Abstract
The urotensin II receptor, bound by the ligand urotensin II, generates second messengers, ie, inositol triphosphate and diacylglycerol, which stimulate the subsequent release of calcium (Ca2+) in vascular smooth muscle cells. Ca2+ influx leads to the activation of Ca2+-dependent kinases (CaMK) via calmodulin binding, resulting in cellular proliferation. We hypothesize that urotensin II signaling in pulmonary arterial vascular smooth muscle cells (Pac1) and primary aortic vascular smooth muscle cells (PAVSMC) results in phosphorylation of Ca2+/calmodulin-dependent kinases leading to cellular proliferation. Exposure of Pac1 cultures to urotensin II increased intracellular Ca2+, subsequently activating Ca2+/calmodulin-dependent kinase kinase (CaMKK), and Ca2+/calmodulin-dependent kinase Type I (CaMKI), extracellular signal-regulated kinase (ERK 1/2), and protein kinase D. Treatment of Pac1 and PAVSMC with urotensin II increased proliferation as measured by 3H-thymidine uptake. The urotensin II-induced increase in 3H-thymidine incorporation was inhibited by a CaMKK inhibitor. Taken together, our results demonstrate that urotensin II stimulation of smooth muscle cells leads to a Ca2+/calmodulin-dependent kinase-mediated increase in cellular proliferation.
Introduction
Hypertrophy, inflammation, and proliferation of vascular cells are major contributors to diseases such as atherosclerosis, arteriosclerosis, and hypertension.Citation1 The combined effects of these major contributors are increases in cell size, migration of immune cells, and abnormal cell growth in affected regions of the vessel.Citation2 Changes in vessel structure due to vascular remodeling result in narrowing of the vessel wall and arterial stiffness.Citation2 Contraction and relaxation of vascular smooth muscle cells are regulated by biologically active mediators which are synthesized and secreted to modulate vascular tone.Citation1 Many of these same mediators also play a pathologic role, such as urotensin II, which can induce abnormal cellular proliferation during disease-related vascular remodeling.Citation1
Urotensin II is similar to somatostatin in both structure and function. Urotensin II is an undecapeptide cleaved from a precursor molecule that stimulates potent vasoconstriction and vascular smooth muscle cell proliferation.Citation3,Citation4 Under nonpathologic conditions, urotensin II influences vascular smooth muscle contraction.Citation5 However, under pathologic conditions, urotensin II promotes cellular migration, and modulates large blood vessels, as shown in studies conducted in human aortic smooth muscle cells.Citation6
Urotensin II is recognized by the orphan G-protein coupled GPR-14, now identified as the urotensin II receptor, resulting in generation of the second messengers, inositol triphosphate and diacylglycerol.Citation7,Citation8 These second messengers trigger the release of Ca2+ from the sarcoplasmic reticulum.Citation9 The urotensin II receptor is expressed in many tissues, including vascular smooth muscle, although the precise mechanisms activated downstream of the urotensin II receptor in vascular smooth muscle cells are largely unknown.Citation10 Studies of other Gq-coupled receptors have shown that stimulation induces intracellular Ca2+ influx and binding of Ca2+ to calmodulin.Citation11,Citation12 Activated calmodulin subsequently binds to and stimulates calmodulin-dependent kinases (CaMK), such as Ca2+/calmodulin-dependent kinase kinase (CaMKK).Citation13 Activation of CaMK members can lead to Ca2+-dependent activation of other protein kinases, such as extracellular signal-regulated kinase (ERK) and protein kinase D (PKD).Citation14–Citation16
ERK phosphorylation is required for proliferation of various cell types and cell lines.Citation17,Citation18 Vascular remodeling, hypertrophy, and proliferative responses are believed to be the result of urotensin II receptor overstimulation.Citation19,Citation20 Studies in thoracic aortic cells demonstrated that urotensin II receptor signaling stimulates the phosphorylation of ERK.Citation5,Citation21 In addition to ERK activation, intracellular Ca2+ influx can also modulate the activity of PKD.Citation14 Vasoactive agents, such as endothelin-1, that bind Gq-coupled receptors, have been demonstrated to mediating PKD phosphorylation in various cell types.Citation22 Here, we show that urotensin II induces intracellular Ca2+ release which stimulates CaMKI phosphorylation in Pac1. Moreover, we demonstrate that urotensin II receptor stimulation leads to CaMK-dependent phosphorylation of ERK and PKD. We go on to reveal that the acute application of urotensin II results in cellular proliferation, which can be blocked by inhibition of CaMKK. Consistent with our hypothesis, we have found that urotensin II-induced CaMKI, ERK, and PKD phosphorylation are also blocked by inhibition of CaMKK in vascular smooth muscle cells. These observations potentially indicate that urotensin II-induced signaling triggers proliferation and may contribute to hypertrophic pathologic conditions.
Materials and methods
Cell culture
Rat pulmonary arterial smooth muscle cells (Pac1) were cultured according to the method described by Rothman A et al.Citation11 In brief, rat Pac1 were cultured in medium 199 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum with gentamicin (Fisher Scientific, Pittsburgh, PA). The cells were carried only through passages 3–15.
