the KIT receptor (wild type and mutant) and PDGFR (CitationCarroll et al 1997; CitationHeinrich et al 2000).
Imatinib is a competitive antagonist of the adenosine triphosphate (ATP) binding site. It blocks the transfer of phosphate groups from ATP to tyrosine residues of the substrates. This causes interruption of the downstream signalling process that leads to cell proliferation, including MAP kinase and Akt (CitationHeinrich et al 2000).
Pharmacology of imatinib mesylate
Pharmacokinetic studies of imatinib in healthy volunteers, patients with CML and GIST have shown good oral absorption and a bioavailability of 98%, regardless of the preparation (solution, capsule or tablet) or dosage strength (100 mg, 400 mg) (CitationPeng et al 2005). Once absorbed, it binds to serum proteins (mainly albumin and alpha 1-acid glycoprotein) and peak concentrations are reached 4 hours after administration (CitationD’Amato et al 2005).
The main circulating metabolite is an N-demethylated piperazine derivative which accounts for 16% of the AUC (area under the curve) for imatinib. This, with imatinib accounts for most of the activity, but there are a number of smaller metabolites. CYP3A4 is the major cytochrome P450 involved in imatinib metabolism. Thus, drugs that are co-administered may alter the pharmacokinetics. Erythromycin, fluconazole, and rifampicin have shown inhibition of imatinib metabolism. Imatinib increases exposure to simvastatin. Alprazolam, caffeine, clindamycin, clonazepam, cortisol, ethinyl oestradiol, and verapamil may cause toxic effects when given with imatinib. St. John’s wort increases imatinib clearance by 43%. Patients should avoid excessive amounts of paracetamol as both are metabolized by CYP3A4 (CitationD’Amato et al 2005; CitationNovartis 2005).
The terminal elimination half-life is approximately 18 hours, which allows for once a day dosage. Excretion is via the feces and urine, the majority being the metabolites. In patients with GIST, steady state exposure was 1.5 fold higher than that observed in CML for the same 400 mg/day doseage. This mainly seems to depend on albumin, white blood count and bilirubin. Hepatic and renal dysfunction and liver metastases may cause variable and reduced metabolism of the drug. Age, race, sex, and bodyweight do not significantly affect the pharmacokinetics of the drug (CitationPeng et al 2005; CitationNovartis 2005).
Preclinical studies of imatinib
Druker and colleagues first recognized the BCR-ABL protein to be an excellent target for imatinib as the BCR-ABL mutation is present in the vast majority of patients with CML. They showed that imatinib inhibited proliferating myeloid cell lines specifically. 95% reductions in concentrations of the abnormal protein were seen. Little toxicity was seen in normal bone marrow cells. Very rapidly in vitro results were tested in vivo, with significant effects of imatinib seen in nude mice and then humans with chronic phase CML (CitationSavage and Antman 2002; CitationDruker et al 1996).
The early success of imatinib in chronic phase CML led investigators to assess its effects on c-KIT receptor tyrosine kinase activity (CitationHeinrich et al 2000). C-KIT autophosphorylation, activation of mitogen activated protein (MAP) kinase and activation of Akt were all inhibited in a c-KIT expressing cell line. In a cell line with an activating mutation of c-KIT, more potent inhibitory effects occurred as compared to the wild-type receptor. These findings led to the use of imatinib in the first patient in 2001 as discussed below (CitationJoensuu et al 2001).
Phase I studies
Following on from the preclinical studies, imatinib was tested in a phase I study in patients with Philadelphia chromosome positive CML (CitationDruker et al 2001). This and subsequent studies established that a continuous dosing schedule of 400 mg/day was safe and effective in CML. The first GIST patient to receive imatinib was a 50-year-old woman, with KIT-positive metastatic GIST associated with a mutation in exon 11. She had progressive metastatic disease despite surgery, combination cytotoxic chemotherapy, thalidomide and interferon alpha. She was treated with 400 mg of imatinib per day and response was evaluated using 18-FDG PET (18-fluorodeoxyglucose positron emission tomography) and CT scanning. After 1 month, the patient had a complete metabolic response and by 8 months many of the liver metastases had disappeared or reduced in size (CitationJoensuu et al 2001).
A formal phase I study was conducted by the EORTC (European Organisation for Research and Treatment of Cancer) Soft Tissue and Bone Sarcoma Group (Citationvan Oosterom et al 2001). 40 patients (36 with GIST and 4 with non-GIST soft tissue sarcoma) were recruited between August and December 2000. The dose groups of imatinib were 400 mg once daily, 300 mg twice daily, 400 mg twice daily, and 500 mg twice daily. Dose limiting toxic effects were nausea, vomiting, oedema and skin rash which were seen at a dose of 500 mg twice daily. The maximum tolerated dose recommended was 400 mg twice daily. No responses were seen in non-GIST sarcomas. Among the GIST patients, responses were seen at all dose levels. Fifty-four percent achieved a partial response, 37% had stable disease, and 5% had progressive disease. After 9 months of therapy, 82% of GIST patients continued to obtain clinically important benefits. In the update, the most common adverse effects for those continuing on therapy were periorbital oedema (40%) and peripheral oedema (37.5%) (Citationvan Oosterom et al 2002).
