Abstract
The dual endothelin receptor antagonist, bosentan, is an orally active therapy, which is effective in the treatment of pulmonary arterial hypertension (PAH). This review critically appraises the evidence for the efficacy of bosentan in idiopathic and familial PAH, in PAH associated with connective tissue disease and in PAH which may develop in association with other conditions. Data from the pivotal placebo controlled studies and their open labeled extensions as well as long term survival and quality of life data is presented. Data is also presented on the potential benefit of bosentan in patients with inoperable chronic thromboembolic pulmonary hypertension. The safety and tolerability of bosentan as well as drug interactions are discussed. Dosage recommendations in adults and pediatrics are presented. An algorithm is provided to guide the reader in monitoring potential increases in alanine and aspartate transaminase levels that may occur with bosentan use and the dose adjustments that are recommended as a result of any increase in the levels of these enzymes are shown. Finally, the role of bosentan as part of combination therapy in PAH is examined.
Introduction
Pulmonary arterial hypertension (PAH) is a condition characterized by dyspnea, fatigue, chest pain and syncope (CitationGaine and Rubin 1998; CitationRuno and Loyd 2003; CitationGalie 2004; CitationRubin 2004). PAH results from narrowing of the small arteries and arterioles resulting in elevation of pulmonary vascular resistance (PVR), which if left untreated, results in the development of right ventricular failure and death.
The pathogenesis that underlies PAH has become better understood. Central to the development of PAH is pulmonary vascular endothelial cell dysfunction. The role of endothelin as a central mediator in the development of PAH has been demonstrated and this has resulted in the development of endothelin receptor antagonists (ERAs), which have favorably impacted on symptoms, exercise capacity and prognosis for patients with PAH (CitationHumbert, Morrell et al 2004).
Bosentan, a dual endothelin receptor antagonist, is the first ERA to be used successfully in the treatment of PAH. This article reviews the evidence for the use of bosentan in PAH including it’s efficacy in various types of PAH, its potential toxicity, and associated drug interactions.
Definition and classification
Pulmonary hypertension is defined as a sustained elevation of mean pulmonary artery pressure to a level greater than 25 mmHg at rest or greater than 30 mmHg during exercise (CitationRuno and Loyd 2003; CitationGalie 2004; CitationRubin 2004).
Previously, pulmonary hypertension was defined as primary (PPH) or secondary (CitationGaine and Rubin 1998). More recently, a series of World Health Organization sponsored expert conferences suggested replacing this simplistic categorization with a new classification () (CitationSimonneau et al 2004). This classification distinguishes pulmonary hypertension via a combination of the underlying mechanisms and etiology, and is more useful when considering the natural history, prognosis and potential therapies of this disorder. Most of the therapies that have been utilized in pulmonary hypertension have been found to be efficacious in group 1 (PAH) of this classification. In this classification, the term primary pulmonary hypertension has been replaced by the terms idiopathic pulmonary arterial hypertension and familial pulmonary arterial hypertension. Patients with PAH (Group 1) have evidence of sustained elevation in pulmonary pressure and additionally have a normal pulmonary artery wedge pressure <15 mmHg. Additionally, this group excludes patients where the underlying cause of the elevated pulmonary pressure is felt to be related to hypoxemic lung disease (Group 3), thromboembolic disease of the pulmonary vasculature (Group 4), or miscellaneous other causes (Group 5).
Table 1 WHO Classifi cation of pulmonary hypertension (abridged) (CitationHumbert, Morrell et al 2004)
Pulmonary hypertension in clinical practice most commonly arises as a result of left heart disease (Group 2). It is important to note that the therapies discussed in this article have not been found to be helpful (and are potentially deleterious) in this group of patients. Pulmonary hypertension may arise from chronic thromboembolic disease (CTEPH) (CitationMoser et al 1990). This condition needs to be excluded when a diagnosis of PAH is being considered because treatment of this condition is predominantly surgical, although medical therapy may be effective in selected cases (CitationBresser et al 2004; CitationBonderman et al 2005; CitationHoeper et al 2005; CitationRubin et al 2006).
Pulmonary hypertension can occur in association with lung disease and/or hypoxemia. In these conditions treatment is directed to the underlying cause and correction of the hypoxemia although it may yet transpire that the PAH specific therapies discussed below may have a role in these patients (CitationJones et al 1989; CitationStrange 2005).
Pathogenesis and rationale for PAH therapies
A detailed discussion of the pathogenesis of PAH is beyond the scope of this article and readers are referred elsewhere (CitationVoelkel et al 1997; CitationHumbert, Morrell et al 2004). In summary, established PAH is characterized by constriction of the pulmonary arterioles resulting predominantly from vascular remodeling, with variable contributions from vasoconstriction and thrombosis. The role of the characteristic plexiform lesions in the pathogenesis and natural history of PAH is unclear.
Within the pulmonary arterioles, the main cellular changes involve the endothelium, platelets, smooth muscle cells and adventitial fibroblast. PAH arises from a complex interplay of molecular and genetic abnormalities resulting in these cellular changes (CitationHumbert, Morrell et al 2004).
Endothelial cell injury and dysfunction is central to the development of the abnormalities seen in PAH, and this results in an imbalance in vaso-active mediators normally produced by the endothelium. Central to the development of PAH is reduced production of prostacyclin (PGI2), nitric oxide (NO) and vasoactive intestinal peptide (VIP), with upregulation of endothelin 1 (ET-1) (CitationHumbert, Sitbon et al 2004). The currently available PAH therapies act at least in part, by helping redress this imbalance.
Epidemiology and genetics
The incidence and prevalence of idiopathic pulmonary arterial hypertension is uncertain. The reported incidence appears to lie somewhere between 1 and 2 per million per year (CitationRubin 2004) to as high as between 2.5 and 4 per million per year (CitationStewart et al 2007). The incidence of PAH is higher in certain families. Familial PAH accounts for between 6%-10% (CitationNewman et al 2001) of all patients.
Underlying the development of both familial and idiopathic PAH are genetic mutations in the TGF-beta receptor families. Mutations in the gene encoding bone morphogenetic protein receptor type 2 (BMPR - II) have been identified as being important in the development of PAH in most, if not all, patients with familial PAH (CitationLane et al 2000) as well as between 10% and 26% of patients with idiopathic PAH (CitationLane et al 2000).
PAH can also be seen in association with a variety of other conditions as described in Group 1 of the World Health Organization Venice classification (). These associated conditions include connective tissue disease and especially systemic sclerosis (particularly limited cutaneous scleroderma), portopulmonary hypertension, certain drugs and toxins, HIV infection, and congenital systemic to pulmonary shunts (Eisenmenger syndrome). It has become increasingly recognized that other conditions, especially certain hematologic, genetic and metabolic disorders are also associated with the development of PAH. The role of genetic mutations in these associated causes of PAH remain unclear.