Primary aortic smooth muscle cells
Rat aortas were isolated from three-month-old Sprague-Dawley rats according to an established protocol.Citation23 The aortas were incubated for five minutes in 10% fetal bovine serum and 199 medium, then incubated at 37°C for 30 minutes in Hanks Balanced Salt Solution (Invitrogen-GIBCO, Carlsbad, CA) with 70 U/mL of collagenase (Millipore-Worthington Biochemical Company, Billerica, MA). Adventitias were stripped from the aortic tissue with watchmaker forceps and discarded. Aortas were digested in Hanks Balanced Salt Solution with collagenase 70 U/mL and elastase 40 U/mL (Sigma Aldrich, St Louis, MO). Aortas were gently agitated at 37°C for 90 minutes. The aortic solution was passed through a strainer with a 70 μm pore size, and centrifuged at 900 rpm for five minutes. Cellular suspensions were dispersed by pipetting the suspensions up and down. Cells were then plated in six-well plates containing 10% fetal bovine serum in 199 medium.
Calcium imaging
Pac1 (1 × 105 cells/mL) were grown on coverslips at 70%–80% confluency, and then bathed in Hanks Balanced Salt Solution (containing NaCl 0.137 M, KCl 5.4 mM, Na2PO4 0.25 mM, KH2PO4 0.44 mM, MgSO4 1.0 mM, NaHCO3 4.2 mM) with and without Ca2+ for measuring intracellular Ca2+. Detection of intracellular Ca2+ was quantified using a ratiometric technique recognized by Fura-2-AM (Invitrogen) involving excitation at 340 nm and 380 nm with emission at 510 nm according to Prasanne et al.Citation24 A Nikon Diaphot microscope using Metafluor software (Universal Imaging, West Chester, PA) was used to measure intracellular Ca2+. Concentration (nM) of intracellular Ca2+ was calculated using the Grynkeiwicz equation. Ethylenediamine tetraacetic acid 0.5 μM, a membrane-impermeable chemical chelating agent that binds Ca2+, was used to reverse the elevated levels of Ca2+. BAPTA-AM (1, 2-bis(2-aminophenoxy)- ethane-N,N,N′, N′-tetra-acetic acid) 10 μM (Invitrogen) was also used as a Ca2+ chemical chelating agent. BAPTA-AM, a membrane-permeable compound, also reverses the elevated levels of intracellular Ca2+.
Immunostaining
Pac1 and PAVSMC were cultured (1 × 105 cells/mL) on glass coverslips and washed twice with phosphate-buffered solution and fixed for 15 minutes with 4% paraformaldehyde at room temperature. Cells were blocked for one hour in blocking solution (3.0% bovine serum albumin in phosphate-buffered solution), and then incubated for one hour in primary antibody smooth muscle cell-specific antimyosin IgG (Biomedical Technologies Incorporated, Stoughton, MA) diluted 1:200 or anti-GPR14R (antibody for the urotensin II receptor, Alpha Diagnostic International Inc., Woodlake Center, TX) antibody 1:200 with blocking solution. The secondary fluorescent labeling was incubated with cells using 1:500 Alexa 488 goat/antirabbit (Molecular Probes- Invitrogen, Eugene, OR) in blocking solution. The cells were washed three times with phosphate-buffered solution. Coverslips were mounted on slides with Pro-Long Gold Anti-Fade and DAPI (Invitrogen). A DP70 Olympus digital camera and AX70 fluorescent microscope was used to visualize stained cells (40 × objective).
3H-thymidine incorporation assay
Pac1 and PAVSMC were seeded (8 × 104 cells/mL) in RPMI (Invitrogen) medium containing 10% fetal bovine serum. After 70% confluency, cells were washed with phosphate-buffered solution and serum starved in serum-free media to induce G1 arrest. Cells were serum-starved for 24 and 48 hours and were incubated in serum-free RPMI media or isotonic artificial CSF (ACSF, 142 mM NaCl, 5 mM KCl, 10 mM glucose, 1.3 mM Mg2+ 10 mM HEPES, and in the presence or absence of 3.1 mM Ca2+ containing 1 μCi/mL of 3H-thymidine, specific activity 48.0 Ci/mmol (GE-Health-care-Amersham Pharmacia, Piscataway, NJ) for four hours in the CO2 incubator at 37°C in the presence and absence of urotensin II 100 nM (Sigma Aldrich) and STO609 250 nM (Sigma Aldrich). After the four-hour incubation period, cells were washed with phosphate-buffered solution. Cells were precipitated with 15% trichloroacetic acid and incubated overnight at 4°C. Cells were then lysed with 1 Normal NaOH and were incubated for 30 minutes at room temperature. Cell lysates were transferred to tubes containing scintillation fluid for analysis using a Beckman 1539 scintillation counter. Proliferation was measured by incorporation of 3H-thymidine.
Western blot analysis
Pac1 were seeded at a density of 4 × 104 cells/mL and cultured until cells were 80% confluent in 100 mm3 dishes. The day prior to treatment, the cells were serum-reduced in 0.1% fetal bovine serum and 199 medium with gentamicin (Invitrogen) or 0.1% fetal bovine serum in ACSF. Twenty-four hours after serum reduction, cells were treated with 100 nM rat urotensin II (Sigma Aldrich), and 250 nM STO609, a synthetic 7-oxo-7H-benzimidazo [2,1-a]benz[de] isoquinoline-3-carboxylic acid-acetic acid peptide (Sigma Aldrich). All treatment conditions were carefully selected based on preliminary dose- and time-dependent studies. Experiments with the use of inhibitors were conducted under optimal conditions, and the concentrations were based on previous studies as referenced earlier. Inhibitors were incubated 30 minutes prior to urotensin II treatments. Dose-response and time-course investigations were conducted with urotensin II treatment. Cells were lysed using RIPA buffer (50 mM TrisHCl, 150 mM NaCl, 2 mM ethylenediamine tetraacetic acid, 1% NP-40, and 0.1% sodium dodecyl sulfate pH 7.4) and a protease inhibitor cocktail (Sigma Aldrich) containing phosphate inhibitors. Protein concentration was measured by the bicinchoninic acid method. Equal amounts of protein (20–30 μg) were loaded and electrophoresed in 10% sodium dodecyl sulfate-acrylamide gel. Proteins were transferred to PVDF membrane (Millipore, Billerica, MA). The membrane was incubated with respective phosphospecific primary antibodies at 4°C overnight and with corresponding horseradish peroxidase-conjugated secondary antibody at room temperature for one hour. After washing with 1 × phosphate-buffered solution three times at room temperature, the phosphorylation state of the proteins were detected by chemiluminescence (GE Healthcare Amersham). Phosphospecific antibodies used for the Western blots were p-ERK1/2 (Cell Signaling-9106S), p-PKD (Cell Signaling, Boston, MA) and pCaMKI (threonine 178, T Soderling Vollum Institute, Oregon Health Science University, Portland, OR). Loading control was determined by stripping the blot and reprobing with anti-beta actin antibody (Santa Cruz Biotech, Santa Cruz, CA). Expression of the urotensin II receptor was measured using anti-GPR14R (antibody for the urotensin II, Alpha Diagnostic International Inc., San Antonios, TX) antibody.