Phase II studies
After the success of the initial studies in GIST and CML, further studies were conceived. The first trial was a US – Finland trial, which was a multicentre, randomized, open label phase II study (CitationDemetri et al 2002). One hundred and forty-seven patients with unresectable or metastatic GIST were randomly assigned to receive 400 mg or 600 mg of imatinib daily, between July 2000 and April 2001. For those who progressed on 400 mg daily, the dose was increased to 600 mg daily. After a median follow up of 21 months, 66% achieved a partial response and 17% had stable disease. No significant differences were seen between the two dose groups. 14% died of progressive disease. Median survival had not been reached, but 85% were estimated to be alive at 19 months. The trial was not powered to distinguish between efficacy at the two dose levels. However a third of those who progressed on the lower dose sustained a subsequent partial response on crossover (CitationBlanke et al 2004).
A phase II trial of the EORTC Soft Tissue and Bone Sarcoma Group confirmed these findings (CitationVerweij et al 2003). This study was designed to test the efficacy of imatinib at the maximum tolerated dose of 400 mg twice daily. A total of 51 patients participated, 27 of whom had confirmed GIST, 24 with other soft tissue sarcomas. In the GIST group, 4% had complete remission, 67% partial remission, 18% stable disease, and 11% progressive disease. In contrast, there were no objective responses in the non-GIST sarcoma group. Among GIST patients, the median time to onset of response was 113 days. 73% of GIST patients were free from disease progression at 1 year. In terms of toxicity, the most frequent adverse effects were anemia (92%), oedema, particularly periorbital (84%), rash (69%), and fatigue (76%). Granulocytopenia was less common at 48%. These studies led to the registration of imatinib for the treatment of locally advanced or metastatic GIST in 2002.
A Japanese phase II randomized study evaluated 74 patients with unresectable or metastatic GIST. Patients received either 400 mg daily or 600 mg daily. The results showed slightly lower response rates of 55% (overall) and a partial response rate of 41%. Interestingly, there were more responses in the higher dose group (61% vs 47%), but this did not reach statistical significance. The higher dose group had more interruptions of therapy as well as dose reductions. Grade 3 or 4 neutropenia and dermatitis was also significantly higher. The investigators therefore recommended 400 mg daily as the optimal dosing schedule (CitationDoi et al 2004).
Response assessment
In clinical trials in patients with advanced GIST treated with imatinib, the median time to objective response is about 13 weeks (CitationDemetri et al 2002). However, some patients have a dramatic improvement in symptoms and signs within days, accompanied by evidence of reduced metabolic activity on PET scans. Interestingly, no cases of tumor lysis syndrome have yet been described. The changes following an imatinib response can be quite distinct. Commonly cystic changes develop that could be confused with progressive disease (CitationLinton et al 2006; CitationRyu et al 2006). Therefore traditional response assessment criteria are not entirely relevant. In the original patient treated with imatinib, biopsy specimens at 1 and 2 months after starting treatment showed a marked decrease in tumor cells as well as myxoid degeneration and scarring. No necrosis or inflammatory reaction was seen. The tumor cells did not stain for Ki-67 (a proliferation marker) suggesting relatively low activity (CitationJoensuu et al 2001).
Many trials have used CT and 18-FDG PET in the assessment of imatinib response. One study compared CT, PET and dual modality PET/CT monitoring the effects of imatinib therapy in 20 patients. PET/CT identified 282 metastases, CT picked up 249 lesions and PET alone showed 135. In evaluating disease at 3 and 6 months of therapy, PET provided 100% accuracy for response, compared to 57%–60% using CT. PET also detected responses earlier than CT (CitationAntoch et al 2004). In a similar study of 28 assessable patients, the absence of FDG uptake after initial treatment was associated with a better prognosis than those with residual uptake. CT detected more lesions, but was less good at predicting prognosis (CitationGoerres et al 2005). A further study showed PET response was associated with a significantly longer progression free survival (CitationStroobants et al 2003; CitationKing 2005).
Choi and colleagues have proposed new response criteria for GISTs. They studied 172 lesions in 40 patients with metastatic disease. Patients were assessed pre-treatment and after 2 months with CT imaging and FDG-PET. A decrease in tumor size of more than 10% or a decrease in tumor density of more than 15% on CT had a specificity of 100% and a sensitivity of 97% in identifying PET responses. This compares to 100% specificity and 52% sensitivity using RECIST criteria. Thus, RECIST criteria are felt to be a poor indicator of clinical benefit, underestimating the effects of treatment (CitationChoi et al 2007). These new criteria have been further evaluated and correlation has been shown with time to tumor progression and disease specific survival. No similar correlation was seen using RECIST criteria (CitationBenjamin et al 2007).
Following initial response to imatinib, resistance often develops. There have been reports of a characteristic new nodule-within-a-mass pattern of recurrence (CitationShankar et al 2005). In this series of 92 patients treated with imatinib, 39 developed progressive disease and 21 of them showed this pattern. It was the first sign of progression in the majority with this feature. The authors postulate that this pattern represents a localized clone of mutant tumor cells developing resistance to c-KIT inhibition. This is important, as traditional response criteria may not identify the first signs of recurrent disease.