Nonetheless, it is evident that one genetic mutation and/or one environmental stimulus cannot explain all forms of PAH. Other candidate genes that are considered relevant to the development of PAH include genes encoding regulating factors in the serotonin pathway as well as other genes related to TGF-beta signaling pathways.
What is clear is that PAH arises from an interplay between multiple genetic and environmental factors. This multi-hit hypothesis (CitationFarber and Loscalzo 2004) suggests that the development of PAH requires primary genetic abnormalities, which when influenced by environmental stimuli lead to the development of clinically evident PAH.
Role of endothelin
One of the most important abnormalities in PAH is an over expression of endothelin-1 (ET-1). This results in abnormally high concentrations in pulmonary arteries (CitationGiaid et al 1993) and an increase in circulating ET-1 (CitationHumbert, Morrell et al 2004; CitationRubin 2004). Upregulation of ET-1 contributes to smooth muscle vasoconstriction and hypertrophy as well as fibrosis and inflammation. This is important in both the development of pulmonary arteriolar constriction and the secondary right ventricular hypertrophy which is seen in PAH. Circulating and pulmonary ET-1 concentrations are strongly correlated with disease severity and prognosis in PAH (CitationGiaid et al 1993; CitationGalie et al 1996; CitationHumbert, Morrell et al 2004; CitationRubin 2004).
Synthesis of ET-1 arises from the cleavage of a 212 residue precursor peptide known as pre pro ET-1, first to the 38 amino acid big ET-1, then by endothelin converting enzyme to the active 21 amino acid isoform.
ET-1 acts upon two receptor sub types, ETA and ETB. These receptors mediate the physiological and pathological effects of ET-1. ETA receptors are found predominantly on vascular smooth muscle cells and induce vasoconstriction. ETB receptors also appear on vascular smooth muscle cells where they stimulate both vasoconstriction and smooth muscle hyperplasia (CitationClozel et al 1992; CitationNeylon et al 1994). However, ETB receptors predominate on endothelial cells where they stimulate the release of vasodilating and anti-proliferative mediators such as prostacyclin (CitationLevin 1995). Under physiological conditions the predominant effect of the ETB receptor is a vasodilatory response (CitationLevin 1995). However, in a pathological state such as PAH, the predominant effect of stimulation of the ETB receptors is that of vasoconstriction and vascular smooth muscle proliferation (CitationBenigni and Ramuzzi 1999). Both ETA and ETB receptors appear to be important in mediating the response to endothelin of fibrosis, vasoconstriction and inflammation (CitationKatwa et al 1993; CitationFilep et al 1995). Furthermore, stimulation of both ETA and ETB receptors by endothelin is important in the myocardial hypertrophy and myocardial fibrosis which is seen in PAH (CitationLevin 1995).
Given the central role of upregulated ET-1 in the pathogenesis of PAH, the role of ET receptor antagonists in PAH received greater attention. The best studied of these endothelin receptor antagonists is bosentan, a dual ETA and ETB receptor antagonist. More recently, the ETA selective receptor antagonists sitaxsentan and ambrisentan have undergone clinical trials in PAH. The relative benefits of dual ETA and ETB receptor antagonism versus selective ETA receptor antagonism remains unclear. Preliminary data suggests that ETA selective antagonism may be as effective, at least in idiopathic PAH and scleroderma associated PAH as dual ETA and ETB receptor antagonism (CitationBenedict 2007).
Treatment of PAH
Conventional therapy for PAH
General measures including management of underlying or contributing factors, avoidance of pregnancy, early treatment of intercurrent respiratory infections with antibiotics and the use of influenza and anti-pneumococcal vaccines are recommended (CitationGalie 2004; CitationRubin 2004). Oxygen is given for hypoxemia and diuretics are efficacious in the treatment of congestive heart failure. Digoxin may be of benefit, especially if the patient is in atrial fibrillation. Non steroidal anti inflammatory medications and decongestant tablets containing pseudoephedrine are usually avoided (CitationGalie 2004; CitationRubin 2004).
Anticoagulation with warfarin is used in most of the patients based on single center, retrospective studies (CitationFuster et al 1984; CitationRich et al 1992).
Improved survival has been seen with high dose calcium channel blockers in the 5%-10% of patients with idiopathic PAH who have demonstrated acute reactivity in response to pulmonary vasodilator testing. The use of high dose calcium channel blockers is indicated in patients with WHO functional class II to III who display acute reactivity and remain stable (CitationGalie et al 2004).
Specific PAH therapies
In recent years, the development of specific PAH therapies has increased the options available to the clinician managing a patient with PAH. There are four main classes acting upon three main intracellular pathways (, ) (CitationHumbert, Sitbon et al 2004).
Figure 1 Consequences of endothelial dysfunction on pulmonary vascular smooth muscle showing potential targets for therapy. Three major pathways and associated therapeutic targets in abnormal proliferation and contraction of smooth muscle cells are shown. Dysfunctional endothelial cells have decreased production of prostacyclin and endogenous nitric oxide and increased production of endothelin-1. This imbalance of mediators along with decreased production of vasoactive intestinal peptide (VIP) results in a condition favouring vasoconstriction and proliferation of pulmonary artery smooth muscle cells. In addition to their actions on smooth muscle, these mediators have other properties including antiplatelet effects of nitric oxide and prostacyclin and profibrotic and proinflammatory effects of endothelin. Plus signs denote an increase in intracellular concentration: minus signs reflect blockage of a receptor, inhibition of an enzyme or a decrease in the intracellular concentration. (Modified and published with permission from CitationHumbert, Sitbon et al (2004)).
Abbreviations: cGMP cyclic guanosine monophosphate; cAMP, cyclic adenosine monophosphate.
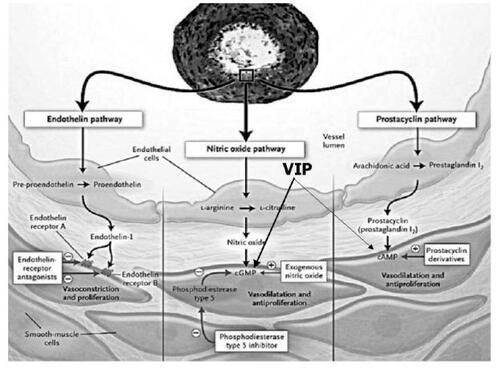
Table 2 Specific PAH therapies and method of delivery
These currently available specific PAH therapies aim to redress the imbalance in mediators which occur as a result of endothelial cell dysfunction. This imbalance consisting of reduced production of prostacyclin and nitric oxide and upregulation of ET-1 resulting in the abnormal proliferation and contraction of pulmonary smooth muscle cells via three pathways, all of which are potential targets for therapy ( and ) (CitationHumbert, Sitbon et al 2004).