Adenoviral infection
Pac1 were seeded into six-well plates. Cells were grown in 199 medium containing 10% serum with penicillin and streptomycin. Cells were allowed to reach near confluency and were infected with the adenovirus Ad- urotensin II and Ad-GO-GFP. The adenovirus-containing (Ad-urotensin II-GFP) and the control adenovirus (Ad-GO-GFP) were both obtained from Walter Thomas (Baker Heart Research Institute, Melbourne, Australia).Citation25 Plaque forming units (PFU) in HEK293 cells were measured to determine the viral titer, which was approximately 1.5 × 107 PFU/mL. The efficiency of infection for viral load was determined by observation of GFP fluorescence under the microscope. Virus-containing media was washed from the cells, and 10% serum-containing media was incubated for 18 hours for expression of virus.
Statistical analysis
Western blot densitometry values were normalized and evaluated relative to control. Ca2+ imaging data were presented relative to control in terms of percentage change. Densitometry, Ca2+, and proliferation data were subjected to one-way analysis of variance (ANOVA, GraphPad-Prism, San Diego, CA) and the Newman-Keuls multiple range test was used for pairwise comparisons of the means. Statistical significance was indicated by P ≤ 0.05.
Results
Urotensin II receptor is expressed in Pac1 and PAVSMC
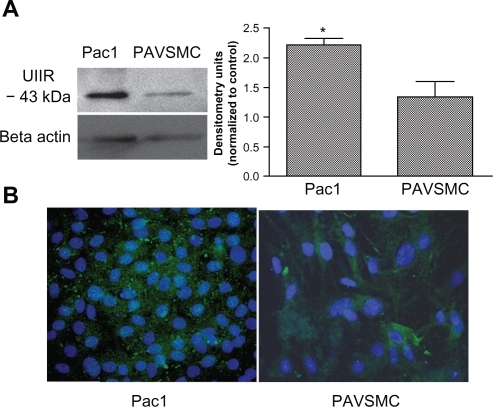
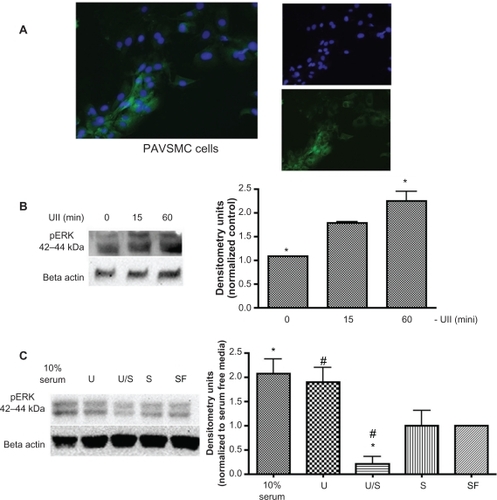
To investigate urotensin II-induced signaling, we used both Pac1 and PAVSMC to verify the expression of the urotensin II receptor. Pac1 are rapidly dividing cells,Citation11 and therefore may not accurately represent the characteristics observed in normal vascular smooth muscle cells. Thus, we incorporated the two cell types to identify the urotensin II receptor signaling mechanisms. Previous studies have identified urotensin II receptor expression in PAVSMC, but urotensin II receptor expression in Pac1 lines have not been examined.Citation10 Western blot analysis confirms that Pac1 have higher levels of the urotensin II receptor compared with PAVSMC (). Immunocytostaining of both cell types with anti-urotensin II receptor antibody indicated the presence of the urotensin II receptor in both Pac1 and PAVSMC ().
Figure 1 Urotensin II is expressed in Pac1 and PAVSMC. A) A representative immunoblot using anti-GPR14 (antibody to urotensin II) demonstrates that urotensin II is expressed in both Pac1 and PAVSMC. Densitometry analysis was conducted on Western blots (n = 3, *P ≤ 0.05). B) Merged image urotensin II immunocytochemistry in Pac1 (image 20 × objective) and PAVSMC (image 40 × objective) cells is highlighted by the green stain using anti-GPR14 antibody (antibody to urotensin II) and the nuclei staining is identified with DAPI, which is shown in blue.
