Abstract
Gaucher disease is a progressive lysosomal storage disorder caused by the deficiency of glucocerebrosidase, and characterized by intralysosomal storage of glucosylceramide that leads to dysfunction in multiple organ systems. Intravenous enzyme replacement with imiglucerase is the accepted standard for treatment of symptomatic patients and has been effective in reducing many of the signs and symptoms of type I Gaucher disease in the majority of patients without serious adverse effects. An alternative therapeutic approach is substrate reduction therapy with N-butyldeoxynojirimycin (NB-DNJ) (miglustat; Zavesca®), an imino sugar that reversibly inhibits glucosylceremide synthase and reduces intracellular storage of glucosylceramide. Miglustat was recently approved in Europe and the United States for symptomatic patients with mild to moderate clinical manifestations for whom enzyme replacement therapy is not an option. This review article discusses the results of clinical studies and use of miglustat as a therapeutic agent in patients with type I Gaucher disease.
First described by Dr. Philippe Gaucher in 1882, Gaucher disease is a lysosomal storage disorder that is caused by the deficiency of glucocerebrosidase, and is characterized by the accumulation of glycosylceramide that leads to dysfunction in multiple organ systems (CitationBeutler and Grabowski 1995). Three types of Gaucher disease have been described, but, actually, these represent different degrees of severity along a spectrum. The clinical features of type I Gaucher disease, the non-neuronopathic form, are splenomegaly, which is more prominent than the hepatomegaly, anemia, thrombocytopenia, and bone lesions. Type II Gaucher disease is the most severe form, which presents with severe central nervous system involvement and is generally fatal within the first 2 years of life. Type III Gaucher disease, the subacute neuronopathic form, presents in early childhood with severe manifestations resembling type I (splenomegaly, hepatomegaly, and bone lesions) and has a more chronic course with onset of neurological disease towards the end of the first decade (CitationBeutler and Grabowski 1995).
In 1991, the advent of targeted enzyme replacement therapy (ERT) using algucerase (Ceredase®; Genzyme Corporation) followed by the introduction of imiglucerase (Cerezyme®; Genzyme Corporation) resulted in huge improvements in the treatment of patients with Gaucher disease. Imiglucerase is a modified form of glucocerebrosidase, created using recombinant DNA technology, and is given as intravenous infusions, usually every other week. Imiglucerase acts like the naturally occurring enzyme glucocerebrosidase to break down the glucosylceramide that has accumulated in Gaucher cells (CitationBarton et al 1991; CitationGrabowski et al 1998). In the majority of patients (>90%), ERT has been effective in reducing many of the signs and symptoms of type I Gaucher disease but has no or limited effect on the neurologic findings of type II and III Gaucher disease because of its inability to cross the blood – brain barrier (CitationWeinreb et al 2002). Side effects (hypersensitivity reactions and neutralizing antibodies) related to ERT have been reported in less than 15% of type I Gaucher patients (CitationBarton et al 1991; CitationGrabowski et al 1998). Life-long intravenous infusion every 2 weeks can be a burden for some patients, particularly in those with poor venous access. The high cost of ERT also precludes its use in some countries and burdens health care cost.
One of the key issues that researchers and pharmaceutical companies are still facing in the treatment of lysosomal disorders including Gaucher disease is the need to develop cheaper new treatments that will decrease the cost, provide easy administration of the drug, and improve and prevent neurological disease. Chemical chaperone therapy and gene therapy hold great promise as future treatment options for all three types of Gaucher disease.
An oral treatment using a small molecule (substrate reduction therapy) is an available form of therapy for type I Gaucher disease (CitationPlatt et al 2001). In 2002, N-butyldeoxynojirimycin (NB-DNJ) (miglustat; Zavesca®, Actelion Pharmaceuticals), a substrate reduction agent, was approved by EPAR and FDA as a treatment option for type I Gaucher disease (CitationEPAR 2002; CitationUS FDA 2003).
Unlike ERT, miglustat is a small molecule and can pass the blood – brain barrier and has the potential to be effective in treating lysosomal storage disorders with neurologic manifestations. CitationCapablo et al (2007) recently reported that combined imiglucerase and miglustat therapy improved some neurological manifestations of type III Gaucher in an adult patient. Miglustat has also been proposed for treatment of other lysosomal diseases like Niemann Pick type C, Tay-Sachs, and Fabry disease (CitationAbe et al 2000; CitationAndersson et al 2004; CitationHear et al 2007; CitationPatterson et al 2007). Miglustat therapy has been unsuccessful in halting the progress of infantile-onset Tay-Sachs disease (CitationBembi et al 2006). CitationPatterson et al (2007) showed that miglustat improved some neurologic symptoms of patients with Nieman Pick type C.