Intravenous epoprostenol (prostacyclin) was the first effective agent to be used for the treatment of PAH, and to date remains the only agent that has been shown to improve survival in a randomized controlled trial of patients with (idiopathic) PAH (CitationBarst et al 1996). Intravenous epoprostenol is limited by the need for a continuous intravenous infusion pump with the potential problem of line sepsis. Further, it is limited by side effects arising from the direct effect of the medication including jaw pain, headaches, flushing and gastrointestinal disturbance.
The prostacyclin analogues treprostinil (which can be given either intravenously or by subcutaneous infusion) and iloprost (which can be given either through the inhaled route or intravenously), have also been shown to improve exercise capacity and WHO functional class (CitationOlschewski et al 2002; CitationSimonneau et al 2002). The current recommendation is that where there are alternatives, the oral prostacyclin analogue, beraprost, should be avoided as it is ineffective in the medium to long term (CitationGalie 2004).
The phosphodiesterase-5 (PDE-5) inhibitor sildenafil has been associated with improvements in WHO functional class, hemodynamics and exercise capacity in a placebo controlled trial (CitationGalie et al 2005). The authors are aware that some clinicians have used the PDE-5 inhibitor, tadalafil, in selected patients due to availability and cost constraints but as yet there is no published data of its long term effectiveness.
Inhaled nitric oxide is efficacious in lowering pulmonary pressures and improving hemodynamics in ventilated patients (CitationZapol et al 1994) but is generally limited to the intensive care unit setting and generally necessitates positive pressure ventilation. Prolonged nitric oxide administration can induce methaemoglobinemia and platelet dysfunction.
Bosentan therapy in PAH
Bosentan was the first endothelin receptor antagonist approved for the use of PAH.
Clinical studies have shown that in PAH, the use of bosentan is associated with improved exercise capacity, WHO functional class, cardiopulmonary hemodynamics, quality of life and delayed time to clinical worsening when compared to placebo. Further, long term studies have demonstrated improved survival with the use of bosentan when compared to historical controls although there is no placebo controlled data confirming a survival benefit ().
Table 3 Impact of bosentan therapy in patients with idiopathic pulmonary arterial hypertension
Placebo-controlled pivotal studies and open labeled extension
In the first placebo controlled study of bosentan in PAH (CitationChannick et al 2001) Channick and co-authors reported on 32 patients with either idiopathic PAH or scleroderma associated PAH who were randomly assigned to receive bosentan (62.5 mg taken twice daily for four weeks then 125 mg twice daily) or placebo for a minimum of 12-weeks. The primary end point was change in exercise capacity. Secondary end points included change in cardiopulmonary hemodynamics, BORG dyspnea index, WHO functional class and withdrawal due to clinical worsening.
In patients given bosentan, the distance walked in 6 minutes improved by 70 meters after 12-weeks compared with baseline where as in those on placebo it worsened by 6 meters (treatment effect 76 m, p = 0.021). There was also a significant improvement in cardiac index and reduction in pulmonary vascular resistance. Further, patients who received bosentan had improved WHO functional class and reduced BORG dyspnea index. Only 3 patients withdrew from the study from clinical worsening and all were within the placebo group (p = 0.033).
In 2002, Rubin and co-authors reported the results of a much larger placebo controlled study (CitationRubin et al 2002). In this double blind study, 213 patients with either idiopathic PAH or PAH associated with connective tissue disease received either placebo or bosentan with an initial dose of 62.5 mg twice daily for 4-weeks followed by either of 2 doses of bosentan (125 or 250 mg twice daily) for a minimum of 12-weeks.
By week 16, the treatment effect of bosentan (improvement in 6 minute walking distance in bosentan group minus improvement in 6 minute walk distance in placebo group) was 44 meters (p < 0.001). Bosentan also improved the WHO functional class and delayed time to clinical worsening.
85 of the 213 patients enrolled in this study were included in an echocardiographic sub study (CitationGalie et al 2003). At baseline, patients had echocardiographic findings consistent with severe PAH. Treatment effects of bosentan compared with placebo after 16 weeks showed improvements in the echocardiographic measures of cardiac index, right ventricular function and right ventricular size and function and reduced frequency of pericardial effusion. Further, left ventricular early diastolic filling was improved in the bosentan group suggesting reduced compression of the left ventricle by a dilated and hypertrophied right ventricle and/or better flow to pulmonary veins secondary to reduced pulmonary pressure allowing earlier and better filling characteristics.
In all three of these studies (CitationChannick et al 2001; CitationRubin et al 2002; CitationGalie et al 2003) patients were in WHO functional class III or IV at baseline.
29 of the 32 patients enrolled in the original study (CitationChannick et al 2001) received bosentan at a dose of 125 mg twice daily in an open labeled extension for an additional year. This one year follow up study (CitationSitbon et al 2003) found that improvements in 6 minute walk distance that were achieved with bosentan in the original study were maintained. 41.4% of patients improved their NYHA functional class, whereas only 1 patient had a deterioration in functional class. Further, 11 patients underwent repeat right heart catheterization between 8 and 22-months after commencement of treatment with bosentan. For these patients, cardiac index and PVR was significantly improved compared to baseline. There was also a small reduction in mean pulmonary artery pressure, which did not reach statistical significance.
Effect of bosentan on survival in idiopathic PAH
None of the placebo-controlled trials of bosentan in PAH were powered to show survival benefit. Therefore the only information available regarding survival benefit from bosentan comes from comparisons with historical controls and National Institute of Health (NIH) registry data.
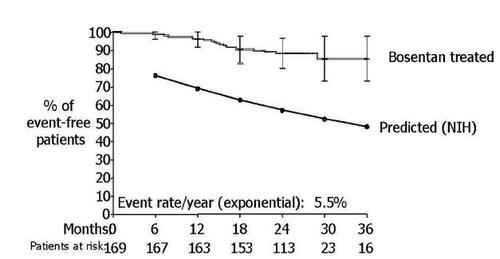
In 2005, CitationMcLaughlin and colleagues (2005) reported the first long term survival data with bosentan in patients with idiopathic PAH. All patients were in WHO functional class III or IV at baseline. The authors reported on 169 patients with idiopathic PAH who received bosentan as first line therapy for their disease. After 12 and 24-months of follow up, 85% and 70% of patients, respectively remained on bosentan monotherapy. Another 7% continued bosentan while receiving additional therapies.
The Kaplan-Meier survival estimate at two years was 89% in this study compared to the predicted two years survival (based on historical cohorts) of 57%. At each six month interval, observed survival was significantly better than predicted. Overall, patients receiving first line bosentan therapy had a 5.5% annual death rate ().
Figure 2 Survival in adult patients with idiopathic pulmonary arterial hypertension treated with first line bosentan therapy compared to predicted survival with conventional therapy according to NIH registry. (Reproduced with permission from CitationMcLaughlin et al (2005)).
