Abstract
Apolipoprotein (apo) AI and apoB are the major apolipoproteins of high-density lipoprotein (HDL) and low-density lipoprotein (LDL), respectively. ApoB assembles the precursor of LDL, very-low-density lipoprotein (VLDL), in the liver. The assembly starts with the formation of a primordial particle, which is converted to VLDL2. The VLDL2 particle is then transferred to the Golgi apparatus and can either be secreted or converted to triglyceride-rich VLDL1. We have reviewed this assembly process, the process involved in the storage of triglycerides in cytosolic lipid droplets, and the relationship between these two processes. We also briefly discuss the formation of HDL. ApoB mediates the interaction between LDL and the arterial wall. Two regions in apoB are involved in this binding. This interaction and its role in the development of atherosclerosis are reviewed. ApoB can be used to measure the number of LDL or VLDL particles present in plasma, as there is one molecule of apoB on each particle. By contrast, the amount of cholesterol and other lipids on each particle varies under different conditions. We address the possibility of using apoAI and apoB levels to estimate the risk of development of cardiovascular diseases and to monitor intervention to treat these diseases.
Introduction
Cardiovascular diseases are increasing in prevalence and represent a major public health burden of the 21st century. Atherosclerosis is the major cause of these diseases and several risk factors have been identified that promote the development of premature atherosclerosis. In a large, international, case-control study (INTERHEART), abnormal plasma lipoproteins have been shown to be a major risk factor for myocardial infarction (CitationYusuf et al 2004).
Several types of lipoprotein have been identified in plasma. They are usually separated according to their hydrated density into high-density lipoprotein (HDL) 2 and 3 , low-density lipoprotein (LDL), intermediate-density lipoprotein, very-low-density lipoprotein (VLDL) 1 and 2, and chylomicrones (the plasma lipoproteins and their composition are reviewed in most modern biochemistry textbooks). VLDL is secreted from the liver and is the precursor of LDL. The conversion of VLDL to LDL is a complex process that has been modeled in vivo in humans both under normal conditions and in diseases (reviewed in (CitationAdiels et al 2006)).
Apolipoproteins are the protein components of plasma lipoproteins and several different apolipoproteins have been identified. The major apolipoprotein of LDL is apoB100, which is synthesized in the liver and enters the plasma with VLDL. There is one apoB100 molecule on each LDL particle, and it is used to estimate the number of LDL and VLDL particles present in plasma. In this aspect, apoB100 is superior to LDL cholesterol or non-HDL cholesterol as the cholesterol content on the LDL and VLDL particles can vary considerably. The two major proteins in HDL are apoAI and apoAII. Of these, apoAI has most frequently been used to estimate the HDL levels but, in contrast to apoB100, it exchanges between lipoproteins and the number of molecules varies between particles.
In this article, we have reviewed how apoB100 assembles VLDL in the liver and how the protein interacts with the arterial wall during the development of atherosclerosis. We also give a brief summary on the assembly of HDL. Moreover, we discuss the possibility of using apoB and apoAI to predict the risk of cardiovascular diseases and to monitor the effect of intervention to treat such diseases.
Composition of VLDL
VLDL is assembled in and secreted from the liver. There are two forms of VLDL: VLDL1, which is large and rich in triglycerides, and VLDL2, which is smaller and contains less triglycerides. The type of particle that is secreted is highly important for the atherogenicity of the plasma lipoproteins. Secretion of VLDL1 increases the levels of so-called small dense LDL and decreases the levels of HDL, and both of these changes have been shown to promote the development of atherosclerosis (for review, see (CitationTaskinen 2003)). Overproduction of VLDL1 is observed in patients with insulin resistance and type 2 diabetes (CitationTaskinen 2003).
As for all lipoproteins, VLDL consists of a core of neutral lipids (triglycerides and cholesterol esters) surrounded by a monolayer of amphipathic structures such as phospholipids, free cholesterol, and proteins. ApoB100 is a large (4536 amino acid long) amphipathic protein that is present on VLDL (CitationSegrest et al 2001). It has a pentapartite structure consisting of a globular N-terminal domain, two domains of amphipathic β-sheets, and two domains of amphipathic α-helices, one between the two β-sheet domains and one near the C-terminus (CitationSegrest et al 2001). The N-terminal domain is of vital importance for the formation of VLDL as it interacts with the microsomal triglyceride transfer protein (MTP), which catalyzes the transfer of lipids to apoB during the formation of lipoproteins (CitationDashti et al 2002). The amphipathic β-sheet domains consist of antiparallel β-sheets with a width of approximately 30 Å. They form very strong lipid-binding structures (CitationSegrest et al 2001), which is generally thought to explain why apoB remains associated with the original core structure rather than equilibrating between different lipoproteins.
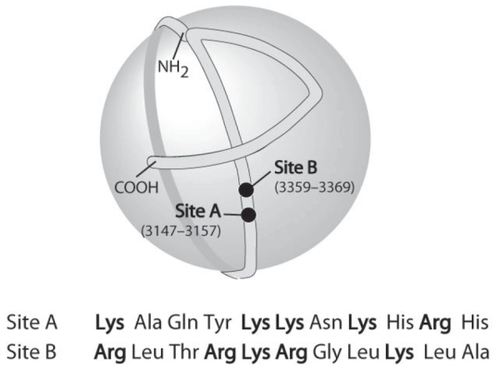
The three-dimensional structure of apoB is not known in detail, but the overall organization on the LDL has been elucidated using immuno-electronmicroscopy (CitationChatterton et al 1995). The protein has an elongated structure that encircles the entire particle (). The C-terminus folds back over the preceding structure; an arginine (residue 3500) binds to a tryptophan (residue 4396), which prevents the C-terminus from sliding over the binding site for the LDL receptor between residues 3359 and 3369 (). Mutation of the arginine (residue 3500) results in reduced binding to the LDL receptor because the arginine–tryptophan interaction is broken (CitationBoren et al 2001a; CitationBoren et al 1998a).
Figure 1 The organization of apoB100 on the LDL particle. Two of the sites (A and B) involved in the binding of apoB100 (and LDL) to proteoglycans are indicated in the figure and their primary sequence is given below. Site B is also the binding site for the LDL receptor.
In several species, an additional apoB, apoB48 is also expressed in the liver, where it forms VLDL of the same type as that formed by apoB100. In humans, apoB48 is formed in the intestine and assembles chylomicrons.
The assembly of VLDL
ApoB100 is a secretory protein and is synthesized on ribosomes attached to the surface of the endoplasmic reticulum (). During its formation, the “nascent” polypeptide is translocated through a channel from the site of synthesis in the ribosome through the membrane to the lumen of the endoplasmic reticulum (ER). Secretory proteins acquire their tertiary structure in the ER by a folding process that depends on so-called chaperone proteins. Correctly folded proteins are sorted into exit sites to leave the ER by transport vesicles. If the correct tertiary structure is not achieved, the protein is retained in the ER and retracted through the membrane channel and sorted to proteasomal degradation (CitationEllgaard and Helenius 2001; CitationEllgaard and Helenius 2003; CitationEllgaard et al 1999; CitationJohnson and Haigh 2000; CitationJohnson and van Waes 1999; CitationKostova and Wolf 2003; CitationLippincott-Schwartz et al 2000).