How effective is miglustat as an alternative treatment to ERT for type I Gaucher disease? The present review assesses published clinical studies of miglustat as a therapeutic agent in patients with type I Gaucher disease.
Miglustat: substrate reduction therapy
Substrate reduction therapy is an alternative approach to treat patients with Gaucher disease. NB-DNJ (miglustat) is one of the N-alkylated iminosugars extracted from plants and microorganisms, and inhibits glycosylceramide synthase, which catalyzes the transfer of glucose from UDP-glucose to ceramide to form glucosylceramide (GlcCer) (). The aim is to decrease the biosynthesis of GlcCer so that patients with significant residual enzyme activity can break down GlcCer more efficiently and thus allow clearance of GlcCer from lysosomes (Platt et al 2007).
Figure 1 Inhibition of glucosylceramide synthesis with miglustat.
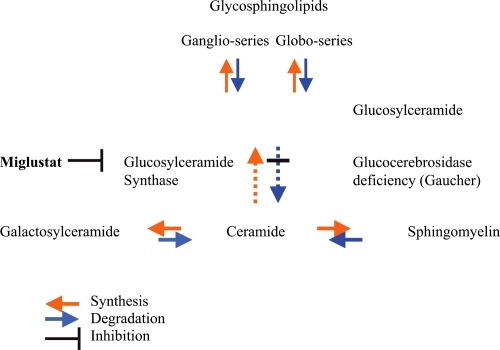
Imino sugars such as NB-DNJ can also target the protein folding and trafficking pathways of glycosidase to assist correction of lysosomal enzyme activity (CitationYam et al 2005) (chaperone mediated therapy). A partial increase in enzyme activity may be sufficient to initiate the metabolic breakdown of glycosphyngolipids (GSL) and decrease the GSL storage in lysosomes. The efficacy of miglustat in Gaucher disease type I probably results from both a decrease in the biosynthesis of GlcCer and an increase in the activity of glucocerebrosidase.
Discovery and development of miglustat
In the 1990s Norman Radin hypothesized that it would be possible to reduce the concentration of glycosphingolipids substrates in the lysosomes by inhibiting their biosynthesis (CitationRadin 1996, Citation2000; CitationPlatt et al 2001). Radin and colleagues synthesized compounds such as 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) to inhibit GlcCer synthase, which is a key enzyme in GlcCer biosynthesis. More recently, several homologues of PDMP have been characterized (CitationAbe et al 1992, Citation2001). However, PDMP and related compounds can inhibit cell growth, and cause undesirable cell death (CitationBarbour et al 1992; CitationAbe et al 1995). In light of PDMP’s undesirable effects, compounds related to PDMP that do not exhibit these effects were chosen and used in several studies (CitationAbe et al 1992). Several animal studies also showed that depletion of GSL could be lethal or lead to neurological disturbance, and substrate reduction therapy should be aimed to reduce biosynthesis of GSL to a degree that can be tolerated by cells (CitationRadin 1996).
NB-DNJ, a compound originally developed as an alpha-glucosidase inhibitor and structurally unrelated to PDMP, is an N-alkylated imino sugar and is an inhibitor of GlcCer synthase. NB-DNJ was initially developed for use as an antiviral agent, because most enveloped viruses use the same pathway for glycoprotein synthesis in infected cells. Using NB-DNJ to alter the glycosylation of envelope glycoproteins could decrease the infectivity in certain viral diseases such as HIV. However, clinical trials on HIV patients were unsuccessful because the antiviral concentrations of NB-DNJ required could not be achieved in humans without a high incidence of adverse effects (CitationTierney et al 1995). These clinical trials provided valuable safety information that helped to guide subsequent studies for SRT in Gaucher disease.
Results of clinical trials of miglustat in patients with type-I Gaucher disease
The current clinical experience with miglustat on type I Gaucher disease is based on the clinical studies undertaken since 2000.