In 2005, CitationSitbon and colleagues (2005) compared the survival of 139 patients with idiopathic PAH in WHO functional class III who received bosentan as first line therapy with historical records of patients who received intravenous epoprostenol as first line therapy. After correcting for differences in severity at baseline, there was no significant difference in one and two year survival between the two groups. This is particularly important because intravenous epoprostenol therapy is the only treatment that has been shown to improve survival in PAH in a prospective placebo controlled study (CitationBarst et al 1996).
In a large single center study, CitationProvencher and colleagues (2006) examined the survival of 103 consecutive patients with NYHA functional class III or IV idiopathic PAH who were treated first line with bosentan. Bosentan was prescribed at a dose of 62.5 mg twice daily for 4-weeks followed by 125 mg twice daily. The mean duration of follow up was 24 ± 15-months and overall survival estimates were 92, 89 and 79% at 1, 2 and 3-years respectively. The predicted survival at 1, 2 and 3-years, based on the equation derived from the NIH registry (CitationD’Alonzo et al 1991) was 71, 61 and 51% respectively. Intravenous epoprostenol was added in 30 patients and inhaled iloprost in an additional 6 patients following a mean duration of exposure of bosentan of 12 ± 10-months. The reasons for the initiation of additional therapy included worsening of NYHA functional class on treatment, a significant fall in 6 minute walk distance or failure of significant improvement in cardiac index.
Bosentan in connective tissue disease associated PAH
PAH can complicate all forms of connective tissue disease. Most commonly it complicates systemic sclerosis (scleroderma) especially the limited cutaneous subtype. Depending on the criteria used, PAH can complicate up to 25% of patients with limited cutaneous scleroderma (CitationHoeper 2002). However, PAH can also complicate SLE, mixed connective tissue disease and less commonly, rheumatoid arthritis (CitationHoeper 2002).
Overall, the results with bosentan (or any other specific PAH therapy) have not been as impressive with scleroderma associated PAH when compared to idiopathic PAH. This most probably reflects a different pathogenesis, older age of patients and that these patients tend to present later in their disease course (CitationHoeper 2002). As a result many groups are recommending that all patients with scleroderma regardless of symptomatology be screened for the presence of PAH by serial echocardiography and lung function (CitationBlack 2005).
CitationDenton and colleagues (2006) analyzed the results of bosentan therapy for PAH related to connective tissue disease from the two placebo-controlled clinical trials and their open labeled extensions. 66 patients with connective tissue disease associated PAH, in WHO functional class III or IV were randomized to participate in the pivotal placebo controlled studies (CitationChannick et al 2001; CitationRubin et al 2002). 44 patients received bosentan and 22 patients received placebo. Baseline characteristics suggested that the group who received bosentan had more severe disease with a lower 6 minute walk distance and higher PVR at baseline. There was a treatment effect of 22 meters favoring bosentan compared with placebo (not significant) although time to clinical worsening was delayed by bosentan, suggesting slower disease progression.
64 of these original 66 patients continued in an open labeled extension. 40 of these patients remained on bosentan monotherapy, one received prostanoids in addition to bosentan and in 23 patients, bosentan was ceased. The mean duration of bosentan therapy in this group of patients was 1.8 ± 0.8-years. Survival was 85.9% at one year and 73.4% at two years and of the 40 patients on bosentan monotherapy, 25% improved in WHO functional class by end of treatment.
CitationWilliams and co-authors (2006) reviewed the data of 45 patients in a routine clinical setting with scleroderma associated PAH who received bosentan monotherapy. Survival in this group was 81% at one year and 71% at two years. This was similar to the survival reported by CitationGirgis and colleagues (2005) who found an 87% one year and 79% two year survival for 17 patients with scleroderma associated PAH who received bosentan as first line therapy.
Effect of bosentan on quality of life of patients with PAH
In an uncontrolled Australian study bosentan monotherapy was associated with significant improvements in quality of life, as measured by the SF-36 questionnaire, which persisted for 12-months (CitationKeogh et al 2007). Patients with idiopathic or familial PAH as well as those with connective tissue disease were included in this study. All patients were in WHO functional class III or IV at baseline and had not had previous treatment with specific PAH therapies. Analysis of a sub group of this study showed that bosentan also improved six minute walk distance over a 12-month period and that quality of life and six minute walk distance were well correlated although improvements in quality of life appeared to be a more sensitive indicator of response to treatment than changes in six minute walk distance (CitationGabbay et al 2005).
Recommendations for role of bosentan monotherapy in PAH
The data from the studies in idiopathic, familial and Scleroderma-associated PAH has led to the recommendation that bosentan monotherapy is indicated for patients in WHO functional class III in these conditions (CitationGalie 2004). Bosentan also appears to be effective in patients in WHO functional class IV but it remains unclear if it is a preferred first line therapy compared to intravenous epoprostenol, where there appears to be more evidence for intravenous epoprostenol therapy (CitationGalie 2004). In patients with idiopathic or familial PAH who present in WHO class IV with evidence of right ventricular dysfunction, the authors and others recommend that intravenous epoprostenol be considered as first line therapy (CitationGalie 2004).
In those patients who are stabilized on intravenous epoprostenol, especially at low doses, it may be possible to switch them to bosentan monotherapy although this needs to be done with a very slow reduction of epoprostenol dose and concomitant monitoring (CitationSteiner et al 2006).
Bosentan has been shown to be effective in a placebo- controlled trial in patients in WHO functional class II (EARLY Study - Actelion press release). However, at the time of writing this study is unpublished and regulatory authorities in most countries have not approved bosentan for use in this class of patients. Therefore its use would be limited by the current prohibitive costs for most individuals in WHO class II (see discussion on cost-effectiveness and availability).
The role of bosentan as part of combination therapy in PAH is discussed later in this article.
Bosentan in other forms of associated PAH
Most of the clinical studies of bosentan in PAH have concentrated on patients with idiopathic and familial PAH as well as those with PAH in association with connective tissue disease especially scleroderma. However, there is also evidence of the efficacy of bosentan in PAH associated with HIV infection and PAH associated with congenital heart disease as well as in the pediatric population ().
Table 4 Impact of bosentan therapy in pulmonary arterial hypertension (PAH) subgroups
In a study on the effects of bosentan on human immunodeficiency virus associated PAH, CitationSitbon and colleagues (2004) described the results of bosentan therapy given for 16-weeks in 16 patients. In patients with HIV infection, the HIV-1 envelope glycoprotein 120 may stimulate the production of endothelin. Endothelin levels are significantly elevated in patients with vascular complications of HIV infection. The authors postulated that endothelin may contribute to the pathogenesis of vasculopathies associated with HIV infection, including PAH. In this open labeled uncontrolled study the authors found that 6 minute walk distance improved by 91 meters (p < 0.001) and there were significant improvements in NYHA functional class, cardiopulmonary hemodynamics, Doppler echocardiographic variables and quality of life. During the study, no patient died and none required other therapies towards their PAH. Furthermore, bosentan did not have a negative impact on control of HIV infection, nor did its use significantly interact with anti-retroviral therapy.