Figure 2 The assembly of VLDL. During its biosynthesis, apoB100 (1) is translocated to the lumen of the endoplasmic reticulum and lipidated by MTP to form a pre-VLDL particle (2). Pre-VLDL is further lipidated to form VLDL2 (5). Alternatively, pre-VLDL and misfolded apoB100 (3) can be retained and degraded in the cell (4). The VLDL2 is transferred to the Golgi apparatus (6, 7) and is either secreted or further lipidated to form VLDL1 (8). Fatty acids are released from cytosolic lipid droplets and used for the formation of triglycerides, which are assembled into VLDL (8).
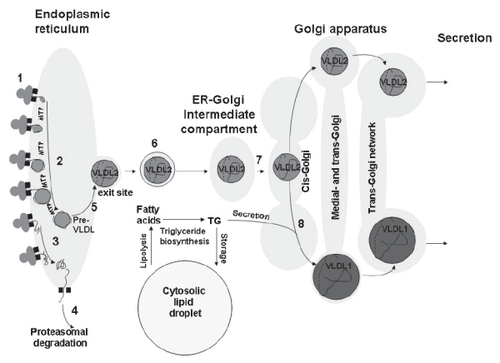
The formation of vesicles at the exit site depends on the GTPase SAR1 and the coat protein coatamere protein II (COPII). These vesicles carry the secretory products out of the ER and form the ER-Golgi intermediate compartment (ERGIC), an organelle that is involved in sorting of proteins. Thus, proteins that should remain in the ER are returned to this organelle from ERGIC. The sorting process involves the formation of vesicles by the GTPase ARF1 and COPI. ERGIC is transported to the cis-Golgi and fuses to continuously form this compartment. ARF1 is of importance for the ability of ERGIC to mature into cis-Golgi (CitationNickel et al 1998; CitationNickel and Wieland 1998; CitationSpang 2002) ().
VLDL is formed by a stepwise lipidation of apoB100 occurring as it is transferred through the secretory pathway (). The process starts during the entry of the N-terminus of apoB100 in the lumen of the ER by an MTP-dependent addition of lipid to the growing apoB protein (). The C-terminal portion of apoB100 breaks this lipidation (CitationStillemark-Billton et al 2005). An underlipidated primordial particle (pre-VLDL) is thus formed that interacts with ER chaperones and is either retained in the cell (and subsequently degraded) or is further lipidated to form VLDL2. We propose that this reflects a need to form a VLDL particle that is large enough to allow apoB to fold correctly in its surface.
The ability of apoB100 to form pre-VLDL is dependent on the sequence between residues 3265 and 4082 (CitationStillemark-Billton et al 2005), which introduces a shift in lipidation of the protein. This sequence is capable of temporarily switching off the lipidation awaiting, for example, the availability of MTP and/or lipids. It is possible that this switch is involved in the lipid-dependent sorting of apoB100 to secretion or degradation. Moreover, it seems that the effect of this sequence is to promote the binding of chaperones to pre-VLDL (CitationStillemark-Billton et al 2005), which are most likely involved in the retention of the particle in the cell. The observation that pre-VLDL is converted to VLDL2 by the addition of more lipids implies that MTP is also active after the completion of apoB100. This fits with our finding that MTP is needed for a period after the completion of apoB100 to allow the formation of a secretable apoB100 containing lipoproteins (CitationRustaeus et al 1998). Thus, during this period, MTP may convert pre-VLDL to VLDL2.
The lipidation of VLDL2 is dependent on the size of apoB, and there is an inverse relationship between the density of the particle formed and the length of the protein (CitationStillemark-Billton et al 2005). A bona fide VLDL2 is only formed by apoB100 while truncated forms of apoB form more dense particles that are VLDL2 analogues. For example, apoB48, which is the natural occuring truncated form of apoB100, forms a dense “HDL-sized” particle that is mature and secreted from the cell, unlike the primordial immature particle formed by apoB100. The apoB48-containing VLDL2 analogue is formed in the ER (CitationStillemark et al 2000), but it is not known whether the apoB100-containing VLDL2 is formed in the ER or after apoB has exited this compartment. Assembly of VLDL2 (and VLDL2 analogues) in the ER would fit with the paradigm that only correctly folded proteins are allowed to be transferred to the later part of the secretory pathway (). (For quality control of folded proteins, see the following reviews (CitationAhner and Brodsky 2004; CitationEllgaard and Helenius 2001; CitationEllgaard and Helenius 2003; CitationKleizen and Braakman 2004; CitationSchroder and Kaufman 2005; CitationSitia and Braakman 2003)). We therefore propose that the primordial particle that is formed by lipidation of the growing apoB100 must be converted to VLDL2 before it can leave the ER, and that VLDL2 must reach the Golgi apparatus before it can be converted to VLDL1. Both VLDL2 and VLDL1 are secreted while the primordial particle (pre-VLDL) is retained and eventually degraded.
The formation of VLDL1 involves a bulk lipidation (ie, the addition of a major load of triglycerides) of VLDL2 or the VLDL2 analogues, and the apoB acceptor is only required to have a minimum size of apoB48 (CitationStillemark-Billton et al 2005). Both apoB and VLDL2 (or its analogues) must be transported to the Golgi apparatus to allow VLDL1 formation (CitationAsp et al 2005; CitationStillemark et al 2000). The time gap necessary for apoB to be transferred to the Golgi apparatus to obtain its major lipidation is in agreement with our recent observation in humans injected with stable amino acid and glycerol isotopes: labeled triglycerides were secreted 15 min before labeled apoB100 (CitationAdiels et al 2005b). Furthermore, we have found evidence of a precursor-product relationship between VLDL2 and VLDL1 in clinical turnover studies using stable isotopes. Thus, in insulin-clamp studies, we observed that insulin decreased the assembly of VLDL1 and at the same time increased the assembly and secretion of VLDL2 (Adiels et al Manuscript in preparation).
Cytosolic lipid droplets
The assembly of VLDL is highly dependent on the amount of triglycerides in the hepatocytes. Several authors have demonstrated that the fatty acids used for the biosynthesis of VLDL triglycerides are derived from triglycerides stored in cytosolic lipid droplets (CitationGibbons et al 2000; CitationSalter et al 1998; CitationWiggins and Gibbons 1992). The droplets are now recognized as dynamic organelles rather than passive stores of lipids (CitationMartin and Parton 2006).
The structure of cytosolic lipid droplets is very similar to that of lipoproteins, with a core of neutral lipids surrounded by a monolayer of amphipathic structures such as phospholipids, cholesterol, and proteins. The most well known of these proteins are the PAT proteins (CitationLondos et al 1999) perilipin, which is present in adipocytes and cells involved in the biosynthesis of steroid hormones, adipocyte differentiation-related protein (ADRP), which is ubiquitously expressed, and tail-interacting protein 47 (TiP47). There are several other proteins with important functions present on the droplets (CitationBrasaemle et al 2004; CitationLiu et al 2004).