Phase III trials
There have been two large randomized controlled phase III trials assessing the optimum dosage of imatinib. The EORTC Soft Tissue and Bone Sarcoma Group conducted the largest randomized phase III trial (CitationVerweij et al 2004). 946 patients with histologically proven advanced or metastatic GIST (c-KIT positive) were randomized to receive imatinib 400 mg or 800 mg daily. The option of crossover to the higher dose was offered to patients with progression on 400 mg daily. The primary end point was progression free survival. The median follow up was 25 months. Response rates were equal in both arms: objective response rate (complete and partial response) was 50% for the 400 mg group and 54% in the 800 mg group. 56% had progression on 400 mg compared to 50% on 800 mg (HR 0.82, 95% CI 0.69–0.98, p = 0.026). Overall survival was 85% at 1 year and 69% at 2 years for the 400 mg group and 86% at 1 year and 74% at 2 years for the 800 mg group. There were more dose reductions and interruptions in the higher dose group. Toxicity was common but manageable. Oedema, anemia, rash, lethargy, nausea, bleeding, diarrhoea, and dyspnoea were statistically more common in the 800 mg group. shows the frequency and grade of toxicity for those receiving 400 mg/day.
Table 3 Frequency of side effects (400 mg/day dose)
The North American Sarcoma Intergroup study (S0033) randomized 746 patients to either 400 mg or 800 mg daily of imatinib. Patients randomized to 400 mg daily were dose escalated to 800 mg daily on disease progression. The primary end point was overall survival. Early results presented in abstract in 2003 showed no difference in overall survival or progression free survival, after a median follow up of 14 months. The proportion of patients achieving an objective response or stable disease was similar in both arms (75% (400 mg) vs 73% (800 mg)). Objective response rates were 43% vs 41% respectively (CitationBenjamin et al 2003). In the 2004 update, after a median follow up of 25 months, the median survival had not been reached. 2 year survival for the 400 mg vs 800 mg group was 73% and 78% respectively, 2 year progression free survival was 50% and 53% respectively. Again these were not statistically different. Eighty-eight patients crossed over to the higher dose on disease progression. Of these, 7% demonstrated partial response and 29% achieved stable disease (CitationRankin et al 2004).
These two studies have shown differing results. The North American study with overall survival as primary end point has to date shown no differences between the two dose groups. In contrast, the EORTC study, with progression free survival as its primary end point, has shown a statistically significant improvement in progression free survival, but requires further follow up to assess overall survival. In both studies, survival results may be influenced by the cross over design.
At ASCO 2007, the GIST Meta-analysis Group presented the results of a meta-analysis of these two randomized studies. After a median follow up of 45 months, a small but significant progression free survival advantage was seen in the high dose arm. Overall survival was identical in the two arms (median survival 4.08 years (bid) vs 4.05 years (od), HR 1.00). Compared to KIT exon 11 mutants, wild type KIT, exon 9 and other mutations conferred a poorer prognosis. The interaction between dose effect on progression free survival and prognostic factors was significant for KIT exon 9 mutations, but not for other factors. The heterogeneity between the two trials is still being evaluated (CitationVan Glabbeke et al 2007).
The results of an interesting phase III trial have recently been reported by the French Sarcoma Group (CitationBlay et al 2007). It was a prospective randomized trial comparing continuous and interrupted use of imatinib beyond 1 year of treatment in patients with advanced GIST. Between May 2002 and April 2004, 182 patients were enrolled. Of these, only 58 were eligible for randomization after 1 year with controlled disease. 26 patients continued with imatinib, whilst 32 discontinued until progression. Documented progression was seen in 8 of 26 patients in the continuous group and 26 of 32 patients in the interrupted group (p < 0.001). However, 24 of the latter subsequently responded to reintroduction of imatinib. There were no differences in survival, resistance or quality of life between the two groups. This study supports the use of continuous imatinib, due to the high level of progression after interruption of therapy. In a further French study, 35 patients free from progression after 3 years of imatinib at 400 mg/day, were randomly offered to continue or interrupt treatment. 9 progressors were reported after a median follow up of 5.3 months, all of whom achieved tumor control (at least stable disease) after rechallenge with imatinib. This study continues (CitationLe Cesne et al 2007).
Neoadjuvant and adjuvant imatinib
The success of imatinib in controlling locally advanced and metastatic GIST has led to interest in the neoadjuvant and adjuvant use of the drug. There have been case reports and series suggesting a role for the neoadjuvant approach (CitationBauer et al 2005; CitationLoughrey et al 2005; CitationRaut et al 2006). In a series of 90 patients reported by Bauer and colleagues, 12 were rendered suitable for surgical resection of residual disease. Eleven of these patients achieved a complete resection, although most had viable tumor cells in the resected specimen. The median overall survival was 46 (range 4–85) months. This report has therefore shown prolonged survival and a significant rate of subsequent resectability after treatment with imatinib induction. In another series, Raut et al evaluated 69 consecutive patients with advanced GIST treated by surgery whilst receiving imatinib or another tyrosine kinase inhibitor, sunitinib malate (SU11248; Sutent®, Pfizer inc, New York). Surgical outcome correlated strongly with pre-surgery disease status. There was no evidence of residual disease in 78% with stable disease before surgery, in 25% with limited progression and in 7% with generalized progression. One-year progression free survival was 80%, 33%, and 0% for stable disease, limited progression and generalized progression respectively. One-year overall survival was 95%, 86%, and 0% for the same groups. These reports suggest a role for primary imatinib therapy and surgery for advanced GIST. Selected patients may experience prolonged survival after this aggressive initial approach. However, the actual benefit of imatinib followed by surgery still requires further evaluation in prospective clinical trials. Such trials are underway and are shown in . Neoadjuvant imatinib is not recommended where a change in tumor size will not affect surgery (CitationBlay et al 2005). It can, however, be considered where a tumor response could permit function-sparing surgery, eg, rectum or oesophagus.