Congenital heart disease characterized by the development of systemic to pulmonary shunting (eg, atrial septal defect, ventricular septal defect, patent ductus arteriosus) may lead to the development of PAH and consequent intracardiac right to left shunting (Eisenmenger syndrome). Galie and co-authors (CitationGalie et al 2006) reported the results of a 16-week double blind placebo controlled study evaluating the effect of bosentan in Eisenmenger syndrome. 54 patients were randomized to receive either bosentan (n = 37) or placebo (n = 17). Despite theoretical concerns that vasodilator therapy may reduce systemic blood pressure more than pulmonary pressures and hence worsen right to left shunt, bosentan did not worsen oxygen saturations. The treatment effect of bosentan when compared to placebo was an increase in six minute walk distance of 53.1 m (p = 0.0079) and a significant fall in PVR and mean pulmonary artery pressure.
In an open label study, Kotlyar and co-authors (CitationKotlyar et al 2006) evaluated the effects of bosentan in 23 patients with Eisenmenger syndrome. There was a mean duration of follow up of 15 ± 10-months, 57% of the patients had improved by at least one functional class (p = 0.016) and mean oxygen saturation at rest increased from 81% to 84% (p = 0.001). Overall, the six minute walk distance did not change from the baseline of 335 m.
Pulmonary hypertension may complicate chronic thromboembolic disease. Although chronic thromboembolic pulmonary hypertension (CTEPH) is a different disease to idiopathic PAH, with different underlying mechanisms, some similarities in the pathogenesis and pathology of these conditions exist. Pulmonary endarterectomy (PEA) surgery is the treatment of choice for patients with severe CTEPH and appropriate disease distribution (CitationJamieson et al 2003). However, a substantial proportion of patients with CTEPH are considered inoperable due to significant distal thromboembolic pathology or due to concomitant morbidity. It has been postulated that in patients with CTEPH who can not undergo PEA or in whom pulmonary hypertension persists following surgery, then specific PAH therapies may be effective.
A summary of the effects of bosentan in either inoperable CTEPH or persistent pulmonary hypertension after PEA is shown in . In summary, bosentan therapy for a treatment duration of up to three years has been associated in these open labeled studies with improvements in NYHA functional class, exercise capacity and cardiopulmonary hemodynamics (CitationBonderman et al 2005; CitationHoeper et al 2005; CitationHughes et al 2005, Citation2006). Further, an open labeled study by CitationMusk and colleagues (2006) found that treatment with Bosentan in CTEPH was associated with improvements in exercise capacity and cardiopulmonary hemodynamics that were similar to the improvements seen in a similarly treated group of patients with idiopathic PAH and with a trend to greater improvements than those seen with scleroderma associated PAH in the same center.
Table 5 Impact of bosentan therapy in chronic thromboembolic pulmonary hypertension (CTEPH)
A multi center randomized trial in patients with inoperable CTEPH (BENEFIT), which includes a four month bosentan/placebo controlled phase, found that bosentan was associated with reduced PVR and prolonged time to clinical worsening when compared to placebo but there was no difference in exercise capacity (as measured by 6 minute walk test) in the two groups (CitationJais et al 2007). The results from the open labeled extension are awaited to help determine if exercise capacity improves in the longer term and this may help to clarify the role of bosentan in CTEPH.
Pediatrics
The data on efficacy and safety of bosentan in pediatric patients with PAH is not as extensive as that in adults. CitationBarst and colleagues (2003) describe three different dosing regimes of bosentan in 19 pediatric patients stratified for body weight and epoprostenol use. They found that the pharmacokinetics of bosentan in pediatric patients with PAH and healthy adults were similar and the treatment with bosentan resulted in hemodynamic improvement. CitationRosenzweig and colleagues (2005) described the use of bosentan in 86 children with either idiopathic PAH or PAH associated with congenital heart disease or connective tissue disease. Median exposure to bosentan was 14-months. In 46% of the patients, WHO functional class improved and there was a significant fall in mean pulmonary artery pressure and PVR. Kaplan-Meier survival estimates at one and two years were 98% and 91% respectively. The authors concluded that bosentan, with or without concomitant epoprostenol therapy, is safe and efficacious for the treatment of PAH in children.
Dose of bosentan in treatment of PAH
The large placebo controlled study of two bosentan dosing regimes in adult patients with PAH (CitationRubin et al 2002) found that the dose of bosentan of 62.5 mg twice daily for 4-weeks followed by 125 mg twice daily was associated with equivalent efficacy as the higher dose of 250 mg twice daily with fewer side effects. Since that study, the dose of bosentan was 62.5 mg twice daily for 4-weeks followed by 125 mg twice daily, and this dosing regimen has been considered standard and hence been used in all future studies of adults with Bosentan in PAH. The dose of bosentan in children is dependent upon age and weight and has been described by CitationBarst et al (2003) and CitationRosenzweig et al (2005).
Recommended dosing regimens for adults and children are summarized in .
Table 6 Recommended dosing regimens for bosentan in adults and children
Safety and tolerability
The most significant adverse event in patients on bosentan treatment is the potential development of abnormal hepatic function and specifically a rise in hepatic amino transaminases. The basis of this is uncertain but it has been postulated that it is related to bosentan interfering with biliary salt excretion. The incidence of abnormal increase in hepatic amino transaminase levels to greater than three times normal varies from 9.7% in the largest placebo controlled study (although there was only a 4% incidence in the group receiving 125 mg twice daily of bosentan) (CitationRubin et al 2002) to as high as 14.9% in the longer observational studies (CitationMcLaughlin et al 2005).
Post marketing surveillance with Bosentan through the internet based (Trax) system has observed the use of bosentan in 4994 patients enrolled between May 2002 and November 2004. Median exposure over the two year observation period was approximately 35-weeks in patients with idiopathic PAH. The frequency of abnormal elevations in hepatic transaminase (ALT/AST) was approximately 8.4%. These elevations are reversible on stopping bosentan. Liver enzyme changes typically occur within the first six weeks of treatment initiation although it appears that they can occur at any time after the initiation of bosentan treatment.
As a result, liver amino transaminase levels must be measured prior to initiation of treatment and monthly thereafter. If elevated amino transaminase levels are seen, then changes in the monitoring and treatment of these patients are made according to a well defined protocol (). If liver amino transaminase elevations are accompanied by clinical symptoms of liver injury or an elevation in bilirubin greater than or equal to two times the upper limit of normal then treatment should be stopped.