In April 2000, CitationCox et al (2000) reported the results of the first clinical trial, a 1-year open label uncontrolled study. Twenty-eight adults with type I Gaucher disease were treated with 100 mg oral miglustat 3 times a day for 12 months (CitationCox et al 2000). Twenty-two patients completed the study. Spleen and liver volumes were lowered by 19% (p < 0.001) and 12% (p < 0.001), respectively, over 12 months (). Hematological markers such as hemoglobin and platelets improved slightly. Chitotriosidase levels fell by 16.4% (p < 0.001) over 12 months (). The most frequent adverse effect was diarrhea, which occurred in 79% of patients. Two patients were withdrawn from the trial due to peripheral neuropathy, which was reported as an adverse effect of miglustat. Based on these results, the EPAR and US FDA approved miglustat (Zavesca®) for adult patients with mild-moderate type I Gaucher disease for whom enzyme replacement therapy is not an option.
Table 1 Changes in clinical parameters in response to miglustat treatment in clinical studies
Eighteen patients who completed the initial 12-month clinical trial were enrolled in an extension study; 14 completed the 24-month extension study. Elstein et al (2004) reported the results of the extension study in 2004. Liver and spleen volumes diminished by 18% and 30%, respectively (). Hemoglobin and platelets counts increased. No new cases of peripheral neuropathy were reported. Diarrhea and weight loss decreased in magnitude and prevalence during the second and third years. This trial suggests that adverse effects diminished when the period of treatment was extended.
In 2002, CitationHeitner et al (2002) reported the result of a 6-month open label uncontrolled study in which 18 adult patients with type I Gaucher received low dose miglustat (50 mg 3 times a day). Seventeen patients completed the 6-month study which reported the significant mean percentage changes in liver (–5.9%) and spleen (–4.5%) volumes and in chitotriosidase levels (–4.6%) (). The lower dose of miglustat (50 mg 3 times a day) did not produce the same beneficial effects delivered by the higher dose (100 mg 3 times a day) while patients suffered similar adverse effects (diarrhea and weight loss)
Sixteen patients were enrolled in a 6-month extension to this study. After a total of 12 months’ treatment, spleen and liver volumes, and chitotriosidase levels decreased by only 10%, 6%, and 15%, respectively (). There were no significant changes in hemoglobin levels or platelets counts.
The third study was undertaken by CitationElstein et al (2002). It was an open label, controlled, 6-month trial to assess the efficacy of miglustat as a maintenance therapy. Thirty-six patients who had received ERT for a minimum 2 years were enrolled in the study and randomized into one of three groups: ERT monotherapy, miglustat monotherapy (100 mg 3 times a day) and ERT and miglustat combined therapy. Thirty-three patients completed the study. No significant differences were noted among the three groups in terms of changes in liver and spleen volumes and hemoglobin levels after 6 months of treatment (). The miglustat monotherapy group had a 33% increase in mean chitotriosidase level compared with both ERT and combined treatment groups, both of which recorded desired stable or decreased levels (). The miglustat monotherapy group showed a mean absolute decrease in platelet count (–21.6 × 109/L) while the other two groups showed some absolute increase in platelet counts (). Twenty-nine patients were enrolled in a 6-month extension to this study. ERT was discontinued and all patients received only miglustat 100 mg 3 times a day. There were no significant changes in liver, spleen volume, or hemoglobin level. Mean platelet counts decreased and chitotriosidase level increased. The authors suggested that miglustat monotherapy might not be sufficient to maintain the same control of disease in all patients (CitationElstein et al 2002; CitationZimran and Elstein 2003).
In 2005, CitationPastores et al (2005) undertook a 2-year open label study and enrolled 10 adults who received 100 mg miglustat 3 times a day. Seven out of 10 patients completed the 2-year study. Chitotriosidase levels decreased by 25.4% over 2 years (). At 18 months, spleen volume decreased by 24.3%, liver volume by 15.1%. Hematological parameters such as the platelet count improved over 2-year period. This study confirmed the findings of CitationElstein et al (2004) that adverse effects such as diarrhea, abdominal pain, distension, weight loss tremors, and paresthesia decrease over the course of the trial. It also produced similar beneficial effects to those reported by CitationCox et al (2000).