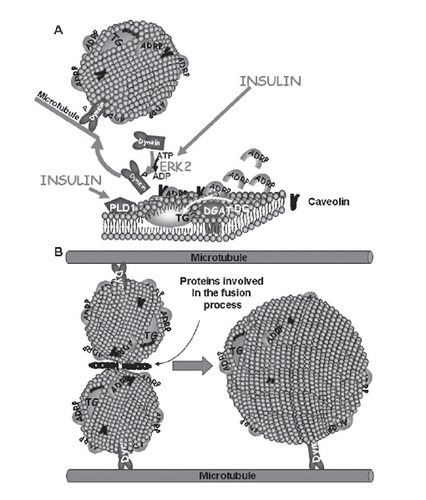
The lipid droplets are formed as small structures (0.1–0.4 μm) in or close to the ER and/or Golgi membranes (CitationMarchesan et al 2003). Insulin promotes the formation of lipid droplets via the activation of phospholipase D1 (PLD1) and extracellular signal-related kinase 2 (ERK2) (CitationAndersson et al 2006). ERK2 phosphorylates the motor protein dynein, which is then targeted to lipid droplets allowing them to transfer to microtubules and increase in size by fusion (CitationAndersson et al 2006; CitationBostrom et al 2005). A model for the assembly of lipid droplets is shown in .
Figure 3 (A) Lipid droplets are formed as primordial droplets at the microsomal membranes. Insulin stimulates the droplet formation by activating PLD1 and ERK2. ERK2 phosphorylates the motor protein dynein, which is then recruited to the droplets to promote their formation and fusion. Diacylglycerol acyltransferase (DGAT) catalyzes the formation of triglycerides (TG) from diglycerides (DG) and fatty acids (FA). It has been suggested that the lipid droplet is initiated by the oiling out of triglycerides between the leaflets of the membrane. (B) Primordial droplets increase in size by fusion. This process depends on dynein and its interaction with microtubules. Moreover it involves specific proteins that catalyze fusion between the droplets.
The importance of triglycerides for the assembly and secretion of VLDL is supported by our turnover studies in vivo, which demonstrate that the secretion of VLDL1 apoB100 increases with increasing concentrations of liver lipids (CitationAdiels et al 2005a). However, the relationship between stored triglycerides and the secretion of VLDL is complex.
On the basis of our results, we conclude that there are at least two ways of influencing the pool of lipid droplets in the cell: (i) by promoting the formation of small primordial droplets and (ii) by promoting the rate of fusion between the droplets. We have demonstrated that both mechanisms also influence the assembly of VLDL. An increase in ADRP results in an increase in the formation of the droplets and the pool of the droplets and a subsequent decrease in the assembly of VLDL, in particular VLDL1 (CitationMagnusson et al 2006). This is because ADRP promotes the storage of newly formed triglycerides in cytosolic droplets, diverting lipids from the VLDL assembly pathway. We also observed that epigallocatechin gallate, which is present in green tea, increases the rate of fusion between droplets, resulting in an increased pool of cytosolic lipid droplets, and decreases the assembly and secretion of VLDL, in particular VLDL1 (CitationLi et al 2006). Together, these results indicate that factors that promote the assembly of lipid droplets and increase their cellular pool can in fact deplete the VLDL assembly pathway of lipids and thus decrease the secretion of the lipoprotein.
It is known that the secretion of VLDL1 is reduced by insulin (see CitationTaskinen 2003) for review). Moreover, a failure of this insulin effect may be of importance for the formation of the dylipidemia seen in insulin resistance and type 2 diabetes. The mechanism is not understood, but it is possible that promotion of lipid droplet assembly by insulin (CitationAndersson et al 2006) promotes a shift of triglycerides from the VLDL assembly pathway to storage in cytosol. There are, however, other tentative mechanisms to explain the influence of insulin on VLDL assembly (for review, see (CitationTaskinen 2003)).
Post-translational regulation of the secretion of apoB100
The secretion of apoB100 is regulated post-transcriptionally by co- and post-translational degradation. It has long been known that apoB100 undergoes intracellular degradation (for review, see (CitationOlofsson et al 1999; CitationDavidson and Shelness 2000; CitationShelness and Sellers 2001)). The intracellular degradation of apoB100 occurs at three different levels (CitationFisher and Ginsberg, 2002; CitationFisher et al 2001):
(i) Close to the biosynthesis of apoB (co- or post-translationally) by a mechanism that involves retraction of the apoB molecule from the lumen of the ER to the cytosol, a ubiquitination and a subsequent proteasomal degradation (CitationMitchell et al 1998; CitationLiang et al 2000; CitationFisher et al 2001; CitationPariyarath et al 2001). The co-translational degradation of apoB is influenced by the availability of lipids and the activity of MTP. Thus, when the amount of lipids or the MTP activity is limited, apoB100 remains associated with the translocon and is sorted to proteasomal degradation (for review, see (CitationFisher and Ginsberg 2002)). It is well known that secretory proteins that misfold undergo proteasomal degradation (see above; ). A lack of lipids may remove one of the prerequisites for the correct folding of apoB, thus promoting its degradation.
(ii) Post-translationally by an unknown mechanism that can be promoted by culturing the cells in the presence of polyunsaturated fatty acids (CitationFisher et al 2001). This degradation seems to occur in a compartment separate from the rough ER, and has therefore been referred to as post-ER presecretory proteolysis (PERPP) (CitationFisher et al 2001). Nothing is known about the enzyme systems involved or the sorting of apoB100 for this degradation. The process is promoted by ω-3 fatty acids and by increased levels of reactive oxygen species (CitationJiang et al 2005). It has also been demonstrated that apoB100 interacts with the protease/chaperone ER 60 (CitationAdeli et al 1997; CitationTaghibiglou et al 2002), and that this interaction is linked to the intracellular degradation of newly synthesized apoB100. The role of this interaction for PERPP has not been elucidated.
(iii) By reuptake from the unstirred water layer around the outside of the plasma membrane (CitationWilliams et al 1990) via the LDL receptor. The LDL receptor has been shown to have an important role in regulation of the secretion of apoB100-containing lipoproteins (CitationHorton et al 1999; CitationTwisk et al 2000). There is even evidence that the effect of the receptor is not only due to the interaction with apoB100 on the cell surface, but that the receptor and apoB100 interact early in the secretory pathway and that this interaction is of importance for the post-translational degradation of apoB100 (CitationGillian-Daniel et al 2002; CitationLarsson et al 2004; CitationTwisk et al 2000)}.