Table 7 Monitoring and management of elevated liver enzymes for patients treated with bosentan
Bosentan treatment is associated with a fall in hemoglobin and hematocrit in up to 10% of patients. It is recommended that hemoglobin concentrations be checked after one and three months and every three months thereafter. The cause of this reduced hemoglobin with bosentan is unclear but in part may be related to dilutional effects as a result of fluid retention, in part as a result of the effects of ETB receptor blockade in the renal glomerulus.
Bosentan is teratogenic in rats and is contraindicated in pregnancy. It is not recommended to be used by lactating women. Other side effects such as headaches, flushing and syncope may occur due to the effects of bosentan on the systemic vasculature and associated systemic vasodilatation.
There is limited data on the safety of suddenly stopping bosentan. In an open labeled Australian study (CitationKeogh et al 2007), 2% of patients died within 1-month of stopping bosentan. These were patients who were deteriorating despite therapy and it is unclear if their death reflected the natural history of the disease rather than due to bosentan withdrawal.
Drug interactions
Bosentan is an inducer of the cytochrome P450 isoenzymes CYP2C9 and CYP3A4. Consequently, plasma concentrations of drugs metabolized by these isoenzymes will be decreased when bosentan is co-administered. Because estrogens and progestogens are partially metabolized by these isoenzymes, there is a possibility of failure of contraception when bosentan is co-administered with oral contraceptive medication.The use of bosentan when co-administered with warfarin may be associated with reduced plasma concentrations of warfarin. In the large placebo controlled study (CitationRubin et al 2002) the frequency of changes in warfarin dose was similar amongst bosentan and placebo treated patients. However, personal experience suggests that the warfarin dose often needs to be increased when bosentan is co-administered. At the very least we would recommend intensified monitoring of international normalized ratio (INR) for patients on warfarin during bosentan initiation and up titration periods.
Co-administration of bosentan and cyclosporin or tacrolimus is contraindicated because of significant reduction in the effectiveness of these immunosuppressive agents. Likewise concomitant glibenclamide (glyburide in the United States) administration is contraindicated both because of an increased risk of elevated hepatic amino transaminase levels as well as a significant increase in glibenclamide concentration during concomitant use.
Bosentan as part of combination therapy
The newer specific PAH therapies such as bosentan have resulted in significant improvements in exercise capacity, cardiopulmonary hemodynamics and survival. However, responses are variable and in many patients, disease progresses despite therapy. As a result an increasing number of patients are being considered for combination therapy (CitationHoeper and Dinh-Xuan 2004).
Current therapies in the treatment of PAH act on the three intracellular pathways, endothelin, nitric oxide and prostacyclin, known to be abnormal in PAH (). A logical extension therefore is to use a combination of two or more therapies each acting in synergy through a different pathway (CitationHoeper and Dinh-Xuan 2004).
The combination of bosentan and sildenafil is of particular interest because both are oral agents, which are generally well tolerated. CitationHoeper and colleagues (2004) reported the clinical course of 9 patients with severe idiopathic PAH, in whom bosentan caused transient clinical improvement, eventually followed by a decline in exercise tolerance, who then received adjunct treatment with sildenafil.
Bosentan was associated with an initial improvement in six minute walk test distance but this effect was not sustained. After an interval of 11 ± five months of bosentan treatment, sildenafil was added. Three months later, the six minute walk test distance had increased by a mean of 115 m and this improvement persisted for a median follow up of nine months (CitationHoeper et al 2004).
In a small German study, involving eleven patients with different forms of pulmonary hypertension, but including idiopathic PAH and congenital heart disease, Lunze and co-authors (CitationLunze et al 2006) found that the combination of sildenafil and bosentan was associated with improvements in exercise capacity and reduction in mean pulmonary artery pressure. They found that the combination was well tolerated although one patient died suddenly for reasons that are not evident in the paper.
There is a pharmacological interaction between bosentan and sildenafil in that bosentan decreases the plasma concentration of sildenafil and sildenafil increases the plasma concentration of bosentan when they are co-prescribed in PAH (CitationPaul et al 2005). However, this interaction does not appear to be associated with significant problems and the combination of bosentan and sildenafil does not appear to be associated with an increased risk of abnormal elevation of liver transaminases when compared to the use of bosentan alone (CitationHoeper, Kiely et al 2005).
Humbert and colleagues reported the results of a double blind placebo controlled prospective study in which 33 patients with PAH were commenced on intravenous epoprostenol treatment and were randomized to receive either bosentan or placebo. Hemodynamics, exercise capacity and functional class improved in both groups at week 16 and in the combination treatment group there was a trend for a greater (although non significant) improvement in all measured hemodynamic parameters. The authors concluded that this was an essentially negative study and that additional information was needed to evaluate the risk/benefit ratio of combined bosentan and epoprostenol therapy in PAH (CitationHumbert, Barst et al 2004).
CitationMcLaughlin and colleagues (2006) reported the results of a randomized multi center double blind trial in 67 patients with PAH (55% idiopathic PAH, 45% associated PAH, 94% NYHA class III), in which inhaled iloprost (5 mcgm) or placebo was added to stable monotherapy with bosentan for 12-weeks. There was a treatment effect of adding iloprost of an increase in six minute walk distance of 26 m (p = 0.051). The addition of iloprost was also associated with significant improvement in NYHA functional class and increased time to clinical worsening when compared with placebo. The authors concluded that within the limitations of the relatively small sample size, the addition of inhaled iloprost in patients with PAH with reduced exercise capacity on bosentan monotherapy was both safe and efficacious.
Goal oriented (combination) therapy
It has become increasingly clear that where it has been used, combination therapy with two or more agents has been associated with improvements in exercise capacity and functional class in patients with PAH. The question of which combination should be used and when remains unclear (CitationHoeper and Dinh-Xuan 2004).
Some authors have recommended adding increasing number of agents in combination, when specific treatment goals have not been met. Hoeper and colleagues have shown (CitationHoeper, Markevych et al 2005) that this goal oriented approach to therapy, based on achieving set functional criteria, has been associated with improved survival and less requirement for transplantation than in a historical cohort treated with monotherapy alone.
In summary, whilst ongoing studies are required in assessing bosentan as part of combination therapy it appears that such a strategy can be associated with improvements in exercise capacity, functional class and possibly survival in patients with PAH (CitationHoeper et al 2004; CitationHoeper and Dinh-Xuan 2004; CitationHumbert, Barst et al 2004; CitationHoeper, Kiely et al 2005; CitationHoeper, Markevych et al 2005; CitationPaul et al 2005; CitationLunze et al 2006; CitationMcLaughlin et al 2006).