Urotensin II induces mobilization of Ca2+ in Pac1 and PAVSMC
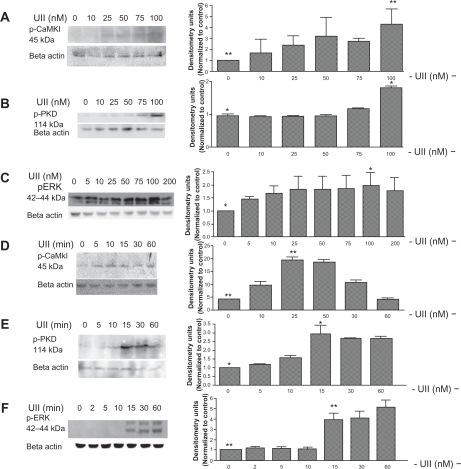
It is well established that binding of urotensin II to the urotensin II receptor leads to mobilization of intracellular Ca2+.Citation26 We measured intracellular Ca2+ transients in urotensin II-treated Pac1 and PAVSMC using Fura-2-AM dye and digital imaging microfluorometry. and demonstrate that urotensin II increases Ca2+ to 800 nM over 30 seconds in Pac1 () and 600 nM in PAVSMC (). Pac1 and PAVSMC () are representative images before and after urotensin II treatment. The Fura-2-AM scale, as shown in , illustrates the level of intracellular Ca2+, demonstrating that urotensin II induces an increase in Ca2+. In we show that urotensin II-induced Ca2+ release does not depend on extracellular Ca2+. The increase in cytoplasmic Ca2+ in response to urotensin II is the same for cells cultured in Hanks Balanced Salt Solution media with or without Ca2+. Ethylenediamine tetraacetic acid (membrane-impermeable) and BAPTA-AM (membrane-permeable) served as our controls in . Taken as a whole, the results in suggest that 100 nM urotensin II causes a release of Ca2+ from internal stores in Pac1 and PAVSMC. Urotensin II induces phosphorylation of CaMKI, ERK, and PKD in Pac1. Western blot analysis was used to assess phosphorylation of CaMKI, PKD, and ERK in response to urotensin II () in Pac1. There was a dose-dependent () and time-dependent () increase in CaMKI phosphorylation (). PKD phosphorylation (), and ERK phosphorylation () in Pac1 treated with urotensin II. Using 100 nM urotensin II, CaMKI phosphorylation was maximal at 10 minutes, and preceded PKD phosphorylation, which peaked at 15 minutes. Phosphorylation of ERK occurred later and was still increasing at 60 minutes.
Figure 2 Urotensin II induces elevated levels of intracellular Ca2+ in Pac1 and PAVSMC. Modulation of intracellular Ca2+ in response to urotensin II treatment in Pac1 (A) and PAVSMC (B) preloaded with Fura-2 and treated with urotensin II. Cells were captured at the 340/380 fluorescence ratio of Fura-2 and cells were monitored to detect changes in Ca2+. Urotensin II concentrations were chosen according to our previous dose response studies. Data are from an average of Ca2+ response in a population of Pac1(A) and PAVSMC (B) stimulated with urotensin II (n = 14), and a representation of three independent experiments of urotensin II treatment in Pac1 and PAVSMC were graphically plotted. Urotensin II-induced mobilization of Ca2+ response was plotted as Ca2+ concentration versus time. D) A representative scale demonstrating the correlation between color and Ca2+ levels. High levels of Ca2+ are shown with bright colors (yellow/red/orange) and low levels correspond to the dark colors (violet/blue/green). C) Fluorescent images of Pac1 and PAVSMC C cells were captured with preloaded Fura-2 AM and urotensin II treatment. Images depict (C-top) control and (C-bottom). E) Pac1 cells were treated with urotensin II and Hanks Balanced Salt Solution ± Ca2+. These treatment groups were compared to determine if intracellular Ca2+ is the predominant source. Pac 1 exposed to membrane-impermeable ethylenediamine tetraacetic acid and membrane-permeable BAPTA-AM were used to reverse the elevated levels of Ca2+. Experiments were conducted in three separate experiments (n = 14, *P ≤ 0.05).
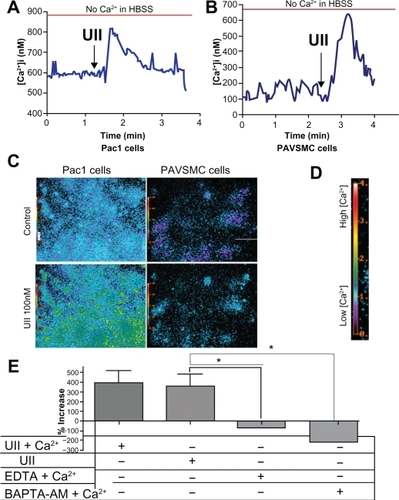
Figure 3 Urotensin II induces phosphorylation of CaMKI, PKD, and ERK in Pac1. Western blot analysis of the time course of CaMKI, PKD, and ERK phosphorylation (A, B, and C) and dose response of urotensin II-stimulated phosphorylation CaMKI, PKD, and ERK (D, E, and F) in Pac1 cells. Densitometry analysis was conducted on Western blots (n = 3, *P ≤ 0.05, **P ≤ 0.01).
