Abstract
The type β transforming growth factors (TGF-βs) are involved in a number of human diseases, including heart failure and myocardial arrhythmias. In fact, during the last 20 years numerous studies have demonstrated that TGF-β affects the architecture of the heart under both normal and pathological conditions. Moreover, TGF-β signaling is currently under investigation, with the aim of discovering potential therapeutic roles in human disease. In contrast, only few studies have investigated whether TGF-β affects electrophysiological properties of the heart. This fact is surprising since electrical remodeling represents an important substrate for cardiac disease. This review discusses the potential role of TGF-β on cardiac excitation-contraction (EC) coupling, action potentials, and ion channels. We also discuss the effects of TGF-β on cardiac development and disease from structural and electrophysiological points of view.
Introduction
The superfamily of type β transforming growth factors (TGF-β) includes TGF-β1, TGF-β2, and TGF-β3, bone morphogenetic proteins (BMPs), growth differentiation factors (GDFs), activins, and inhibins. At least 35 members of this superfamily have been discovered in vertebrates. These factors regulate a diverse array of cellular processes, including tissue development and repair. Their effects are mediated by binding to two types of functional receptors (TBRI and TBRII), which are single-pass transmembrane serine/threonine kinases that phosphorylate specific signal-transducing molecules termed Smads (). Activated Smads can be translocated into the nucleus, bind to the DNA, and regulate the transcription of specific genes. The TGF-βs bind to and activate the type II receptor (TBRII), which in turn binds to and phosphorylates the type I receptor (TBRI). This yields to formation of a tetrameric complex of proteins, formed by two TBRIs and two TBRIIs. Once activated, the TBRI phosphorylates the C-terminus of receptor-associated Smads (or R-Smads, eg, Smad 3); see CitationChang and colleagues (2002) for an excellent and comprehensive review regarding signaling pathways and in vivo effects of TGF-β.
Figure 1 TGF-β1 and its general mechanism of action. Binding of TGF-β1 with its receptor (TGF-βR) activates intracellular signaling proteins termed Smads receptors (R-Smads, eg, Smad3), which are translocated into the nucleus following interaction with co-activator proteins termed co-Smads. Once in the nucleus, the Smads bind to the DNA and regulate transcription of specific genes. Inhibitory Smads, like Smad 6 and 7 prevent activation of R-Smads, by competitively inhibiting either its activation by the receptor, or its association with Co-Smads. Smads independent signaling pathways can also contribute to diversify responses to TGF-β1 (eg, MAPK kinases, and Rho GTPases).
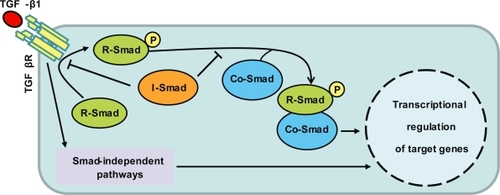
Even though C-terminal phosphorylation is the key event in Smads activation, other kinase pathways also regulate the Smad signaling. For example, both tyrosine kinase receptors to epidermal growth factor (EGF) and hepatocyte growth factor phosphorylate Smad2, and induce its nuclear translocation (Citationde Caestecker et al 1998). In fact, the activation of Smads can be also induced by at least the following signaling pathways: the Erk mitogen-activated protein kinase (MAPK) and the Ca2+/calmodulin-dependent protein kinase II (CamKII). Thus, similar to other signaling pathways, the TGF-β signaling exhibits cross-talk with a number of second messengers. Moreover, other molecules apart from Smad proteins interact with and regulate the activaty of TBRs, without apparent direct activation of Smads (eg, FK-506 binding protein). Finally, the activated receptor can also activate non-Smad signaling pathways, such as PP2A, Erk, JNK, PI3K, and p38MAPK (CitationDerynck and Zhang YE 2003).