The apoB100 primordial particle (pre-VLDL) is not secreted from the cell to any significant extent, but appears to be retained and degraded (unless converted to VLDL2; see above) (CitationStillemark-Billton et al 2005)
The development of atherosclerosis: different hypotheses
Although it is well documented that elevated levels of LDL and other apoB-containing lipoproteins cause increased atherosclerosis, the molecular and cellular mechanisms for the pathobiological changes that lead to the disease are still poorly understood. Several hypotheses have been articulated to explain the events that initiate atherogenesis: the response-to-injury hypothesis states that endothelial injury leads to an inflammatory response as a part of a healing process in the arterial wall (CitationRoss 1999; CitationRoss et al 1977); the response-to-oxidation hypothesis proposes that lipoprotein oxidation is the important link in atherosclerosis (CitationSteinberg et al 1989); and the response-to-retention hypothesis suggests that subendothelial retention of lipoproteins is the initiating step and leads to oxidation, inflammation, and endothelial dysfunction (CitationWilliams and Tabas 1995; CitationWilliams and Tabas 1998). This hypothesis is based on pioneering work from the 1970s and 1980s showing that lipoproteins can interact with the arterial wall (CitationIverius 1972; CitationCamejo et al 1975; CitationVijayagopal et al 1981).
While these hypotheses are by no means mutually exclusive, and may even be considered mutually compatible with differences in emphasis, a growing body of recent evidence supports the response-to-retention hypothesis. This hypothesis states that the subendothelial retention of lipoproteins determines the concentration and the susceptibility to modification of lipoproteins in the arterial wall. Several lines of evidence indicate that the retention of arterial lipoproteins involves the extracellular matrix, and proteoglycans in particular appear to play an important role.
Mechanism for retention of atherogenic lipoproteins in the artery wall
The binding of atherogenic lipoproteins to artery wall proteoglycans is mediated by ionic interactions between clustered basic amino acids in the apoB100 that bind to the negatively charged glycosaminoglycans (GAGs) of the proteoglycans. In vitro studies of delipidated apoB100 identified eight specific regions of clustered basic amino acids in the apoB moiety of LDL that bind to the negatively charged GAGs (CitationHirose et al 1987; CitationWeisgraber and Rall Jr, 1987; CitationCamejo et al 1988). However, it was not known which of these sites were functional on LDL particles. To identify the main proteoglycan-binding sites on LDL, specifically mutated forms of the human apoB gene were expressed in transgenic mice, and the ability of the recombinant human LDL isolated from the mice to bind to artery wall proteoglycans in vitro was assessed (CitationBoren et al 1998b). These experiments implicated the basic amino acids in Site B (residues 3359–3369; ), the site in apoB100 that binds to the LDL receptor, as the main site on apoB100 that interacts with proteoglycans (CitationBoren et al 1998b). In addition, a mutant recombinant LDL with a single amino acid mutation (K3363E) was generated that bound normally to the LDL receptor but defectively to proteoglycans (CitationBoren et al 1998b). This finding showed that it is possible to discriminate between the LDL receptor-binding activity and proteoglycan-binding activity, even though their binding sites coincide (CitationBoren et al 1998a; CitationBoren et al 1998b). These and other experiments also indicate that the LDL receptor binding is conformational dependent, whereas proteoglycan-binding is charge dependent.
Atherosclerosis studies in mice expressing recombinant LDL
The atherogenic potential of the interaction between apoB100 and artery wall proteoglycans was investigated in transgenic mice expressing human control LDL and recombinant proteoglycan-binding-defective LDL (ie, LDL with a Site B mutation that abolishes the binding to proteoglycan) (CitationSkalen et al 2002). The study was designed to ensure that any differences in atherosclerosis were due to weak binding of the proteoglycan-binding-defective LDL to proteoglycans and not to some other attribute of the mutated LDL, such as the inability to bind to LDL receptors.
The transgenic mice were fed a Western diet for 20 weeks. The results showed that the extent of the vessel wall covered by atherosclerotic lesions correlated with the plasma cholesterol level in both groups of transgenic mice. However, the extent of atherosclerosis differed dramatically. Transgenic mice expressing the proteoglycan-binding-defective LDL had strikingly less atherosclerosis than mice expressing wild-type recombinant LDL. These findings show that proteoglycan-binding-defective LDL has a greatly reduced atherogenic potential and provide direct experimental evidence that binding of LDL to artery wall proteoglycans is an early step in atherogenesis (CitationSkalen et al 2002).
A tentative explanation for why some atherosclerosis developed in mice that express proteoglycan-binding-defective apoB is that LDL from mice on a high-fat diet contains apoE, which can mediate binding to proteoglycans and thus retention. Indeed, proteoglycan-binding-defective LDL isolated from the mice in the atherosclerosis study contained apoE and displayed approximately 20% of normal proteoglycan binding (CitationSkalen et al 2002).
Is it possible to modulate the proteoglycan binding of LDL?
The conformation of apoB100 on the surface of the LDL particle is dependent on the composition of the core lipids, the surface phospholipid content, and the diameter of the lipoprotein particle. Thus, binding sites other than Site B may become functional in modified LDL. LDL that has been modified by secretory group IIA phospholipase A2 (sPLA2), a strong risk factor for coronary heart disease (CitationKugiyama et al 1999), binds to proteoglycans more avidly than unmodified LDL (CitationSartipy et al 1998). Recent studies have shown that Site A (residues 3147–3157 in apoB100) becomes functional in modified forms of LDL and that it then acts cooperatively with Site B in the association with proteoglycans (CitationFlood et al 2004). Furthermore, the core lipids of LDL also influence the folding of apoB100 on the LDL particle. Recent studies have shown that the triglyceride content of LDL influences the conformation of apoB and decreases the affinity for GAGs. This mechanism is mediated by a conformational change of Site B and is, in contrast to sPLA2-modified LDL, independent of Site A (CitationFlood et al 2004). The finding that the triglyceride content of LDL decreases the binding of LDL to artery wall proteoglycans may explain why triglyceride-rich apoB LDL are not as atherogenic as the triglyceride poor (CitationWillner et al 2003).
Indirect interactions of LDL to the extracellular matrix
In addition to the direct interaction between apoB100 and artery wall proteoglycans, retention of LDL also appears to involve an indirect interaction between LDL and artery wall proteoglycans facilitated by “bridging molecules”. ApoE functions as a potent bridging molecule between recombinant LDL and artery wall proteoglycans, and apoE enrichment of proteoglycan-binding-defective LDL substitutes for the defective direct interaction between RK3359–3369SA LDL and artery wall proteoglycans (CitationSkalen et al 2002).
In addition to apolipoproteins, several lipases have been shown to mediate bridging between lipoproteins and heparin sulfate proteoglycans (HSPGs) on the cell surface, which results in increased cellular uptake and degradation of lipoproteins. Lipoprotein lipase (LPL), which is secreted by smooth muscle cells and macrophages in atherosclerotic lesions (CitationBoren et al 2001b), has been shown to act as a bridge between GAG and extensively oxidized LDL, which is sufficiently depleted of positive charges to inhibit direct binding to GAG (CitationOlin et al 1999; CitationPentikainen et al 2000).