Urotensin II-induced phosphorylation of CaMKI, PKD, and ERK reduced in presence of CaMKK inhibitor in Pac1
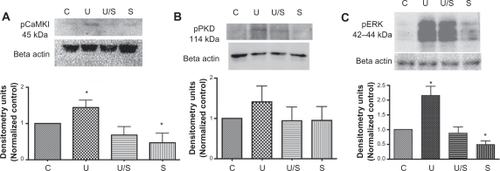
To test if CaMKK is an integral target affecting the activation of several downstream effectors in the urotensin II signaling cascade, we used the CaMKK inhibitor prior to the addition of 100 nM urotensin II for 10 minutes, and measured CaMKI phosphorylation via Western blot analysis (). Our results in revealed that STO609 blocked the urotensin II-induced phosphorylation of CaMKI. We also pretreated Pac1 with 250 nM STO609 for 30 minutes, followed by a treatment of 100 nM urotensin II for 15 minutes to measure PKD phosphorylation (). Our findings demonstrated a reduction in urotensin II-stimulated PKD phosphorylation in the presence of STO609 (). We then used 250 nM STO609 for a 30-minute pretreatment, followed by a treatment of 100 nM urotensin II for 30 minutes. Under these treatment conditions, STO609 reduced urotensin II-induced ERK phosphorylation in Pac1 (). By and large, the inhibition of CaMKK reduces urotensin II-induced phosphorylation of CaMKI, PKD, and ERK (). Therefore, our results with STO609 establish CaMKK’s involvement in urotensin II receptor signaling ().
Figure 4 Urotensin II receptor-induced phosphorylation of CaMKI, PKD, and ERK is inhibited by CaMKK inhibitor in Pac1. Urotensin II receptor-induced phosphorylation of A. CaMKI, B. PKD, and C. ERK inhibited by CaMKK. Densitometry analysis was conducted on Western blots (n = 4, *P ≤ 0.05).
Characterization of primary cell cultures
Urotensin II-induced signaling mechanisms are poorly understood in vascular smooth muscle cells, therefore we looked at urotensin II receptor signaling in both a cell line and a primary culture. We verified isolation of primary rat aortas by immunostaining the cells with a smooth muscle cell-specific antibody-antimyosin primary antibody for smooth muscle cells. As shown in , more than 70% of PAVSMC are smooth muscle cell-positive. Western blot analysis was used to determine if urotensin II treatment of PAVSMC induces ERK phosphorylation. Our results demonstrate that PAVSMC exposed to 100 nM urotensin II induces phosphorylation of ERK maximally after 15 minutes (). Furthermore, 30 minutes of pretreatment with 250 nM STO609 blocked urotensin II-induced ERK phosphorylation in PAVSMC (). Therefore, our results demonstrate that urotensin II induces the phosphorylation of ERK in PAVSMC, and CaMKK exposure to PAVSMC blocks urotensin II-induced phosphorylation of ERK ().
Figure 5 Characterization of PAVSMC. A) PAVSMC were immunostained with antimyosin smooth muscle cell-specific antibody to identify if cultured cells are composed of vascular smooth muscle cells. Merged image of cultured PAVSMC depicts positive antimyosin smooth muscle cell staining (left image). Antimyosin smooth muscle cell antibody is highlighted by the green stain (top image), and the nuclei staining is identified with DAPI, which is shown in blue (bottom image). B) A representative immunoblot demonstrates that urotensin II induces ERK phosphorylation in a time-dependent manner. Densitometry analysis was conducted on Western blots (n = 3, *P ≤ 0.05. C) and D) A representative Western blot demonstrating that CaMKK blocks urotensin II receptor-induced phosphorylation of ERK (10% serum and 10% fetal bovine serum in media (n = 2, *P ≤ 0.05, #P ≤ 0.05).
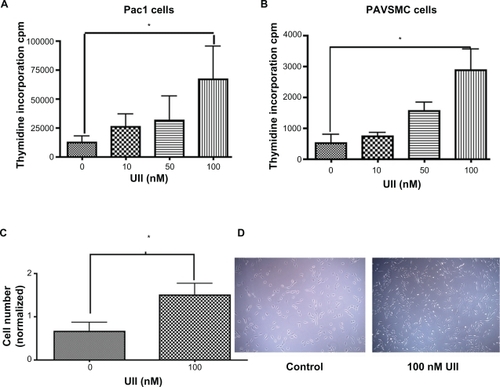
Urotensin II stimulates proliferation of Pac1 and PAVSMC
Studies have linked urotensin II-induced ERK phosphorylation with cellular proliferation in thoracic aortic cells.Citation21 Therefore, we tested whether urotensin II induces cellular proliferation in Pac1 and PAVSMC. We measured 3H-thymidine uptake as a proliferative assay in Pac1 () and PAVSMC (). Cellular proliferation was measured by counting numbers of Pac1, and we found that the cell number is greater with urotensin II treatment (). Representative images of urotensin II-treated Pac1 were taken to show differences in cell numbers with urotensin II treatment (). Urotensin II induced proliferation of both cell types in a dose-dependent manner ().
Figure 6 Urotensin II-induced cellular proliferation in a dose-dependent manner. 3H-thymidine uptake in A. Pac1 and B. PAVSMC treated with urotensin II in a dose-dependent manner stimulates proliferation (n = 4, *P ≤ 0.01). C) Cell counting was used as a measure of cellular proliferation in Pac. Experiment was blinded and six independent experiments were conducted with triplicates (n = 18, *P ≤ 0.05). D) A representative image of control and 100 nM urotensin II (image 20 × objective) in Pac1.
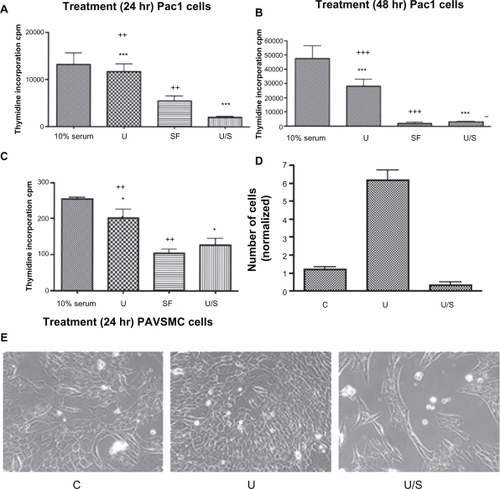
CaMKK inhibitor blocks urotensin II-induced cellular proliferation in Pac1 and PAVSMC
Pretreatment with STO609 to inhibit CaMKK blocks urotensin II-induced proliferation of PAVSMC () and Pac1 (). Representative images were taken (), and cellular proliferation was measured by counting cell numbers to show that STO609 blocks urotensin II-induced proliferation in Pac1 ().