The TGF-β signaling is involved in a number of human pathologies, including lung fibrosis (CitationWillis and Borok 2007), renal and liver injury (CitationBreitkopf et al 2005; CitationBöttinger 2007), Alzheimer (CitationMasliah et al 2001), cancer (CitationRoberts and Wakefield 2003), and cardiac remodeling (CitationBujak and Frangogiannis 2007; CitationBurstein and Nattel 2008). The role of TGF-β on cardiac pathophysiology began to be elucidated ∼20 years ago by CitationThompson and colleagues (1988). Basically, what they found was that ventricular myocytes from the infarcted myocardium overexpresses TGF-β1 protein and mRNA. Soon thereafter, CitationPotts and Runyan (1989) suggested a role for TGF-β signaling in promoting development of the heart. To this date, a great deal of information regarding to the effects of TGF-β on cardiac architecture has been accumulated. In fact, significant efforts are currently been made to discover potential therapeutic roles for TGF-β signaling in cardiac pathology (CitationNarine et al 2004; CitationNg et al 2004; CitationLi et al 2005; CitationLiao 2005; CitationOkada et al 2005).
Role of TGF-β in cardiac development
The TGF-β signaling is essential to epithelial–mesenchymal transformation (EMT). This is an embryonic phenomenon that determines formation of cardiac valves and the septa. Specifically, the EMT involves endothelial cells that migrate into an expanded extracellular matrix (or the cardiac jelly) where they proliferate and differentiate into mesenchymal cells. Subsequently, locally expanded swellings of cardiac jelly and mesenchymal cells form what is known as endocardial cushion tissue, which undergoes an extensive remodeling from bulbous swellings to eventual thinly tapered heart valves (CitationNakajima et al 2000).
A number of studies show that the TGF-β superfamily signaling is essential for heart development. For example, TGF-β1, -β2, and -β3, as well as BMP-2, -4, -6, and -7, are all expressed at specific regions and stages of development of the immature heart. In addition, several receptors (ALK2, ALK3, and ALK5) and downstream molecules (Smad5 and Smad6) are important in cardiac morphogenesis. Moreover, BMP-2-null mice either do not have a heart, or develop a very retarded and malformed heart, mice carrying mutations in BMP-5 or -7 die before birth with multiple defects in heart development, and mice deficient in BMP-6 or -7 have delayed cardiac cushion formation, which results in subsequent valve and septation defects, as well as a premature death due to heart failure (CitationChang et al 2002). Isoforms of the TGF-β subfamily are also important for heart development. For example, specific antibodies against the corresponding receptors inhibit EMT (CitationPotts and Runyan 1989; CitationBoyer et al 1999; CitationBrown et al 1999), the heart of TGF-β2-null mouse embryos have specific defects in the development of the valves and septa (CitationSandford et al 1997), and both TGF-β2 and TGF-β3 are critical to the initiation and regulation of EMT (CitationChang et al 2002). Thus, several TGF-βs are required to achieve proper valve morphogenesis, and according to this, there are numerous of non-compensated functions between the three different isoforms of the TGF-β subfamily. For example, TGF-β2 null-mice exhibit multiple defects that are not overlapping with TGF-β1- and TGF-β3-null mice (CitationSandford et al 1997), and TGF-β2 promotes cardiac cushion formation by activating a unique set of downstream mediators (ie binding to a third type receptor or TBRIII, which in turn modifies the functional TBRI-TBRII signaling complex (CitationBrown et al 1999). Moreover, TGF-β2 and TGF-β3 exert distinct effects in EMT during valve formation (CitationBoyer et al 1999), and TGF-β1 plays a critical role in adult cardiac structural remodeling (CitationBujak and Frangogiannis 2007; CitationBurstein and Nattel 2008). Below we describe the major alterations associated to cardiac remodeling, as well as the corresponding TGF-β effects.
Structural remodeling of the ventricle
Cardiac structural remodeling stands for several morphological changes that can be induced by the following physiological and pathophysiological factors, cytokine and neurohumoral stimuli, hemodynamic load, an altered function of mutant proteins, and a loss of contractile mass from prior infarction (). The involved changes include alterations on the expression levels of structural proteins, as well as an increase on the following parameters: proliferation rate of fibroblast, deposition of extracellular matrix (ECM) constituents or fibrosis, and the size of cardiac myocytes or hypertrophy. Ventricular hypertrophy accompanies many forms of heart disease, such as ischemic disease, hypertension, heart failure, and valvular disease. Initially, hypertrophy helps the heart to meet the needs of the body (compensated hypertrophy). However, in response to a more severe, or prolonged stimuli, the hypertrophy becomes inadequate (decompensated hypertrophy), and contributes per se to the genesis of ischemia, arrhythmia, and heart failure (see the vicious circles that are indicated with gray arrows in ,). Some alterations in gene expression that are involved in hypertrophy are switching from adult α-myosin heavy chain (α-MHC) to fetal β-myosin heavy chain (β-MHC), and reexpression of skeletal α-actin. Altogether, this pattern of gene expression mimics that seen during embryonic development and is thus called re-induction of the “fetal-gene program”. Thus, hypertrophy seems to be a programmed reversion toward a more fetal phenotype (CitationBers 2001; CitationKatz 2002; CitationHill 2003).