Thus, the proteoglycan-binding activity of LDL depends on the diameter and lipid composition of the LDL particle and on the presence of apolipoproteins other than apoB on the LDL particle. Several pathological conditions with increased risk of cardiovascular diseases, such as diabetes and rheumatoid arthritis, are characterized by LDL particles with enhanced interaction with the subendothelium (CitationHurt-Camejo et al 2001; CitationNesto and Rutter 2002)
The assembly and metabolism of HDL
Another important player in atherogenesis is HDL and its major apolipoprotein apoA1. Amphipatic α-helices anchor apoAI in the surface of the HDL particles. In contrast to apoB100, apoAI can transfer between particles.
The assembly and metabolism of HDL have recently been extensively reviewed (see for example (CitationLewis and Rader 2005; CitationYokoyama 2005 CitationKrimbou et al 2006)), and here we will only deal briefly with this subject. A groundbreaking event in the understanding of the assembly of HDL was the identification of the gene for Tangiers disease (CitationBodzioch et al 1999; CitationBrooks-Wilson et al 1999; CitationMarcil et al 1999; CitationRemaley et al 1999; CitationRust et al 1999), a condition in which HDL is almost completely lacking. This gene was shown to encode the ATP-binding cassette transporter (ABCA1), which is involved in supplying lipid-poor apoAI or pre-β-HDL with cholesterol.
It is now believed that apoAI is secreted from liver and intestine as lipid-poor apoAI (for recent reviews, see for example (CitationKrimbou et al 2006; CitationLewis and Rader 2005; CitationYokoyama 2005)) . Lipid-poor apoAI seems to be the major target for the lipidation by ABCA1; the transporter is critical for the initial lipidation of apoAI, protecting it from being rapidly degraded in the liver, kidney, and steroidogenic cells. The lipidation of apoAI by ABCA1 is a complex process that is not understood in detail, but is thought to involve an interaction between the apolipoprotein and the transporter and the formation of a high-affinity binding site for apoAI from which the nascent pre-β-HDL is formed. The process involves intracellular cAMP release induced by apoAI and phosphorylation of ABCA1 by protein kinase A. It also appears that the apoAI–ABCA1 complex can be internalized and transported through early and late endosomes (and possibly through other unknown compartments), during which time nascent HDL is formed and secreted.
In addition to ABCA1, transporters of the ABCG family play an essential role in the efflux of cellular cholesterol to HDL (CitationKlucken et al 2000; CitationKennedy et al 2005). ABCG1 has been reviewed recently (CitationBaldan et al 2006b; CitationOram and Vaughan 2006) and it seems to primarily influence the cholesterol content of macrophages. In ABCG1-/- mice, high levels of neutral lipids accumulated particularly in pulmonary macrophages, but also in Kupffer cells and hepatocytes; by contrast, little or no accumulation was observed in these cells in human ABCG1 transgenic mice (CitationBaldan et al 2006b).
Interestingly, ABCG1 is important for the exit of cholesterol to HDL but not to lipid-poor apoA-1 (for review, see (CitationBaldan et al 2006b)). Indeed, it has been suggested that ABCA1 and ABCG1 work in concert. Thus, ABCA1 is suggested to lipidate lipid-poor apoA-I while ABCG1 promotes the efflux of cholesterol to the already preformed lipid–protein complexes (CitationGelissen et al 2006; CitationVaughan and Oram 2006). However, there are suggestions that ABCA1 and ABCG1 control separate cholesterol efflux pathways (for review, see (CitationBaldan et al 2006b)). There are diverging opinions about the importance of ABCG1 for the development of atherosclerosis (CitationBaldan et al 2006a; CitationOut et al 2006; CitationRanalletta et al 2006), but a recent paper seems to favour the idea that ABCG1 protects against the development of this disease (CitationOut et al 2007).
Nascent HDL is modified in plasma by several factors. The most well known is lecithin cholesterol acyltransferase (LCAT), which esterifies cholesterol allowing the formation of a core in the lipoprotein. The core of HDL is also modified by cholesterol ester transfer protein (CETP), which exchanges cholesterol and triglycerides between HDL and triglyceride-rich lipoproteins such as VLDL. Phospholipids are also transferred from the surface of triglyceride-rich lipoprotein (during triglyceride hydrolyses) to HDL.
The catabolism of HDL can occur in at least two ways: (i) by selective removal of cholesterol and other lipids without uptake of the whole particle (this involves the scavenger receptor B1); and (ii) by uptake and degradation of the whole particle, which involves endocytosis and lysosomal degradation.
ApoAI and HDL are highly linked to the so-called reverse cholesterol transport, ie, the transfer of cholesterols from peripheral cells to the liver to be secreted as bile acids. The roles of ABCA1, ABCG1 and the scavenger receptor B1 in the turn over of HDL seem to fit very well into such a role of HDL. However, the process is far from elucidated and may be complex. In humans, radiolabeled HDL cholesterol that is eventually secreted into the bile is almost entirely transported to apoB-containing lipoproteins (presumably through CETP).
Apolipoprotein levels in plasma as a tool for clinical judgements
In most clinical trials on lipid lowering for prevention of cardiovascular disease, LDL cholesterol has been used both as a target for treatment and for the identification of treatment success. This relates to trials using drugs as well as dietary intervention. Consequently, the different guidelines for prevention of cardiovascular disease have LDL levels as their primary target (2005; CitationConroy et al 2003; CitationGrundy et al 2004). With the increased efficiency of lipid-lowering drugs, it has also become evident that targets far below the earlier suggested goals for LDL cholesterol may further reduce the morbidity and mortality in cardiovascular disease.
Recently, apoB as a target for treatment has been widely discussed and also supported by authorities in some guidelines (CitationGrundy 2002; CitationGenest et al 2003). ApoB in plasma is regarded as a marker for the total number of atherogenic lipoproteins in plasma, and the equivalent in lipid terms would be non-HDL cholesterol. Non-HDL cholesterol is also given as an alternative treatment goal in the National Cholesterol Education Program (NCEP) (CitationClearfield 2003). Although strongly correlated, however, apoB and non-HDL cholesterol are not identical, and several studies have found that apoB is a better predictor of risk for cardiovascular disease than non-HDL cholesterol (for review, see (CitationSniderman et al 2003).
It has been suggested that apoB could substitute for LDL cholesterol and non-HDL cholesterol for risk prediction (CitationPischon et al 2005). However, this idea has been questioned because the currently available population and disease correlation data and the cholesterol education program are based on LDL cholesterol and non-HDL cholesterol (CitationDenke 2005). It is important to note that apoB is a direct measure of particle number rather than lipid content, which is a crucial point for patients with the metabolic syndrome or type 2 diabetes. These subjects often have increased levels of small dense LDL, which are not detected by a simple lipid analysis. Thus, the combination of hypertriglyceridemia and high apoB is a new important risk algorithm (CitationSniderman 2004). Few intervention studies have been designed to use apoB levels for inclusion or as treatment target. However, data from some of the large statin trials indicate that apoB could also serve as an important treatment goal. For example, in the AFCAPS/TexCAPS trial, apoB in subjects on treatment was a strong predictor for major coronary events, both in the actively treated and the placebo groups (CitationGotto et al 2000).