Figure 7 CaMKK inhibitor reduces urotensin II-induced cellular proliferation in Pac1 and PAVSMC. Proliferation studies involved serum starvation for A 24 hours and B 48 hours. Pac1 were treated with urotensin II and were measured via 3H-thymidine incorporation assay. Inhibitor studies using CaMKK inhibitor in the presence of urotensin II were conducted in Pac1 (A and B) and PAVSMC (C), and were analyzed through 3H-thymidine incorporation as a measurement of cellular proliferation (10% serum and 10% fetal bovine serum in media (n = 4, *P ≤ 0.05, ++P ≤ 0.01, +++P ≤ 0.001, ***P ≤ 0.001). C) Cell counting was performed as a method to measure cellular proliferation (three independent experiments, n = 18, P ≤ 0.05). E) A representative image was taken comparing cellular proliferation in Pac1 cells (image 40 × objective).
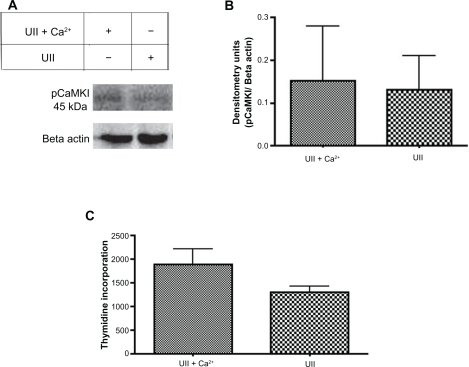
Extracellular Ca2+ does not alter urotensin II-induced phosphorylation of CaMKI and cellular proliferation
To determine if 100 nM urotensin II in the presence of extracellular Ca2+ alters CaMKI phosphorylation, the urotensin II-induced phosphorylation status of CaMKI was measured in 0.1% fetal bovine serum in ACSF media in the presence and absence of Ca2+ via Western blot analysis (). The results indicated no statistical difference between treatment groups (ACSF + urotensin II) and (ACSF + urotensin II + Ca2+, n = 3, ). To address the role of extracellular Ca2+ and 100 nM urotensin II-induced cellular proliferation, we treated cells for 24 hours with reduced serum ACSF in the presence and absence of Ca2+ and measured cellular proliferation via 3H-thymidine incorporation assay (). Results in demonstrate that urotensin II treatment in ACSF media, in the presence or absence of Ca2+, did not alter the levels of cellular proliferation.
Figure 8 Intracellular Ca2+ is the predominant source of Ca2+ involved in urotensin II-induced phosphorylation of CaMKI and cellular proliferation in Pac1. A) A representative immunoblot demonstrates that urotensin II-induced phosphorylation of CaMKI is independent of extracellular Ca2+. Pac1 were treated with ACSF + urotensin II ± Ca2+. B) Densitometry analyses of treatment groups are compared in pairwise t-tests. The y-axis data is plotted as a fraction of control (n = 2). C) Proliferation studies conducted in Pac1 were measured via 3H-thymidine incorporation in Pac1. Treatment groups for ACSF + urotensin II ± Ca2 were compared in a pairwise t-test (n = 4).
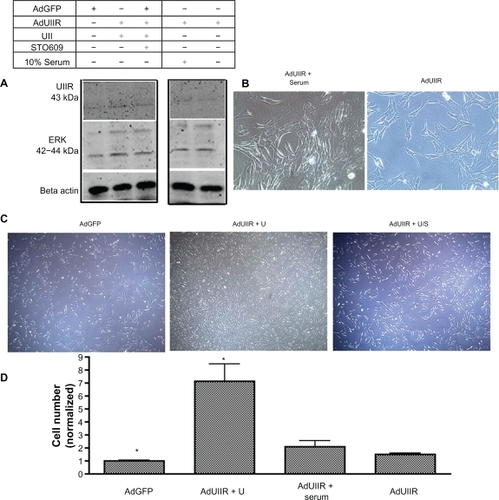
Urotensin II overexpression enhances ERK phosphorylation and proliferation of Pac1
In order to delineate whether urotensin II-induced signaling occurs through the urotensin II receptor, cells were kept in reduced serum for treatment groups (Ad-GoGFP, Ad-urotensin II receptor, Ad-urotensin II receptor + urotensin II, Ad-urotensin II receptor + urotensin II + STO609). We overexpressed the urotensin II receptor by infecting Pac1 with a urotensin II receptor adenovirus (Ad-urotensin II) in the presence and absence of urotensin II or STO609. Ten percent of serum-treated cells in the presence of the Ad-urotensin II receptor were compared with the reduced serum treatment groups to determine if urotensin II or full serum specifically affects the urotensin II receptor proliferation signaling pathway. We measured downstream targets, such as ERK phosphorylation (), and cellular proliferation (). Our results reveal that urotensin II receptor overexpression enhances urotensin II-induced ERK phosphorylation and cellular proliferation of Pac1.