Atrial septostomy and lung transplantation
Atrial septostomy and lung transplantation are potential lifesaving therapies, which are reserved for a small number of patients who are deemed likely to benefit from them and in whom the selective PAH therapies discussed above are ineffective. A detailed discussion of these therapies is beyond the scope of this article and the reader is referred elsewhere for a detailed discussion of surgical therapies in PAH including atrial septostomy and lung transplantation (CitationASTP et al 1998; CitationDoyle et al 2004).
Sitbon and colleagues have described the criteria that they use to categorize patients as failing to respond to 3-months of intravenous epoprostenol therapy (CitationSitbon et al 2002). These patients were then considered for lung transplantation. Although no such criteria have been described for bosentan, alone or in combination with other agents, the reader is referred to the general principles prescribed by Sitbon and others when considering timing for referral for transplantation (CitationASTP et al 1998; CitationSitbon et al 2002; CitationDoyle et al 2004).
Cost-effectiveness and access to specific PAH therapies
There is limited data on the cost-effectiveness of bosentan or other specific PAH therapies (CitationWlodarczyk et al 2006), despite the importance that health authorities appropriately place on such data. These drugs are generally expensive and availability is limited in most countries. Almost all of the patients in the previously described clinical trials were in WHO (or NYHA) class III or IV at enrolment. This may reflect the resistance of some health authorities to subsidise therapy for patients in other functional classes.
It is our view that part of the responsibility of health care professionals is to advocate for reduced costs and greater availability of these therapies especially in developing countries.
Summary
The dual endothelin receptor antagonist bosentan given at a dose (in adults) of 62.5 mg twice daily for four weeks followed by 125 mg twice daily is a safe and efficacious therapy in PAH. The use of bosentan as part of a comprehensive management plan has resulted in improvements in exercise capacity, functional class, quality of life and survival. Patients require regular monthly monitoring of liver function tests and clear guidelines are in place in terms of reducing or stopping bosentan therapy depending upon the results of these liver function tests. Because of bosentan’s ability to induce the cytochrome p450 family, patients must be advised to contact their treating physician before initiation of other prescription medicines including oral contaraceptives and antibiotics. Bosentan has been extensively used as monotherapy in PAH especially in patients with idiopathic PAH and scleroderma associated PAH but also appears to be efficacious in other forms of pulmonary hypertension including other connective tissue disease associated PAH, HIV associated PAH and Eisenmenger syndrome as well as in selected cases of chronic thromboembolic pulmonary hypertension. Bosentan may also have a role as part of combination therapy in patients who have responded sub optimally to monotherapy.
References
- ASTP, ATS, ERS, ISHLT joint statementInternational guidelines for the selection of lung transplant patientsAm J Respir Crit Care Med199815833599655748
- BarstRJIvyDDingemanseJPharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertensionClin Pharmacol Ther2003733728212709727
- BarstRJRubinLJLongWAA comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study GroupN Engl J Med19963342963028532025
- BenedictNJSitaxsentan in the management of pulmonary arterial hypertensionAm J Health-System Pharmacy2007643638
- BenigniARamuzziGEndothelin antagonistsLancet1999353133810023915
- BlackCPulmonary arterial hypertension: are we doing enough to identify systemic sclerosis patients at high risk of this rare condition?Rheumatology (Oxford)200544141215681336
- BondermanDNowotnyRSkoro-SajerNBosentan Therapy for Inoperable Chronic Thromboembolic Pulmonary HypertensionChest2005128259960316236930
- BresserPFedulloPFAugerWRContinuous intravenous epoprostenol for chronic thromboembolic pulmonary hypertensionEur Respir J20042359560015083760
- ChannickRNSimonneauGSitbonOEffects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled studyLancet200135811192311597664
- ClozelMGrayGABreuVThe endothelin ETB receptor mediates both vasodilation and vasoconstriction in vivoBiochem Biophys Res Commun1992186867731323294
- D’AlonzoGEBarstRJAyresSM1991Survival in patients with primary pulmonary hypertension. Results from a national prospective registryAnn Intern Med11534391863023
- DentonCPHumbertMRubinLBosentan therapy for pulmonary arterial hypertension related to connective tissue disease: a subgroup analysis of the pivotal clinical trials and their open-label extensionsAnn Rheum Dis20066513364016793845
- DoyleRLMcCroryDChannickRNJournal of American College of Chest Physicians: Surgical treatments/interventions for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelinesChest20041261S63S71S15249495
- FarberHWLoscalzoJPulmonary arterial hypertensionN Engl J Med200435116556515483284
- FilepJGFournierAFoldes-FilepEAcute pro-inflammatory actions of endothelin-1 in the guinea pig lung: Involvement of ETA and ETB receptorsBr J Pharmacol1995115227367670725
- FusterVSteelePMEdwardsWDPrimary pulmonary hypertension: natural history and the importance of thrombosisCirculation19847058076148159
- GabbayEMcNeilKWilliamsTJBosentan for pulmonary arterial hypertension; the relationship between 6MWT and quality of lifeAm J Resp Crit Care Med2005169A175
- GaineSPRubinLJPrimary pulmonary hypertensionLancet1998352719259729004
- GalieNGuidelines on diagnosis and treatment of pulmonary arterial hypertension: The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of CardiologyEur Heart J20042522437815589643
- GalieNBeghettiMGatzoulisMABosentan therapy in patients with Eisenmenger syndromeCirculation2006114485416801459
- GalieNGhofraniHATorbickiASildenafil citrate therapy for pulmonary arterial hypertensionN Engl J Med200535321485716291984
- GalieNGrigioniFBacchi-RggianinKRelation of endothelin-1 to survival in patients with primary pulmonary hypertensionEur J Clin Invest199626273
- GalieNHinderliterALTorbickiAEffects of the oral endothelin-receptor antagonist Bosentan on echocardiographic and Doppler measures in patients with pulmonary arterial hypertensionJ Am Coll Cardiol2003411380612706935
- GalieNSeegerWNaeijeRComparative analysis of clinical trials and evidence-based treatment algorithm in pulmonary arterial hypertensionJ Am Coll Cardiol20044381S88S15194183
- GiaidAYanagisawaMLanglebenDExpression of Endothelin-1 in the lungs of patients with pulmonary hypertensionN Engl J Med1993328173298497283