Levels of ApoAI in plasma are strongly correlated with HDL levels. In agreement with the studies on HDL, low apoAI has been shown to be equivalent or better than low HDL cholesterol as a risk marker for atherosclerosis or cardiovascular events (CitationFrancis and Frohlich 2001; CitationLuc et al 2002; CitationWalldius and Jungner 2006). Several mechanisms to explain the strong association between low apoAI and risk have been suggested (CitationBarter and Rye 2006). The most common is that apoAI has an important role in the reversed cholesterol transport, and an alternative suggestion is that apoAI is anti-inflammatory or an antioxidant (for review see (CitationBarter and Rye 2006). However, the process is not convincingly understood.
It is more complex to determine whether elevation of apoAI also can be used as a target for treatment. ApoAI levels in plasma reflect both production and clearance. Interference in these processes may have opposing effects on the reverse cholesterol transport. In recent studies in humans and animal models, infusions of HDL or apoAI resulted in reduced atherosclerosis (CitationNissen et al 2003; CitationNicholls et al 2005), suggesting that increased plasma levels of apoAI or HDL have the potential to treat and prevent atherosclerosis.
A lot of enthusiasm has recently been focused on the apoB/apoAI ratio as a risk marker or treatment target. Several large epidemiological studies strongly support this concept by finding the apoB/apoAI ratio superior to lipid parameters as a risk marker (for review see (CitationWalldius and Jungner 2006). Whether the ratio apoB/apoAI also can be used as a general treatment target still has to be proven. It seems unlikely that a high ratio caused by low apoAI should be treated the same way as a low ratio caused by high apoB. Furthermore, we do not know if a change in the ratio caused by apoB reduction is equivalent to an increase in apoAI. This has to be further studied, but the treatment should be based on levels of both apoB and apoAI, and probably also on LDL and HDL lipid levels.
Acknowledgements
We are indebted to Dr Rosie Perkins for expert editing of the manuscript. This work was supported by grants from the Swedish research council, the Swedish foundation for strategic research, the Swedish Heart and Lung foundation and NovoNordic Foundation
References
- AdeliKMacriJMohammadiAApolipoprotein B is intracellularly associated with an ER-60 protease homologue in HepG2 cellsJ Biol Chem199727222489949278400
- AdielsMBorenJCaslakeMJOverproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemiaArterioscler Thromb Vasc Biol2005a
- AdielsMOlofssonSOTaskinenMRDiabetic dyslipidaemiaCurr Opin Lipidol2006172384616680028
- AdielsMPackardCCaslakeMJA new combined multicompartmental model for apolipoprotein B-100 and triglyceride metabolism in VLDL subfractionsJ Lipid Res2005b46586715489544
- AhnerABrodskyJLCheckpoints in ER-associated degradation: excuse me which way to the proteasome?Trends Cell Biol200414474815350974
- AnderssonLBostromPEricsonJPLD1 and ERK2 regulate cytosolic lipid droplet formationJ Cell Sci200611922465716723731
- AspLMagnussonBRutbergMRole of ADP ribosylation factor 1 in the assembly and secretion of ApoB-100-containing lipoproteinsArterioscler Thromb Vasc Biol2005255667015618550
- BaldanAPeiLLeeRImpaired development of atherosclerosis in hyperlipidemic Ldlr-/- and ApoE-/- mice transplanted with Abcg1-/- bone marrowArterioscler Thromb Vasc Biol2006a262301716888235
- BaldanATarrPLeeRATP-binding cassette transporter G1 and lipid homeostasisCurr Opin Lipidol2006b172273216680026
- BarterPJRyeKAThe rationale for using apoA-I as a clinical marker of cardiovascular riskJ Intern Med20062594475416629850
- BodziochMOrsoEKluckenJThe gene encoding ATP-binding cassette transporter 1 is mutated in Tangier diseaseNat Genet1999223475110431237
- BorenJEkstromUAgrenBThe molecular mechanism for the genetic disorder familial defective apolipoprotein B100J Biol Chem2001a2769214811115503
- BorenJLeeIZhuWIdentification of the low density lipoprotein receptor-binding site in apolipoprotein B100 and the modulation of its binding activity by the carboxyl terminus in familial defective apo-B100J Clin Invest1998a1011084939486979
- BorenJLookeneAMakoveichukEBinding of low density lipoproteins to lipoprotein lipase is dependent on lipids but not on apolipoprotein BJ Biol Chem2001b276269162211331277
- BorenJOlinKLeeIIdentification of the principal proteoglycan-binding site in LDL A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor bindingJ Clin Invest1998b1012658649637699
- BostromPRutbergMEricssonJCytosolic lipid droplets increase in size by microtubule-dependent complex formationArterioscler Thromb Vasc Biol20052519455116051877
- BrasaemleDLDoliosGShapiroLProteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytesJ Biol Chem2004279468354215337753
- Brooks-WilsonAMMarcilSMCleeMutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiencyNat Genet1999223364510431236
- CamejoGLopezAVegasHThe participation of aortic proteins in the formation of complexes between low density lipoproteins and intima-media extractsAtherosclerosis19752177911131302
- CamejoGOlofssonS-OLopezFIdentification of apo B-100 segments mediating the interaction of low density lipoproteins with arterial proteoglycansArteriosclerosis19888368773395272
- ChattertonJEPhillipsMLCurtissLKImmunoelectron microscopy of low density lipoproteins yields a ribbon and bow model for the conformation of apolipoprotein B on the lipoprotein surfaceJ Lipid Res1995362027378558090
- ClearfieldMBThe national cholesterol education program adult treatment panel ill guidelinesJ Am Osteopath Assoc2003103S1512572622
- ConroyRMPyoralaKFitzgeraldAPEstimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE projectJ Eur Heart2003249871003
- DashtiNGandhiMLiuXThe N-terminal 1000 residues of apolipoprotein B associate with microsomal triglyceride transfer protein to create a lipid transfer pocket required for lipoprotein assemblyBiochemistry20024169788712033930
- DavidsonNOShelnessGSApolipoprotein B: mRNA editing lipoprotein assembly and presecretory degradationAnn Rev Nutr2000201699310940331
- DenkeMAWeighing in before the fight: low-density lipoprotein cholesterol and non-high-density lipoprotein cholesterol versus apolipoprotein B as the best predictor for coronary heart disease and the best measure of therapyCirculation200511233687016316961
- EllgaardLHeleniusAER quality control: towards an understanding at the molecular levelCurr Opin Cell Biol200113431711454449
- EllgaardLHeleniusAQuality control in the endoplasmic reticulumNat Rev Mol Cell Biol200341819112612637
- EllgaardLMolinariMHeleniusASetting the standards:Quality control in the secretory pathwayScience19992861882810583943