Table 4 Neoadjuvant and adjuvant trials of imatinib in GIST
There has also been significant interest in the use of imatinib in the adjuvant setting. This stems from the successes of treatment of advanced disease with the rationale that it may be more effective in minimal residual disease and may be curative in this setting. In high risk patients, the rates of recurrence after primary surgery are about 50%. Prospective trials are running to assess the role of imatinib following surgery for high risk GIST and are shown in (CitationNCI, 2007). The ACOSOG Z9001 study randomized 708 patients following complete resection of a primary GIST of at least 3 cm diameter, to either 1 year of imatinib 400 mg/day or placebo. Upon recurrence, patients could be crossed over from placebo to imatinib, or their imatinib dose increased to 800 mg/daily. The trial was stopped early after a planned interim analysis, with a median follow up of 1.2 years. The primary endpoint of 1-year relapse free survival was 97% in the imatinib arm and 83% in the placebo arm, with a hazard ratio of 0.325 (p = 0.0000014). It appears the significant benefit occurs in the first 18 months, but then the recurrence rate is similar in both arms. This has brought concern that early treatment may lead to the early development of resistance. Longer follow up required to see if there is an improvement in overall survival, although contamination of the placebo group may jeopardise this analysis (CitationDeMatteo et al 2007). The EORTC study (62024) of adjuvant imatinib selects patients on the basis of intermediate or high risk of recurrence, rather than tumor size alone. Patients are randomized to 2 years treatment with imatinib 400 mg/day or observation. The primary endpoint is overall survival, and the study is expected to complete accrual by January 2008.
Imatinib resistance
Two types of resistance to imatinib treatment exist: primary resistance (on initial use of the drug) and secondary resistance (after an initial response). Ten to fifteen percent of patients are primarily resistant to treatment with imatinib. Fletcher and associates evaluated 3 patients with primary and 13 patients with secondary resistance (CitationFletcher et al 2003). Four mechanisms of imatinib resistance were identified: 1) Target resistance due to mutation – a new KIT or PDGFRα point mutation and protein activation, superimposed on the original mutation in that gene. 2) KIT genomic amplification with overexpression of the KIT oncoprotein, without a new point mutation. 3) Target modulation – activation of an alternate receptor tyrosine kinase protein, accompanied by loss of KIT oncoprotein expression. 4) Functional resistance – KIT or PDGFRα activation, outside the juxtamembrane hotspot regions, in the absence of a secondary point mutation. All these mechanisms were involved in late resistance, but only 4) was seen in primary resistance. At progression, all GISTs showed activation of essential downstream pathways dependent on KIT or PDGFRα oncogenic stimulation in untreated tumors. In PDGFRα exon 18 (90% of all PDGFRα), the point mutations have been shown to be resistant to imatinib. The in-frame deletions are, however, sensitive to imatinib. A further study showed secondary mutations in almost half of patients with acquired resistance (CitationAntonescu et al 2005). These all had a primary exon 11 mutation and mostly exon 17 secondary mutations. Primary resistant tumors did not contain secondary mutations. Other studies have also shown new KIT mutations as a cause for secondary resistance to imatinib (CitationWardelmann et al 2005; CitationBertucci et al 2006; CitationWardelmann et al 2006). RNA interference has revealed imatinib resistant GIST cells are still dependent on KIT kinase activity for activation of essential downstream signalling pathways. HSP 90 inhibition has been shown, in preclinical studies, to be a possible solution to imatinib resistance (CitationBauer et al 2006).
As part of the EORTC phase III study, various co-factors were studied to assess if they played a part in early or late resistance (CitationVan Glabbeke et al 2005). Initial resistance occurred in 12%. This was independently predicted for by the presence of lung metastases and the absence of liver metastases. Predictors of late resistance were high baseline granulocyte count, a non-stomach primary tumor, large primary size, and low initial imatinib dose.
There may also be a pharmacological reason behind imatinib resistance (CitationDe Giorgi and Verweij 2005). Toxic effects of imatinib decrease over time as does the exposure of the drug (CitationJudson et al 2005). Dose related effects have been seen in some of the clinical studies which may suggest a change in the pharmacokinetics during drug exposure could result in a change in the efficacy of the drug.
Other clinical studies
There have been many studies in the imatinib era which have been evaluating the effects of imatinib treatment in clinical practice. In the pre-imatinib era, surgery was the mainstay of treatment. For example, 5-year overall survival in surgical series have ranged between 21% and 95%, partly depending on the grade of tumor included (CitationWu et al 2003, CitationBucher et al 2006). GISTs of the jejunum and ileum treated surgically have been shown to have a 39% tumor related mortality, which was twice that of gastric GISTs (CitationMiettinen et al 2006). Case series assessing the use of imatinib in advanced disease have shown comparable responses to the trial data, with overall responses in excess of 80% (CitationSchindler et al 2005; CitationKasper et al 2006,).
The role of surgery is now changing in the face of imatinib prolonging survival of advanced disease. The ESMO consensus meeting in 2004, suggested that surgical resection of disease after an imatinib response (usually after 4–12 months) still remains experimental (CitationBlay et al 2005). Global progression after imatinib is certainly a contraindication to radical surgery (CitationRaut et al 2006). The use of surgery for limited intra-abdominal metastatic disease and locally advanced disease after imatinib is unproven, but appealing. Resection of isolated recurrent disease has not been shown to confer a significant survival benefit (CitationNeuhaus et al 2005). However, with the use of imatinib, liver metastatectomy may produce long term survival, in excess of 24 months (CitationSakakura et al 2006). Careful selection may improve results, with data suggesting a disease free interval of more than 18 months, particularly for peritoneal or liver recurrence (CitationKosmadakis et al 2005). Good clinical responses have been seen with the use of imatinib in recurrent and metastatic disease, but the real benefits in association with surgery will have to be gauged in the light of several ongoing clinical trials (CitationWu et al 2003; CitationBenjamin et al 2006).