Figure 9 Overexpression of the urotensin II receptor in the presence of urotensin II enhances ERK phosphorylation and cellular proliferation in Pac1 cells. A)A representative immunoblot demonstrates that the Ad-urotensin II receptor in the presence of urotensin II, induces ERK phosphorylation in Pac1. B) A representative image of proliferating Pac1 were taken of Ad-urotensin II receptor + serum, Ad-urotensin II receptor, (image 40 × objective) and C) Ad-GFP, Ad-urotensin II receptor + urotensin II, Ad-urotensin II receptor + urotensin II + STO609 (image 20 × objective). D) Cellular proliferation determined by cell counting in Pac1, measures Ad-urotensin II receptor in the presence of urotensin II (n = 2, P ≤ 0.01).
Discussion
The results reported here demonstrate that urotensin II induces Ca2+/calmodulin-dependent kinase-dependent proliferation of Pac1 and PAVSMC. Furthermore, our present study identified that, in vascular smooth muscle cells, urotensin II-induced phosphorylation of several urotensin II receptor downstream targets can be blocked using a CaMKK inhibitor. These findings suggest that CaMKK inhibition may have therapeutic relevance by blocking cellular proliferation in Pac1 and PAVSMC.
The urotensin II receptor signaling pathway was initially thought to be activated by the single agonist, urotensin II.Citation5 However, recent evidence identified urotensin II-related peptide as an additional agonist of the urotensin II receptor, similarly activating urotensin II-induced downstream targets.Citation27 Both agonists have been observed to play a role in disease processes, although the scope of our study did not include urotensin II-related peptide effects on urotensin II receptor signaling.Citation27,Citation28 Hirose et al demonstrated that during heart failure, urotensin II-related peptide expression is increased in the atrium and right ventricle of the heart.Citation29 Within the past decade, studies have observed the effects of urotensin II binding of the urotensin II receptor in various tissues including the brain, kidney, heart, and vessels.Citation30 Cellular proliferation in all these tissue types, except for heart, has shown a close correlation with increases in urotensin II levels.Citation30 Previous studies involving human umbilical vein cells show that urotensin II induces ERK phosphorylation and cellular proliferation.Citation20 ERK has been recognized as a target for several G-protein coupled receptors, and its activation is correlated with proliferation.Citation18,Citation31 ERK activation is dependent on several intracellular signals, including an increase in intracellular Ca2+.Citation32 Tamura et al has shown that urotensin II receptor signaling stimulates ERK phosphorylation via the classical pathways involving Ras and Raf in primary thoracic aortic cells.Citation21 Conversely, Sauzeau et al revealed via BrdU and cell counting assays that urotensin II stimulates human aortic smooth muscle cell proliferation through RhoA and Rho kinase.Citation33 Although our studies indicate that activation of cell proliferation by the urotensin II receptor and its Gq-coupled receptor requires Ca2+ activation of CaMK, it is possible that induction of Ras/Raf or Rho kinase pathways may also be involved. Schmidt et al had shown in hippocampal cells that CaMKI stimulates ERK phosphorylation, and conversely reduced ERK phosphorylation using a dominant negative CaMKI or pharmacologic inhibitors to CaMKI, which is consistent with our findings.Citation20
Previous studies examining cellular proliferation identified the sarcoplasmic reticulum as the predominant source of Ca2+ release during proliferation. Dramatic changes in Ca2+ flux have been linked to cardiovascular disease, such as arteriosclerosis and renal disease.Citation34,Citation35 However, the mechanisms inducing intracellular Ca2+ flux are not yet well defined. Others have demonstrated that Ca2+/ATPase and sarco/endoplasmic reticulum Ca2+-ATPase pumps could modulate cytoplasmic Ca2+ increases during proliferation.Citation37 Recent proliferation studies have shown that the intracellular store involved in sarcoplasmic reticulum release (via Ca2+-induced Ca2+ release) may occur through the ryanondine receptor.Citation36,Citation37 We have not identified the source or the specific mechanism of intracellular Ca2+ release, although future studies will entail delineating which intracellular Ca2+ store is involved in the urotensin II-induced signaling pathway. Currently it is our belief that the primary mechanism of this Gq signaling-mediated event occurs through inositol triphosphate and modulates the release of Ca2+.
Although our studies indirectly identify Ca2+/calmodulin kinases as the target signaling kinases in the urotensin II-urotensin II receptor pathway, future studies are needed to elucidate urotensin II receptor signaling mechanisms using a urotensin II receptor-specific antagonist. We have used an adenovirus to overexpress the urotensin II receptor, although using siRNA to the urotensin II receptor or a direct inhibitor will determine the involvement of Ca2+/calmodulin kinases in the urotensin II-induced proliferation pathways of vascular smooth muscle cells.
In summary, Pac1 and PAVSMC cultures exposed to urotensin II result in cellular proliferation, and CaMKK inhibitor treatment blocks cellular proliferation. Understanding CaMKK’s role in urotensin II-induced proliferation may provide further insight into potential therapeutic targets for vascular smooth muscle cell proliferation.
Disclosure
The authors report no conflicts of interest in this work.