- FisherEAGinsbergHNComplexity in the secretory pathway: the assembly and secretion of apolipoprotein B-containing lipoproteinsJ Biol Chem2002277173778012006608
- FisherEAPanMChenXThe triple threat to nascent apolipoprotein B Evidence for multiple distinct degradative pathwaysJ Biol Chem2001276278556311285257
- FloodCGustafssonMPitasREMolecular mechanism for changes in proteoglycan binding on compositional changes of the core and the surface of low-density lipoprotein-containing human apolipoprotein B100Arterioscler Thromb Vasc Biol2004245647014726411
- FrancisMCFrohlichJJCoronary artery disease in patients at low risk – apolipoprotein AI as an independent risk factorAtherosclerosis20011551657011223438
- GelissenICHarrisMRyeKAABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-IArterioscler Thromb Vasc Biol2006265344016357317
- GenestJFrohlichJFodorGRecommendations for the management of dyslipidemia and the prevention of cardiovascular disease: summary of the 2003 updateCmaj2003169921414581310
- GibbonsGFIslamKPeaseRJMobilisation of triacylglycerol stores BiochimBiophys Acta200014833757
- Gillian-DanielDLBatesPWTebonAEndoplasmic reticulum localization of the low density lipoprotein receptor mediates presecretory degradation of apolipoprotein BProc Natl Acad Sci USA2002994337211904390
- GottoAMWhitneyEJrSteinEARelation between baseline and on-treatment lipid parameters and first acute major coronary events in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS)Circulation20001014778410662743
- GrundySMLow-density lipoprotein non-high-density lipoprotein and apolipoprotein B as targets of lipid-lowering therapyCirculation20021062526912427645
- GrundySMCleemanJIMerzCNImplications of recent clinical trials for the national cholesterol education program adult treatment panel III guidelinesJ Am Coll Cardiol2004447203215358046
- HiroseNBlankenshipDTKrivanekMAIsolation and characterization of four heparin binding cyanogen bromide peptides of human plasma apolipoprotein BBiochemistry1987265505123676266
- HortonJDShimanoHHamiltonRLDisruption of LDL receptor gene in transgenic SREBP-1a mice unmask hyperlipidemia resulting from production of lipid-rich VLDLJ Clin Invest199910310677610194480
- Hurt-CamejoEParedesSMasanaLElevated levels of small low-density lipoprotein with high affinity for arterial matrix components in patients with rheumatoid arthritis: possible contribution of phospholipase A2 to this atherogenic profileArthritis Rheum2001442761711762936
- IveriusP-HThe interaction between human plasma lipoproteins and connective tissue glycosaminoglycansJ Biol Chem19722472607134336380
- JiangXCLiZLiuRPhospholipid transfer protein deficiency impairs apolipoprotein-B secretion from hepatocytes by stimulating a proteolytic pathway through a relative deficiency of vitamin E and an increase in intracellular oxidantsJ Biol Chem2005280183364015734742
- JohnsonAEHaighNGThe ER translocon and retrotranslocation: is the shift into reverse manual or automatic?Cell20001027091211030614
- JohnsonAEvan WaesMAThe translocon: a dynamic gateway at the ER membraneAnnu Rev Cell Dev Biol19991579984210611978
- Joint British Societies’JBS 2: guidelines on prevention of cardiovascular disease in clinical practiceHeart200591Suppl 5v15216365341
- KennedyMABarreraGCNakamuraKABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulationCell Metab200511213116054053
- KleizenBBraakmanIProtein folding and quality control in the endoplasmic reticulumCurr Opin Cell Biol200416343915261665
- KluckenJBuchlerCOrsoEABCG1 (ABC8) the human homolog of the Drosophila white gene is a regulator of macrophage cholesterol and phospholipid transportProc Natl Acad Sci USA2000978172210639163
- KostovaZWolfDHFor whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connectionEmbo J20032223091712743025
- KrimbouLMarcilMGenestJNew insights into the biogenesis of human high-density lipoproteinsCurr Opin Lipidol2006172586716680030
- KugiyamaKOtaYTakazoeKCirculating levels of secretory type II phospholipase A(2) predict coronary events in patients with coronary artery diseaseCirculation19991001280410491371
- LarssonSLSkogsbergJBjorkegrenJThe Low Density Lipoprotein Receptor Prevents Secretion of Dense ApoB100-containing Lipoproteins from the LiverJ Biol Chem2004279831614583618
- LewisGFRaderDJNew insights into the regulation of HDL metabolism and reverse cholesterol transportCirc Res20059612213215976321
- LiLStillemark-BilltonPBeckCEpigallocatechin gallate increases the formation of cytosolic lipid droplets and decreases the secretion of apoB-100 VLDLJ Lipid Res200647677716227197
- LiangJ-SWuXFisherEAThe amino-terminal domain of apolipoprotein B does not undergo retrograde translocation from the endoplasmic reticulum to the cytosol Proteasomal degradation of nascent apolipoprotein B begins at the carboxyl terminus of the protein while apolipoprotein B is still in its original transloconJ Biol Chem2000275320031010922368
- Lippincott-SchwartzJRobertsTHHirschbergKSecretory protein trafficking and organelle dynamics in living cellsAnnu Rev Cell Dev Biol2000165578911031247
- LiuPYingYZhaoYChinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane trafficJ Biol Chem200427937879214597625
- LondosCBrasaemleDLSchultzCJPerilipins ADRP and other proteins that associate with intracellular neutral lipid droplets in animal cellsSemin Cell Dev Biol19991051810355028
- LucGBardJMFerrieresJValue of HDL cholesterol apolipoprotein A-I lipoprotein A-I and lipoprotein A-I/A-II in prediction of coronary heart disease: the PRIME Study Prospective Epidemiological Study of Myocardial InfarctionArterioscler Thromb Vasc Biol20022211556112117731
- MagnussonBAspLBostromPAdipocyte differentiation-related protein promotes fatty acid storage in cytosolic triglycerides and inhibits secretion of very low-density lipoproteinsArterioscler Thromb Vasc Biol20062615667116627799
- MarchesanDRutbergMAnderssonLA phospholipase D-dependent process forms lipid droplets containing caveolin adipocyte differentiation-related protein and vimentin in a cell-free systemJ Biol Chem20032782729330012730229
- MarcilMBrooks-WilsonACleeSMMutations in the ABC1 gene in familial HDL deficiency with defective cholesterol effluxLancet19993541341610533863
- MartinSPartonRGLipid droplets: a unified view of a dynamic organelle Nat Rev Mol Cell Biol20067373816550215
- MitchellDMZhouMPariyarathRApoprotein B 100 har a prolonged interaction with the translocon during which its lipidation and translocation change from dependence on the microsomal triglyceride transfer protein to independenceProc Natl Acad Sci USA1998951473389843958
- NestoRWRutterMKImpact of the atherosclerotic process in patients with diabetesActa Diabetol200239Suppl 2S22812222624
- NichollsSJCutriBWorthleySGImpact of short-term administration of high-density lipoproteins and atorvastatin on atherosclerosis in rabbitsArterioscler Thromb Vasc Biol2005252416116141405