Future perspectives
The role of imatinib in GIST is now well established. It has become a standard of care for advanced and metastatic disease. Its role in neoadjuvant and adjuvant therapy is currently the subject of clinical trials. Evidence for the use of mutational analysis is increasing. This could be used to give information on tumor aggressiveness and potential response to therapy. Debiec-Rychter and colleagues have shown the presence of KIT exon 9 activating mutations and the absence of detectable KIT or PDGFRα predict for a worse outcome. However, expression of exon 9 KIT oncoprotein was shown to be associated with an improved progression free survival with the higher dose schedule of imatinib. There was no difference according to dose in those with the exon 11 mutation (CitationDebiec-Rychter et al 2006). This suggests that those GISTs with an exon 9 mutation should be commenced on the higher 800 mg/day dose of imatinib. Others could safely be started on 400 mg/day and increased to 800 mg on evidence of progression.
Gene expression patterns have been assessed using DNA microarray techniques. The gene FLJ10261, encoding for the DOG1 protein is specifically expressed in GISTs, irrespective of KIT or PDGFRA mutation status (CitationWest et al 2004). Its function is not known, although it seems to be fairly specific to GIST, rarely being expressed in other soft tissue tumors. It may have a future role in diagnosis, especially in PDGFR mutants failing to express the KIT antigen.
There are now various newer agents being investigated in GIST. Sunitinib, an oral, multitargeted receptor tyrosine kinase inhibitor has shown promise in tumor models. Specifically, it has been shown to inhibit KIT, PDGFR, FMS-like tyrosine kinase 3 and VEGF. Its role has now been investigated in a randomized controlled trial against placebo for imatinib resistant GIST patients (CitationDemetri et al 2006). The study was unblinded early, as sunitinib gave a significantly longer time to tumor progression (27.3 weeks) compared with placebo (6.4 weeks). The difference was statistically significant (HR 0.33, p < 0.0001). However, the trial had a crossover design and patients who progressed on the placebo arm were offered the active drug. These patients were analysed by intention-to-treat as part of the placebo group, which may have reduced the observed benefit of the sunitinib treatment. The design of this trial has been questioned (CitationJoensuu 2006). There is evidence that the discontinuation of imatinib may cause enhanced growth of previously controlled metastases and tumor flare. This may be because patients on imatinib have high concentrations of stem cell factor. Thus, for those patients discontinuing imatinib, an inferior outcome may have occurred than if they continued on imatinib treatment. As a result, a larger difference in time to progression was seen. The question that needs to be asked now is how does sunitinib compare with continued imatinib use in imatinib resistant patients?
The most common side effect of sunitinib was fatigue. There was no difference in the incidence of grade 3 or 4 toxicity between the groups, which may reflect the level of disease related symptoms. The rate of hypertension was increased in the sunitinib group and this may be due to the drugs antiangiogenic properties. There is also a small risk of hypothyroidism with long term usuage (CitationDesai et al 2006).
There has been growing interest in the use of VEGF inhibitors such as Bevacizumab (a monoclonal antibody targeted against the VEGF receptor) in advanced GIST. A large phase III trial proposed by SWOG (South Western Oncology Group) will be testing its use with or without imatinib. A phase II study is currently evaluating the use of Sorafenib, a small molecular inhibitor of Raf kinase, PDGFR and VEGFR receptor kinase, in patients previously progressing on imatinib and sunitinib. PTK787/ZK222584, a novel tyrosine kinase inhibitor, is being assessed in another phase II trial. Additional multitargeted tyrosine kinase inhibitors, including AZD2171 and AMG706 are also being investigated. The chemotherapeutic agents docetaxel and gemcitabine in combination are to be given in a proposed phase II trial. Other new agents are PKC 412 (inhibitor of protein kinase C, KIT, PDGFR, and VEGF), BMS-354825 (tyrosine kinase inhibitor of KIT, PDGFR, abl and src), oblimerson sodium (an antisense oligonucleotide inhibiting BCL-2), and CCI 779 (a rapamycin analogue inhibitor of the protein kinase mammalian target of rapamycin). There are a few phase I trials proposed or underway: IPI-504 (an inhibitor of heat shock protein 90) and perifosine and sunitinib in combination are two for the future (CitationDe Giorgi and Verweij 2005; CitationNCI 2007).
Other drugs that have been tested in GIST and found inactive include ET-743 (trabectedin) and brostallicin.
Conclusion
There has been rapid progress in the management of GIST in the 15 years since they were recognized as a distinct tumor entity. This has led to changes in the surgical and oncological approach to GIST patients. Their management by specialist teams has facilitated multicentre international research, leading to the participation of astonishingly high proportions of patients in high quality clinical trials. The introduction of imatinib mesylate has revolutionized the treatment of patients with locally advanced and metastatic GIST, leading to important gains in quality of life and survival. The process of discovery, development and evaluation of this drug acts as a new paradigm for the introduction of other biologically targeted agents in cancer.