In 2007, CitationElstein et al (2007) reported the results of a 2-year open label trial in which 36 clinically stable patients on enzyme replacement therapy were enrolled and the effects of combined enzyme and miglustat therapy compared with miglustat monotherapy. This was an extension of the previously reported 6-month clinical trial, CitationElstein et al (2002). Miglustat was well tolerated by both groups, and the safety profile was consistent with the previous trials. They suggested that miglustat could be an effective maintenance therapy in stabilized patients with type 1 Gaucher.
Departing from previous clinical trials with their focus on hematologic markers, and liver and spleen volumes, CitationPastores et al (2007) evaluated the effects of miglustat on bone manifestations and bone mineral density. They recruited 72 patients with type I Gaucher disease, who had previously participated in 3 multinational, open-label clinical trials of miglustat. The data were collected prospectively over an observation period of 2 years. At study entry, osteoporosis (68%) and bone pain (63%) were the most common bone-related findings. They showed that 83% of the patients reported no bone pain and BMD Z-scores were improved from baseline at both the lumbar spine and femoral neck at each time point (months 6, 12, and 24). The authors suggested that the beneficial effect of miglustat on bone symptoms might be explained by its wide tissue distribution and by a direct effect on bone cells.
In 2007, CitationGiraldo et al (2006) reported the results of an open label 12-month study. Twenty-five patients (10 therapy naïve, 15 previously treated with ERT) with mild to moderate Gaucher disease type 1 were treated with miglustat. At 6 months, the previously untreated group showed a mean increase in hemoglobin of 0.77 g/dL, platelet count of 41.5 × 109/L or remained stable, and a mean decrease in their chitotriosidase levels of 38.2% (). Their results were similar to those treated with ERT only. They concluded that the efficacy of miglustat therapy after 6 months was comparable to that of ERT.
Adverse reactions/safety/precautions
Gastrointestinal symptoms
Diarrhea and weight loss were common in the clinical studies (13, 15, 18, 20, 23, 24). Diarrhea is probably due to inhibition of disaccharidase activity by miglustat in the intestine that results in osmotic diarrhea. Diarrhea was noted to improve over time whether or not the dose of miglustat was decreased. Some patients were treated with anti-diarrheal medications such as loperamide. The cause of weight loss is unknown. The incidence of weight loss was more common in the first 12 months of treatment and most of patients recovered from the weight loss by 24 months of treatment.
Neurological symptoms
Tremor was reported in about 30% of patients (CitationCox et al 2003). It was more common within the first month of treatment and improved between 1 to 3 months during treatment and resolved spontaneously, or after reduction of dose or discontinuation of miglustat. Parasthesias and burning sensation due to peripheral neuropathy were reported in 15%–20% of patients, and resolved after the discontinuation of the medication or dose reduction (CitationCox et al 2000; CitationPastores et al 2005). Cognitive dysfunction was reported in 5 patients, but no causal relationship was found (CitationElstein et al 2005). Baseline and repeat neurological evaluations at about 6-month intervals are recommended for all patients who will be treated with miglustat.
Reproduction toxicity/pregnancy/breast feeding
Animal studies showed that miglustat adversely affects spermatogenesis and sperm parameters and reduces fertility (CitationWalden et al 2006). In contrast to the observations in mice, the recently published study showed that miglustat has no apparent effect on spermatogenesis in man (CitationAmory et al 2007).
Reproductive effects in females could be increased post-implantation loss and early embryonic death, and dystocia. It is not clear if these toxic effects will be reversible after long-term treatment. Animal studies showed that miglustat could cause fetal harm when administered to a pregnant animal. Miglustat is contraindicated in women who are or may become pregnant. Male patients are recommended to use a contraceptive method while taking miglustat, and for 3 months after its discontinuation.
It is unknown if miglustat is excreted in human milk. Because of potential serious adverse reactions of miglustat in nursing infants, it should not be used in nursing mothers.
Conclusion
Miglustat is an oral medication that inhibits glucosylceremide synthase (substrate reduction therapy) and may partially increase glucocerebrosidase enzyme activity (chaperone mediated therapy). Studies showed that miglustat did improve the biochemical outcomes of patients with mild to moderate type 1 Gaucher during clinical trials ranging from 6 to 36 months, but there is insufficient evidence to show if treatment will maintain both clinical and biochemical effects over a long-term period, or on its efficiency in treating the bones and preventing serious aspects such as pulmonary hypertension.