- NickelWBruggerBWielandFTProtein and lipid sorting between the endoplasmic reticulum and the Golgi complexSemin Cell Dev Biol199894935019835636
- NickelWWielandFTBiosynthetic protein transport through the early secretory pathwayHistochem Cell Biol1998109477869681629
- NissenSETsunodaTTuzcuEMEffect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trialJama2003290229230014600188
- OlinKLPotter-PerigoSBarrettLipoprotein lipase enhances the binding of native and oxidized low density lipoproteins to versican and biglycan synthesized by cultured arterial smooth muscle cellsJ Biol Chem199927434629610574927
- OlofssonS-OAspLBorénJThe assembly and secretion of apolipoprotein B-containing lipoproteinsCurr Opin Lipidol199910341610482137
- OramJFVaughanAMATP-Binding cassette cholesterol transporters and cardiovascular diseaseCirc Res20069910314317095732
- OutRHoekstraMHildebrandRBMacrophage ABCG1 deletion disrupts lipid homeostasis in alveolar macrophages and moderately influences atherosclerotic lesion development in LDL receptor-deficient miceArterioscler Thromb Vasc Biol200626229530016857950
- OutRHoekstraMMeursITotal Body ABCG1 Expression Protects Against Early Atherosclerotic Lesion Development in MiceArterioscler Thromb Vasc Biol2007
- PariyarathRWangHAitchisonJDCo-translational interactions of apoprotein B with the ribosome and translocon during lipoprotein assembly or targeting to the proteasomeJ Biol Chem20012765415011022045
- PentikainenMOOorniKKovanenPTLipoprotein lipase (LPL) strongly links native and oxidized low density lipoprotein particles to decorin-coated collagen Roles for both dimeric and monomeric forms of LPLJ Biol Chem2000275569470110681554
- PischonTGirmanCJSacksFMNon-high-density lipoprotein cholesterol and apolipoprotein B in the prediction of coronary heart disease in menCirculation200511233758316316964
- RanallettaMWangNHanSDecreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with Abcg1-/- bone marrowArterioscler Thromb Vasc Biol20062623081516917103
- RemaleyATRustSRosierMHuman ATP-binding cassette transporter 1 (ABC1): genomic organization and identification of the genetic defect in the original Tangier disease kindredProc Natl Acad Sci USA199996126859010535983
- RossRAtherosclerosis—an inflammatory diseaseN Engl J Med1999340115269887164
- RossRGlomsetJHarkerLResponse to injury and atherogenesisAm J Pathol19778667584842616
- RustSRosierMFunkeHTangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1Nat Genet199922352510431238
- RustaeusSStillemarkPLindbergKThe microsomal triglyceride transfer protein catalyzes the post-translational assembly of apolipoprotein B-100 very low density lipoprotein in McA-RH7777 cellsJ Biol Chem199827351962039478974
- SalterAMWigginsDSessionsVAThe intracellular triacylglycerol/fatty acid cycle: a comparison of its activity in hepatocytes which secrete exclusively apolipoprotein (apo) B100 very-low-density lipoprotein (VLDL) and in those which secrete predominantly apoB48 VLDLJ Biochem1998332Pt 366772
- SartipyPBondjersGHurt-CamejoEPhospholipase A2 type II binds to extracellular matrix biglycan: modulation of its activity on LDL by colocalization in glycosaminoglycan matrixesArterioscler Thromb Vasc Biol1998181934419848887
- SchroderMKaufmanRJThe Mammalian Unfolded Protein ResponseAnnu Rev Biochem2005747398915952902
- SegrestJPJonesMKDe LoofHStructure of apolipoprotein B-100 in low density lipoproteinsJ Lipid Res20014213466711518754
- ShelnessGSSellersJAVery-low-density lipoprotein assembly and secretionCurr Opin Lipidol200112151711264986
- SitiaRBraakmanIQuality control in the endoplasmic reticulum protein factoryNature2003426891414685249
- SkalenKGustafssonMRydbergEKSubendothelial retention of atherogenic lipoproteins in early atherosclerosisNature2002417750412066187
- SnidermanADApplying apoB to the diagnosis and therapy of the atherogenic dyslipoproteinemias: a clinical diagnostic algorithm Curr Opin Lipidol200415433815243216
- SnidermanADFurbergCDKeechAApolipoproteins versus lipids as indices of coronary risk and as targets for statin treatmentLancet20033617778012620753
- SpangAARF1 regulatory factors and COPI vesicle formationCurr Opin Cell Biol200214423712383792
- SteinbergDParthasarathySCarewTEBeyond cholesterol Modifications of low-density lipoprotein that increase its atherogenicityN Engl J Med1989320915242648148
- StillemarkPBorénJAnderssonMThe assembly and secretion of apolipoprotein-B48-containing very low density lipoproteins in McA-RH7777 cellsJ Biol Chem2000275105061310744742
- Stillemark-BilltonPBeckCBorenJRelation of the size and intracellular sorting of apoB to the formation of VLDL 1 and VLDL 2J Lipid Res2005461041415520448
- TaghibiglouCRashid-KolvearFVan IderstineSCHepatic very low density lipoprotein-ApoB overproduction is associated with attenuated hepatic insulin signaling and overexpression of protein-tyrosine phosphatase 1B in a fructose-fed hamster model of insulin resistanceJ Biol Chem200227779380311598116
- TaskinenMRDiabetic dyslipidaemia: from basic research to clinical practiceDiabetologia2003467334912774165
- TwiskJGillian-DanielDLTebonAThe role of the LDL receptor in apolipoprotein B secretionJ Clin Invest20001055213210683382
- WalldiusGJungnerIThe apoB/apoA-I ratio: a strong new risk factor for cardiovascular disease and a target for lipid-lowering therapy—a review of the evidenceJ Intern Med200625949351916629855
- VaughanAMOramJFABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDLJ Lipid Res20064724334316902247
- WeisgraberKHRallSCJrHuman apolipoprotein B100 heparin-binding sitesJ Biol Chem1987262110971033301850
- WigginsDGibbonsGFThe lipolysis/esterification cycle of hepatic triacylglycerol Its role in the secretion of very-low-density lipoprotein and its response to hormones and sulphonylureasJ Biochem199228445762
- VijayagopalPSrinivasanSRRadhakrishnamurthyBInteraction of serum lipoproteins and a proteoglycan from bovine aortaJ Biol Chem19812568234417263647
- WilliamsKJBrociaRWFisherEAThe unstirred water layer as a site of control of apolipoprotein B secretionJ Biol Chem19902651674142170353
- WilliamsKJTabasIThe response-to-retention hypothesis of early atherogenesisArterioscler Thromb Vasc Biol199515551617749869
- WilliamsKJTabasIThe response-to-retention hypothesis of atherogenesis reinforcedCurr Opin Lipidol1998947149812202
- WillnerELTowBBuhmanKKDeficiency of acyl CoA:cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein E-deficient miceProc Natl Acad Sci USA20031001262712538880
- YokoyamaSAssembly of high density lipoprotein by the ABCA1/apolipoprotein pathwayCurr Opin Lipidol2005162697915891387
- YusufSHawkenSOunpuuSEffect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control studyLancet20043649375215364185