References
- AntochGKanjaJBauerSComparison of PET, CT, and dual-modality PET/CT imaging for monitoring of imatinib (STI571) therapy in patients with gastrointestinal stromal tumorsJ Nucl Med2004453576515001674
- AntonescuCRBesmerPGuoTAcquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutationClin Cancer Res20051141829015930355
- BauerSHartmannJTDe WitMResection of residual disease in patients with metastatic gastrointestinal stromal tumors responding to treatment with imatinibInt J Cancer20051173162515900603
- BauerSYuLDemetriGDHeat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumorCancer Res20066691536116982758
- BenjaminRSBlankeCDBlayJYManagement of gastrointestinal stromal tumors in the imatinib era: selected case studiesOncologist20061192016401709
- BenjaminRSChoiHMacapinlacHAWe should desist using RECIST, at least in GISTJ Clin Oncol2007251760417470866
- BenjaminRSRankinCFletcherCPhase III dose-randomized study of imatinib mesylate (STI571) for GIST: Intergroup S0033 early resultsProc Am Soc Clin Oncol200322
- BerindoagueRTargaronaEMFeliuXLaparoscopic resection of clinically suspected gastric stromal tumorsSurg Innov200613231717227921
- BertucciFGoncalvesAMongesGAcquired resistance to imatinib and secondary KIT exon 13 mutation in gastrointestinal stromal tumourOncol Rep2006169710116786129
- BlankeCJoensuuHDemetriGLong-term follow up of advanced gastrointestinal stromal tumor (GIST) patients treated with imatinib mesylateProc Am Soc Clin Oncol GI Cancer Symposium20042
- BlayJYBonvalotSCasaliPConsensus meeting for the management of gastrointestinal stromal tumorsAnn Oncol200516Report of the GIST Consensus Conference of 20–21 March 2004, under the auspices of ESMO56678 erratum appears in Ann Oncol. 2005 Jun; 16(6):993 Note: Mac Clure, J [corrected to McClure, J15781488
- BlayJYLe CesneARay-CoquardIProspective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma GroupJ Clin Oncol20072511071317369574
- BucherPEggerJFGervazPAn audit of surgical management of gastrointestinal stromal tumours (GIST)Eur J Surg Oncol200632310416414236
- BurgerHDen BakkerMAKrosJMActivating mutations in c-KIT and PDGFRalpha are exclusively found in gastrointestinal stromal tumors and not in other tumors overexpressing these imatinib mesylate target genesCancer Biol Ther2005412704 see comment16294026
- CarneyJAShepsSGGoVLThe triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondromaN Engl J Med197729615178865533
- CarrollMOhno-JonesSTamuraSCGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteinsBlood1997904947529389713
- ChenHPruittANicolTLComplete hepatic resection of metastases from leiomyosarcoma prolongs survivalJ Gastrointest Surg1998215159834411
- ChoiHCharnsangavejCFariaSCCorrelation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteriaJ Clin Oncol2007251753917470865
- CorlessCLFletcherJAHeinrichMCBiology of gastrointestinal stromal tumorsJ Clin Oncol20042238132515365079
- D’AmatoGSteinertDMMcAuliffeJCUpdate on the biology and therapy of gastrointestinal stromal tumorsCancer Control200512445615668652
- DeGiorgiUVerweijJImatinib and gastrointestinal stromal tumors: Where do we go from here?Mol Cancer Ther2005449550115767559
- Debiec-RychterMLasotaJSarlomo-RikalaMChromosomal aberrations in malignant gastrointestinal stromal tumors: correlation with c-KIT gene mutationCancer Genet Cytogenet2001128243011454425
- Debiec-RychterMSciotRLe CesneAKIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumoursEur J Cancer200642109310316624552
- DeMatteoROwzarKMakiRACOSOG AIAGSAdjuvant imatinib mesylate increases recurrence free survival (RFS) in patients with completely resected localized primary gastrointestinal stromal tumor (GIST): North American Intergroup Phase III trial ACOSOG Z900120072007 ASCO Annual Meeting, 10079
- DeMatteoRPHeinrichMCEl-RifaiWMClinical management of gastrointestinal stromal tumors: before and after STI-571Hum Pathol2002334667712094371
- DeMatteoRPLewisJJLeungDTwo hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survivalAnn Surg200023151810636102
- DemetriGDVan OosteromATGarrettCREfficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trialLancet200636813293817046465
- DemetriGDVon MehrenMBlankeCDEfficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumorsN Engl J Med20023474728012181401
- DesaiJYassaLMarquseeEHypothyroidism after sunitinib treatment for patients with gastrointestinal stromal tumorsAnn Intern Med2006145660417088579
- DoiTNishidaTHirotaSPhase II clinical study of STI571 in Japanese (Jpn) patients (pts) with malignant gastrointestinal stromal tumors (GIST): Results of the B 1201 studyJ Clin Oncol2004224078
- DrukerBJTalpazMRestaDJEfficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemiaN Engl J Med20013441031711287972
- DrukerBJTamuraSBuchdungerEEffects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cellsNat Med1996256168616716
- ErnstSIHubbsAEPrzygodzkiRMKIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumorsLab Invest199878163369881963
- FletcherCDBermanJJCorlessCDiagnosis of gastrointestinal stromal tumors: A consensus approachHum Pathol2002334596512094370
- FletcherJACorlessCLDimitrijevicSMechanisms of resistance to imatinib mesylate (IM) in advanced gastrointestinal stromal tumor (GIST)Proc Am Soc Clin Oncol200322