Bone manifestations are considered among the most painful and debilitating components of type I Gaucher. ERT can improve the bone symptoms but it takes much longer to achieve satisfactory results (CitationCharrow et al 2007). However, recently published data shows that miglustat may well improve bone density and prevent bone crisis (CitationPastores et al 2007). Long-term studies are needed to support this finding.
While some splenectomized patients participated in the clinical trials, no studies have yet evaluated the impact of splenectomy on the effectiveness of miglustat. Splenectomy is thought to impact adversely the severity of bone disease.
Currently, miglustat has limited indication for use in only adults with mild, moderate type I Gaucher disease in the USA and Europe, due to lack of information about the beneficial effects on severe cases (CitationCox et al 2003). Potential advantages of miglustat should be carefully weighed in individual patients, and the treatment decision should be made by physicians with expertise in Gaucher disease. Patients with some residual enzyme activity and mild symptoms of type I Gaucher disease can be treated with miglustat if ERT is not an option. It is essential to monitor the disease progression by checking levels of hemoglobin, platelets, chitotriosidase every 3–6 months and liver and spleen volumes and bone studies including dual energy X-ray absorptiometry (DXA) scan and bone MRI once a year.
The adverse effects such as weight loss, diarrhea, and tremors are dose related and reversible, but close neurologic follow up is recommended due to possible complications such as peripheral neuropathy and cognitive decline. None of the authors discussed how much the adverse effects interfered with the patients’ daily activities of living.
The potential long-term neurologic adverse effects of miglustat are difficult to assess from available studies. There seems to be a direct association between using miglustat and developing tremors. The cause of peripheral neuropathy was attributed to vitamin B12 deficiency in some patients but the use of miglustat was not totally excluded as a cause of peripheral neuropathy. Patients who were on miglustat did perform significantly poorer than patients who were not on miglustat on tests measuring verbal learning and memory on psychologist administrated tests, but this difference between two groups was not observed in computer assessed tests (CitationElstein et al 2005). It is not clear if patients on miglustat are at greater risk for verbal and memory difficulties.
None of the studies addresses cardiac and pulmonary status especially regarding pulmonary hypertension, and quality of life.
Additional controlled longitudinal studies are needed to be undertaken with a larger number of patients not only to determine the biochemical but also clinical long term outcomes of patients with type I Gaucher disease on miglustat therapy.
Disclosures
The author has no conflicts of interest to disclose.
Acknowledgements
The author would like to thank Dr. Paige Kaplan for critical reading of the manuscript.
References
- AbeAInokuchiJJimboMImproved inhibitors of glucosylceramide synthaseJ Biochem199211119161533217
- AbeAGregorySLeeLReduction of globotriaosylceramide in Fabry disease mice by substrate deprivationJ Clin Invest20001051563710841515
- AbeARadinNShaymanJAStructural and stereochemical studies of potent inhibitors of glucosylceramide synthase and tumor cell growthJ Lipid Res199536611217775872
- AbeAWildSLeeWAgents for the treatment of glycosphingolipid storage disordersCurr Drug Metab20012331811513334
- AmoryJKMullerCHPageSTMiglustat has no apparent effect on spermatogenesis in normal menHum Reprod200722702717067996
- AnderssonUSmithDJeyakumarMImproved outcome of N-butyl deoxy- galactonojirimycin-mediated substrate reduction therapy in a mouse model of Sandhoff diseaseNeurobiol Dis2004165061515262262
- BarbourSEdidinMFeldingHarbermannBGlycolipid depletion using a ceramide analogue (PDMP) alters growth, adhesion, and membrane lipid organization in human A431 cellsJ Cell Physiol199215061091537889
- BartonNWBradyRODambrosiaJMReplacement therapy for inherited enzyme deficiency: macrophage-targeted glucocerebrosidase for Gaucher’s diseaseN Engl J Med19913241464702023606
- BembiBMarchettiFGuerciVISubstrate reduction therapy in the infantile form of Tay-Sachs diseaseNeurology2006662788016434676