References
- JefferyTKMorrellNWMolecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertensionProg Cardiovasc Dis200245317320212525995
- RudijantoAThe role of vascular smooth muscle cells on the pathogenesis of atherosclerosisActa Med Indones2007392869317933075
- MatsushitaMShichiriMFukaiNUrotensin II is an autocrine/paracrine growth factor for the porcine renal epithelial cell line, LLCPK1Endocrinology200314451825183112697688
- CohenJDOverview of physiology, vascular biology, and mechanisms of hypertensionJ Manag Care Pharm200713Suppl 5S6S817605504
- WatanabeTKanomeTMiyazakiAKatagiriTHuman urotensin II as a link between hypertension and coronary artery diseaseHypertens Res200629637538716940699
- MatsusakaSWakabayashiIEnhancement of vascular smooth muscle cell migration by urotensin IINaunyn-Schmiedebergs Arch Pharmacol2006373538138616896801
- MatsushitaMShichiriMImaiTCo expression of urotensin II and its receptor (GPR14) in human cardiovascular and renal tissuesJ Hypertens200119122185219011725162
- PapadopoulosPBousetteNGiaidAUrotensin II and cardiovascular remodelingPeptides200829576476917988761
- HidalgoCNunezMTCalcium, iron and neuronal functionIUBMB Life2007594–5280285 Available at : http://www3.interscience.wiley.com/journal/117879563/abstract?CRETRY=1&SRETRY=0. Accessed on Jul 15, 2010.17505966
- WangYXDingYJZhuYZShiYYaoTZhuYCRole of PKC in the novel synergistic action of urotensin II and angiotensin II and in urotensin II induced vasoconstrictionAm J Physiol Heart Circ Physiol20072921H348H35916951045
- RothmanAKulikTJTaubmanMBBerkBCSmithCWNidel-GinardBDevelopment and characterization of a cloned rat pulmonary arterial smooth muscle cell line that maintains differentiated properties through multiple subculturesCirculation1992866197719861333373
- CobbMHHeplerJEChengMRobbinsDThe mitogen activated protein kinases, ERK1 and ERK2Semin Cancer Biol1994542612687803762
- TokumitsuHTakahashiNEtoKYanoSSoderlingTRMuramatsuMSubstrate recognition by Ca2+/Calmodulin dependent protein kinase kinase. Role of the arg pro rich insert domainJ Biol Chem199927422158031581010336483
- McKinseyTADerepression of pathological cardiac genes by members of the CaM kinase superfamilyCardiovasc Res200773466767717217938
- RozengurtEMitogenic signaling pathways induced by G protein-coupled receptorsJ Cell Physiol2007213358960217786953
- ShiLDingWLiDProliferation and anti apoptotic effects of human urotensin II on human endothelial cellsAtherosclerosis2006188226026416343502
- BroseNRosenmundCMove over protein kinase C, you’ve got company: Alternative cellular effectors of diacylglycerol and phorbol estersJ Cell Sci2002115Pt 234399441112414987
- RaiesdanaALoscalzoJPulmonary arterial hypertensionAnn Med20063829511016581695
- NakagawaYKuwaharaKHaradaMClass II HDACs mediate CaMK dependent signaling to NRSF in ventricular myocytesJ Mol Cell Cardiol20064161010102217011572
- SchmittJMGuireESSaneyoshiTSoderlingTRCalmodulin dependent kinase kinase/calmodulin kinase I activity gates extracellular regulated kinase dependent long term potentiationJ Neurosci20052551281129015689566
- TamuraKOkazakiMTamuraMIsozumiKTasakiHNakashimaYUrotensin II induced activation of extracellular signal regulated kinase in cultured vascular smooth muscle cells: Involvement of cell adhesion mediated integrin signalingLife Sci20037291049106012495783
- GuibertCMarthanRSavineauJPModulation of ion channels in pulmonary arterial hypertensionCurr Pharm Res2007132424432455
- TravoPBarrettGBurnstockGDifferences in proliferation of primary cultures of vascular smooth muscle cells taken from male and female ratsBlood Vessels19801721101167362876
- DibasAPrasannaGYorioTLocalization of endothelin converting enzyme in bovine optic nerve and retinaJ Ocul Pharmacol Ther200521428829716117692
- OnanDPipoloLYangEHannahRDThomasWGUrotensin II promotes hypertrophy of cardiac myocytes via mitogen activated protein kinasesMol Endocrinol20041892344235415205471
- AmesRSSarauHMChambersJKHuman urotensin II is a potent vasoconstrictor and agonist for the orphan receptor GPR14Nature1999401675028228610499587
- JarryMDialloMLecointreCThe vasoactive peptides urotensin II and urotensin II related peptide regulate astrocyte activity through common and distinct mechanisms: Involvement in cell proliferationBiochem J2010428111312420192922
- KristofASYouZHansYSGiaidAProtein expression of urotensin II, urotensin related peptide, and their receptor in the lungs of patients with lymphangioleiomyomatosisPeptides2010528 [Epub ahead of print]
- HiroseTTakahashiKMoriNIncreased expression of urotensin II, urotensin II related peptide and urotensin II receptor mRNAs in the cardiovascular organs of hypertensive rats: Comparison with endothelin 1Peptides20093061124112919463745
- YoshimotoTMatsushitaMHirataYRole of urotensin II in peripheral tissue as an autocrine/paracrine growth factorPeptides200425101775178115476945
- SoderlingTRThe Ca calmodulin dependent protein kinase cascadeTrends Biochem Sci199924623223610366852
- HarrisonBCKimMSvan RooijERegulation of cardiac stress signaling by protein kinase d1Mol Cell Biol200626103875388816648482
- SauzeauVLe MellionecEBertiglioJScalbertEPacaudPLoirandGHuman urotensin II induced contraction and arterial smooth muscle cell proliferation are mediated by RhoA and Rho kinaseCirc Res200188111102110411397774
- InoueRHaiLHondaAPathophysiological implications of transient receptor potential channels in vascular functionCurr Opin Nephrol Hypertens200817219319818277154
- MenePTransient receptor potential channels in the kidney: Calcium signaling, transport and beyondJ Nephrol2006191212916523421
- Berra RomaniRRaqueebAAvelino-CruzJECa2+ signaling in injured in situ endothelium of rat aortaCell Calcium200844329830918276005
- Berra RomaniRMazzocco-SpezziaAPulinaMVGolovinaVACa2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in cultureAm J Physiol Cell Physiol20082953C779C79018596214