- GoerresGWStuppRBarghouthGThe value of PET, CT and in-line PET/CT in patients with gastrointestinal stromal tumours: long-term outcome of treatment with imatinib mesylateEur J Nucl Med Mol Imaging2005321536215690223
- GunawanBBergmannFHoerJBiological and clinical significance of cytogenetic abnormalities in low-risk and high-risk gastrointestinal stromal tumorsHum Pathol2002333162111979372
- HeinrichMCCorlessCLDemetriGDKinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumorJ Clin Oncol2003a214342914645423
- HeinrichMCCorlessCLDuensingAPDGFRA activating mutations in gastrointestinal stromal tumorsScience2003b2997081012522257
- HeinrichMCGriffithDJDrukerBJInhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitorBlood2000969253210910906
- HirotaSIsozakiKMoriyamaYGain-of-function mutations of c-kit in human gastrointestinal stromal tumorsScience1998279577809438854
- HirotaSOhashiANishidaTGain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumorsGastroenterology2003125660712949711
- IshidaTWadaIHoriuchiHMultiple small intestinal stromal tumors with skeinoid fibers in association with neurofibromatosis 1 (von Recklinghausen’s disease)Pathol Int199646689958905879
- JoensuuHSunitinib for imatinib-resistant GISTLancet20063681303417046443
- JoensuuHFletcherCDimitrijevicSManagement of malignant gastrointestinal stromal tumoursLancet Oncol200236556412424067
- JoensuuHRobertsPJSarlomo-RikalaMEffect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumorN Engl J Med20013441052611287975
- JudsonIMaPPengBImatinib pharmacokinetics in patients with gastrointestinal stromal tumour: a retrospective population pharmacokinetic study over time. EORTC Soft Tissue and Bone Sarcoma GroupCancer Chemother Pharmacol2005553798615592836
- KasperBKallinowskiBHerrmannTTreatment of gastrointestinal stromal tumor with imatinib mesylate: a retrospective single-center experience in HeidelbergDig Dis2006242071116699280
- KindblomLGRemottiHEAldenborgFGastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of CajalAm J Pathol19981521259699588894
- KingDMThe radiology of gastrointestinal stromal tumours (GIST)Cancer Imaging20055150616361144
- KosmadakisNVisvardisEEKartsaklisPThe role of surgery in the management of gastrointestinal stromal tumors (GISTs) in the era of imatinib mesylate effectivenessSurg Oncol200514758415993051
- LasotaJJasinskiMSarlomo-RikalaMMutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomasAm J Pathol199915453609916918
- Le CesneARay-CoquardIBuiMContinuous versus interruption of imatinib (IM) in responding patients with advanced GIST after three years of treatment: A prospective randomized phase III trial of the French Sarcoma GroupJ Clin Oncol2007252007 ASCO Annual Meeting Proceedings, Part I18S June 20 Supplement10005
- LiSQO’LearyTJSobinLHAnalysis of KIT mutation and protein expression in fine needle aspirates of gastrointestinal stromal/smooth muscle tumorsActa Cytol200044981611127756
- LintonKMTaylorMBRadfordJAResponse evaluation in gastrointestinal stromal tumours treated with imatinib: misdiagnosis of disease progression on CT due to cystic change in liver metastasesBr JRadiol200679e40416861316
- LoughreyMBMitchellCMannGBGastrointestinal stromal tumour treated with neoadjuvant imatinibJ Clin Pathol2005587798115976351
- MavligitGMZukwiskiAAEllisLMGastrointestinal leiomyosarcoma metastatic to the liver. Durable tumor regression by hepatic chemoembolization infusion with cisplatin and vinblastineCancer199575208387697597
- MazurMTClarkHBGastric stromal tumors. Reappraisal of histogenesisAm J Surg Pathol19837507196625048
- MiettinenMLasotaJGastrointestinal stromal tumors – definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosisVirchows Arch200143811211213830
- MiettinenMMajidiMLasotaJPathology and diagnostic criteria of gastrointestinal stromal tumors (GISTs): a reviewEur J Cancer200238S395112528772
- MiettinenMMakhloufHSobinLHGastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-upAm J Surg Pathol2006304778916625094
- MiettinenMSobinLHLasotaJGastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-upAm J Surg Pathol200529526815613856
- MiettinenMVirolainenMMaarit SarlomoRGastrointestinal stromal tumors – value of CD34 antigen in their identification and separation from true leiomyomas and schwannomasAm J Surg Pathol199519207167530409
- NCIGIST clinical trials search [online]2007 Accessed 5 May 2007. URL: http://www.cancer.gov/search/ResultsClinicalTrials.aspx?protocolsearchid=2096061
- NeuhausSJClarkMAHayesAJSurgery for gastrointestinal stromal tumour in the post-imatinib eraANZ J Surg2005751657215777399
- NgEHPollockREMunselLMFPrognostic factors influencing survival in gastrointestinal leiomyosarcomas. Implications for surgical management and stagingAnn Surg199221568771731651
- NilssonBBummingPMeis-KindblomJMGastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era – a population-based study in western SwedenCancer2005103821915648083
- NovartisGlivec (imatinib) Summary of Product Characteristics2005
- NovitskyYWKercherKWSingRFLong-term outcomes of laparoscopic resection of gastric gastrointestinal stromal tumorsAnn Surg200624373845 discussion 745–716772777