- BeutlerEGrabowskiGAScriverCRBeaudetALSlyWSChildsBKinzlerKWVogelsteinBGaucher’s diseaseThe Metabolic and Molecular Bases of Inherited Disease19958New YorkMcGraw-Hill264170
- CapabloJLFrancoRde CabezonASNeurologic improvement in a type 3 Gaucher disease patient treated with imiglucerase/miglustat combinationEpilepsia2007481406817433057
- CharrowJDulisseBGrabowskiGAThe effect of enzyme replacement therapy on bone crisis and bone pain in patients with type1 Gaucher diseaseClin Genet2007712051117309642
- CoxTLachmannRHollakCNovel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesisLancet20003551481510801168
- CoxTMAertsJMAndriaGAdvisory council to the European working group on Gaucher disease The role of the iminosugar N-butyldeoxynojirimycin (Miglustat) in the management of type I (non-neuronopathic) Gaucher disease: a position statementJ Inherit Metab Dis2003265132614605497
- ElsteinDDwekAAttiasDA randomized study of OGT 918 as an oral therapy inpatients previously treated with enzyme replacement for type 1 Gaucher disease2002Presented at the Fifth Workshop of the European Working Group on Gaucher DiseasePrague
- ElsteinDDwekAAttiasDOral maintenance clinical trial with Miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacementBlood2007110229630117609429
- ElsteinDGuedaliaJDonigerGMComputerized cognitive testing in patients with type I Gaucher disease: effects of enzyme replacement and substrate reductionGenet Med200571243015714080
- ElsteinDHollakCAertsJMSustained therapeutic effects of oral Miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher diseaseJ Inherit Metab Dis2004277576615505381
- [EPAR] European Public Assessment ReportZavesca [Miglustat]2002
- Committee for Proprietary Medicinal Products EPARCPMP/3795/02. London, England: The European Agency for the Evaluation of Medicinal Products [online] URL: http://www.emea.eu.int/humandocs/Humans/EPAR/zavesca/zavesca.htm
- GiraldoPLatrePPilareAShort-term effect of miglustat in every day clinical use in treatment-naïve or previously treated patients with type 1 Gaucher’s diseaseHeamatologica2006917036
- GrabowskiGALeslieNWenstrupREnzyme therapy for Gaucher disease: the first 5 yearsBlood Rev199812115339661800
- HeareTAlpNjPierstmanDASevere endothelial dysfunction in the aorta of a mouse model of Fabry disease; partial prevention by N-butyldeoxynojirimycin treatmentJ Inherit Metab Dis200730798717189993
- HeitnerRElsteinDAertsJLow-dose N-butyldeoxy- nojirimycin (OGT 918) for type I Gaucher diseaseBlood Cells Mol Dis2002281273312064906
- PastoresGMBarnettNLKolodnyEHAn open-label, noncomparative study ofmiglustat in type I Gaucher disease: Efficacy and tolerability over 24 months of treatmentClin Ther20052712152716199246
- PastoresGMElsteinDHrebicekMEffect of miglustat on bone disease in adults with type 1 Gaucher disease: a pooled analysis of three multinational, open-label studiesClin Ther20072916455417919546
- PattersonMCVecchioDPradyHMiglustat for treatment of Niemann-Pick C disease: a randomized controlled studyLancet Neurol200767657217689147
- PlattFMJeyakumarMAnderssonUInhibition of substrate synthesis as a strategy for glycolipid lysosomal storage disease therapyJ Inherit Metab Dis2001242759011405346
- RadinNSTreatment of Gaucher disease with enzyme inhibitorGlyco J1996131537
- RadinNSTreating glucosphingolipid disorders by chemotherapy: Use of approved drugs and over-the-counter remediesJ Inherit Metab Dis2000237677711196103
- TierneyMPottageJKesslerHThe tolerability and pharmacokinetics of N-butyl-deoxynoirimycin in patients with advanced HIV disease (ACTG100). The AIDS Clinical Trials Group (ACTG) of the National Institute of Allergy and Infectious DiseasesAcquir Immune Defic Syndr Hum Retrovirol19951054953
- US FDA Medical Review2003 [online] URL: http://www.fda.gov/cder/foi/nda/2003/21-348_Zavesca.htm
- WaldenCMButtersTDDwekRALong-term non-hormonal male contraception in mice using N-butyldeoxynojirimycinHum Reprod20062113091516396932
- WeinrebNJCharrowJAnderssonHCEffectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher RegistryAm J Med20021131121912133749
- YamGHZuberCRothJA synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorderFASEB J200519121815629890
- ZimranAElsteinDGaucher disease and the clinical experience with substrate reduction therapyPhilos Trans R Soc Lond B Biol Sci2003358961612803930