- GirgisREMathaiSCKrishnanJALong-Term outcome of Bosentan treatment in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with the scleroderma spectrum of diseasesJ Heart Lung Transplantation200524162631
- HoeperMMPulmonary hypertension in collagen vascular diseaseEur Respir J200219571611936539
- HoeperMMDinh-XuanATCombination therapy for pulmonary arterial hypertension: still more questions than answersEur Respir J2004243394015358685
- HoeperMMFaulenbachCGolponHCombination therapy with bosentan and sildenafil in idiopathic pulmonary arterial hypertensionEur Respir J20042410071015572546
- HoeperMMKielyDGCarlsenJSafety profile of pulmonary arterial hypertension patients treated with bosentan and sildenafil: results from the European surveillance programAm J Resp Crit Care Med2005169A135
- HoeperMMKrammTWilkensHBosentan Therapy for Inoperable Chronic Thromboembolic Pulmonary HypertensionChest20051282363716236895
- HoeperMMMarkevychISpierkerkoetterEGoal-oriented treatment and combination therapy for pulmonary arterial hypertensionEur Respir J2005268586316264047
- HughesRGeorgePParameshwarJBosentan in inoperable chronic thromboembolic pulmonary hypertensionThorax20056070716061720
- HughesRJJaisXBondermanDBosentan in inoperable chronic thromboembolic pulmonary hypertension: efficacy at 1 yearEur Respir J2006281384316611652
- HumbertMBarstRJRobbinsIMCombination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2Eur Respir J200424353915358690
- HumbertMMorrellNWArcherSLCellular and molecular pathobiology of pulmonary arterial hypertensionJ Am Coll Cardiol20044313S24S15194174
- HumbertMSitbonOSimonneauGTreatment of pulmonary arterial hypertensionN Engl J Med200435114253615459304
- JaisXGhofraniAHoeperMMBosentan for inoperable chronic thromboembolic pulmonary hypertension (CTEPH): a randomized, placebo-controlled trialAm J Resp Crit Care Med2007173A896
- JamiesonSWKapelanskiDPSakakibaraNPulmonary endarterectomy: experience and lessons learned in 1,500 casesAnn Thorac Surg20037614576414602267
- JonesKHigenbottamTWallworkJPulmonary vasodilation with prostacyclin in primary and secondary pulmonary hypertensionChest19899678492507232
- KatwaLCGuardaEWeberKTEndothelin receptors in cultured adult rat cardiac fibroblastsCardiovasc Res199327212598313418
- KeoghAMMcNeilKDWlodarczykJQuality of life in pulmonary arterial hypertension: improved and maintained with bosentanJ Heart Lung Transplantation2007261817
- KotlyarESyRKeoghAMBosentan for the treatment of pulmonary arterial hypertension associated with congenital cardiac diseaseCardiol Young2006162687416725066
- LaneKBMachadoRDPauciuloMWHeterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH ConsortiumNat Genet20002681410973254
- LevinEREndothelinsN Engl J Med1995333356637609754
- LunzeKGilbertNMebusSFirst experience with an oral combination therapy using bosentan and sildenafil for pulmonary arterial hypertensionEur J Clin Invest200636S332816919008
- McLaughlinVVOudizRJFrostARandomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertensionAm J Respir Crit Care Med200617412576316946127
- McLaughlinVVSitbonOBadeschDBSurvival with first-line bosentan in patients with primary pulmonary hypertensionEur Respir J200525244915684287
- MoserKMAugerWRFedulloPFChronic major-vessel thromboembolic pulmonary hypertensionCirculation1990811735432188751
- MuskMChambersDLawrenceSBosentan improves WHO functional class, exercise capacity and RV size in inoperable chronic thromboembolic pulmonary hypertension (CTEPH)J Heart Lung Transplantation200625S77
- NewmanJHWhellerLLaneKBMutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindredN Engl J Med20013453192411484688
- NeylonCBAvdoninPVDilleyRJDifferent electrical responses to vasoactive agonists in morphologically distinct smooth muscle cell typesCirc Res199475733417522987
- OlschewskiHSimonneauGGalieNAerosolized Iloprost Randomized Study Group: Inhaled iloprost for severe pulmonary hypertensionN Engl J Med2002347322912151469
- PaulGAGibbsSRBoobisARBosentan decreases the plasma concentration of sildenafil when coprescribed in pulmonary hypertensionBr J Clin Pharmacol2005601071215963102
- ProvencherSSitbonOHumbertMLong-term outcome with first line bosentan therapy in idiopathic pulmonary arterial hypertensionEur Heart J2006275899516431875
- RichSKaufmannELevyPSThe effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertensionN Engl J Med199232776811603139
- RosenzweigEBIvyDDWilditzAEffects of long-term bosentan in children with pulmonary arterial hypertensionJ Am Coll Cardiol20054669770416098438
- RubinLJACCP evidence-based clinical practice guidelines: Diagnosis and management of pulmonary arterial hypertensionChest20041261S92S
- RubinLJBadeschDBBarstRJBosentan therapy for pulmonary arterial hypertensionN Engl J Med200234689690311907289
- RubinLJHoeperMMKlepetkoWCurrent and future management of chronic thromboembolic pulmonary hypertension: from diagnosis to treatment responsesProc Am Thorac Soc20063601716963541
- RunoJRLoydJEPrimary pulmonary hypertensionLancet200336115334412737878
- SimonneauGBarstRJGalieNContinuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trialAm J Respir Crit Care Med2002165800411897647
- SimonneauGGalieNRubinLJClinical classification of pulmonary hypertensionJ Am Coll Cardiol2004435S12S15194173
- SitbonOBadeschDBChannickRNEffects of the dual endothelin receptor antagonist Bosentan in patients with pulmonary arterial hypertension: A 1-year follow-up studyChest20031242475412853530
- SitbonOGressinVSpeichRBosentan in pulmonary arterial hypertension associated with HIV infectionAm J Respir Crit Care Med200417012121715317666
- SitbonOHumbertMNunesHLong-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survivalJ Am Coll Cardiol200240780812204511
- SitbonOMcLaughlinVVBadeschDBSurvival in patients with class III idiopathic pulmonary arterial hypertension treated with first-line oral bosentan compared with an historical cohort of patients started on iv epoprostenolThorax20056010253016055621
- SteinerMKPrestonIRKlingerJRConversion to bosentan from prostacyclin infusion therapy in pulmonary arterial hypertension: a pilot studyChest200613014718017099026
- StewartSMurphyNFMcMurrayJJVA population-based analysis of pulmonary arterial hypertension in Scotland (1996-2001)Eur Respir J2007in press
- StrangeCTreatment for Secondary Pulmonary HypertensionChest20051281897816236830
- VoelkelNFTuderRMWeirEKRubinLRichSPathophysiology of primary pulmonary hypertension: From physiology to molecular mechanismsPrimary Pulmonary Hypertension1997New York, NYMarcel Deckerp83129
- WilliamsMHDasCHandlerCE2006Systemic sclerosis associated pulmonary hypertension: improved survival in the current eraHeart929263216339813
- WlodarczykJHClelandLGKeoghAM2006Public funding of Bosentan for the treatment of pulmonary arterial hypertension in AustraliaPharmacoeconomics249031516942124
- ZapolWMFalkeKJHurfordWEInhaling nitric oxide: a selective pulmonary vasodilator and bronchodilatorChest199410587S91S8131625