- PengBLloydPSchranHClinical pharmacokinetics of imatinibClin Pharmacokinetics20054487994
- PlaatBEHollemaHMolenaarWMSoft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: differences in clinical outcome and expression of multidrug resistance proteinsJ Clin Oncol20001832112010986053
- RajanDKSoulenMCClarkTWSarcomas metastatic to the liver: response and survival after cisplatin, doxorubicin, mitomycin-C, Ethiodol, and polyvinyl alcohol chemoembolizationJ Vasc Interv Radiol2001121879311265882
- RankinCVon MehrenMBlankeCDose effect of imatinib (IM) in patients (pts) with metastatic GIST – Phase III Sarcoma Group Study S0033Proc Am Soc Clin Oncol200422
- RautCPPosnerMDesaiJSurgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitorsJ Clin Oncol20062423253116710031
- RubinBPSingerSTsaoCKIT activation is a ubiquitous feature of gastrointestinal stromal tumorsCancer Res20016181182111719439
- RutkowskiPNoweckiZIMichejWRisk criteria and prognostic factors for predicting recurrences after resection of primary gastrointestinal stromal tumorAnn Surg Oncol20071420182717473953
- RyuMHLeeJLChangHMPatterns of progression in gastrointestinal stromal tumor treated with imatinib mesylateJpn J Clin Oncol200636172416418188
- SakakuraCHagiwaraASogaKLong-term survival of a case with multiple liver metastases from duodenal gastrointestinal stromal tumor drastically reduced by the treatment with imatinib and hepatectomyWorld J Gastroenterol2006122793716718773
- SanbornREBlankeCDGastrointestinal stromal tumors and the evolution of targeted therapyClin Adv Hematol Oncol200536475716167051
- SavageDGAntmanKHImatinib mesylate – a new oral targeted therapyN Engl J Med20023466839311870247
- SchindlerCGArmbrustTGunawanBGastrointestinal stromal tumor (GIST) – single center experience of prolonged treatment with imatinibZ Gastroenterol2005432677315765299
- ShankarSVansonnenbergEDesaiJGastrointestinal stromal tumor: new nodule-within-a-mass pattern of recurrence after partial response to imatinib mesylateRadiology2005235892815833985
- SihtoHSarlomo-RikalaMTynninenOKIT and platelet-derived growth factor receptor alpha tyrosine kinase gene mutations and KIT amplifications in human solid tumorsJ Clin Oncol200523495715545668
- SteigenSEEideTJWasagBMutations in gastrointestinal stromal tumors – a population-based study from Northern NorwayAPMIS20071152899817504295
- StroobantsSGoeminneJSeegersM18FDG-Positron emission tomography for the early prediction of response in advanced soft tissue sarcoma treated with imatinib mesylate (Glivec)Eur J Cancer200339201220 see comment12957455
- TakahashiHSaitohKKishiHImmunohistochemical localisation of stem cell factor (SCF) with comparison of its receptor c-Kit proto-oncogene product (c-KIT) in melanocytic tumoursVirchows Arch199542728387496598
- TaniguchiMNishidaTHirotaSEffect of c-kit mutation on prognosis of gastrointestinal stromal tumorsCancer Res199959429730010485475
- Van Der ZwanSMDematteoRPGastrointestinal stromal tumor: 5 years laterCancer20051041781816136600
- Van GlabbekeMVerweijJCasaliPGInitial and late resistance to imatinib in advanced gastrointestinal stromal tumors are predicted by different prognostic factors: a European Organisation for Research and Treatment of Cancer-Italian Sarcoma Group-Australasian Gastrointestinal Trials Group studyJ Clin Oncol200523579580416110036
- Van GlabbekeMMOwzarKRankinCMETAGISTGM-AGComparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors (GIST): A meta-analyis based on 1,640 patients (pts)J Clin Oncol2007252007 ASCO Annual Meeting Proceedings, Part I18S June 20 Supplement10004
- Van OosteromATJudsonIRVerweijJSafety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I studyLancet20013581421311705489
- Van OosteromATJudsonIRVerweijJEuropean Organisation for Research and Treatment of Cancer Soft Tissue and Bone SarcomaUpdate of phase I study of imatinib (STI571) in advanced soft tissue sarcomas and gastrointestinal stromal tumors: a report of the EORTC Soft Tissue and Bone Sarcoma GroupEur J Cancer200238S83712528778
- VerweijJCasaliPGZalcbergJProgression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trialLancet2004364112734 see comment15451219
- VerweijJVan OosteromABlayJYImatinib mesylate (STI-571 Glivec, Gleevec) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II studyEur J Cancer200339200611 see comment12957454
- WardelmannEHrychykAMerkelbach-BruseSAssociation of platelet-derived growth factor receptor alpha mutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumorsJ Mol Diagn2004619720415269295
- WardelmannEMerkelbach-BruseSPaulsKPolyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylateClin Cancer Res20061217439 see comment16551858
- WardelmannEThomasNMerkelbach-BruseSAcquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutationsLancet Oncol200562495115811621
- WentPTDirnhoferSBundiMPrevalence of KIT expression in human tumorsJ Clin Oncol20042245142215542802
- WestRBCorlessCLChenXThe novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation statusAm J Pathol20041651071315215166
- WuPCLangermanARyanCWSurgical treatment of gastrointestinal stromal tumors in the imatinib (STI-571) eraSurgery20031346566514605627