Figure 2 Role of TGF-β on ventricular remodeling. Certain human diseases provoke cardiac work-overload, which in turn promotes several changes known as structural and electrical remodeling. Initially, these changes help the heart to meet the needs of the body, by means of optimizing cardiac output (acute remodeling). However, in response to prolonged stimuli the remodeling becomes inadequate and contributes per se to cardiac dysfunction (vicious circles indicated by gray arrows). Interestingly, work-overload also increases the expression levels of TGF-β1. In fact, TGF-β1 per se reproduces most of the hallmarks associated to structural remodeling (asterisks), including its own overexpression. TGF-β1 may also provoke electrical remodeling (dotted arrow), but the corresponding effects have not been thoroughly investigated.
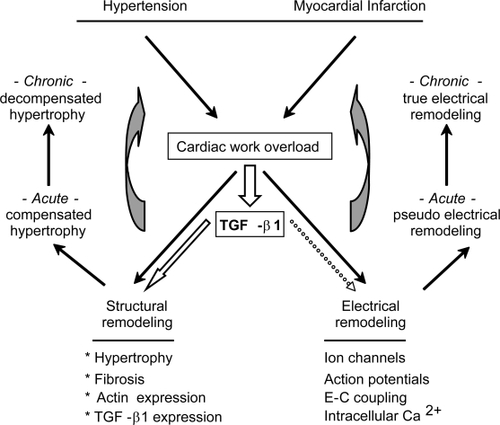
Activation of the renin–angiotensin system (RAS) has been implicated in the generation of cardiac remodeling (hypertrophy and fibrosis). In fact, inhi bition of angiotensin II (or Ang II, the effector molecule of the RAS), by angiotensinconverting enzyme (ACE) inhibitors or Ang II type 1 (AT1) receptor antagonists prevents structural remodeling of the ventricle. Interestingly, while Ang II directly promotes growth in neonatal cells, it does not in adult cardiomyocytes, suggesting Ang II indirectly promotes hypertrophy. In fact, Ang II exerts its in vivo profibrotic and hypertrophic effects by increasing the expression levels of TGF-β1 (CitationRosenkranz 2004).
Role of TGF-β in structural remodeling of the ventricle
The proposed role for TGF-β1 in cardiac disease is illustrated in . Both neonatal and adult cardiomyocytes synthesize and release the three mammalian isoforms of the TGF-β subfamily (ie, TGF-β1, TGF-β2, and TGF-β3), although it is the TGF-β1 and TGF-β3 isoforms that predominate (CitationLong 1996). The cardiac myocytes increase expression levels of TGF-β1 in response to myocardial stress in an animal model of myocardial infarction (CitationThompson et al 1988). Accordingly, expression levels of TGF-β1 mRNA are also increased in left ventricular myocardium of patients with either idiopathic hypertrophic cardiomyopathy (CitationLi et al 1997) or dilated cardiomyopathy (CitationPauschinger et al 1999), as well as in animal models of norepinephrine-induced hypertrophy (CitationBhambi and Eghbali 1991), progressive coronary artery occlusion (CitationWünsch et al 1991), and pressure overload (CitationVillarreal and Dillmann 1992). In both experimental models and humans TGF-β1 is particularly overexpressed in myocardium during transition from stable hypertrophy to heart failure. Thus, TGF-β1 is considered an essential mediator of cardiac adaptation to work overload. In fact, TGF-β1 represents one of the few markers that discriminate between compensated and decompensated hypertrophy (in addition to increased collagen content) (CitationRosenkranz 2004).
Perhaps more importantly, TGF-β1 reproduces most of the hallmarks seen in structural remodeling (, asterisks). Specifically, TGF-β1 induces expression levels of ECM constituents by cardiac fibroblasts (ie, fibrillar collagen, fibronectin, and proteoglycans), self-amplifies its own expression in both cardiac myocytes and fibroblast (CitationDesmouliére et al 1993; CitationLong 1996), stimulates the proliferation of fibroblasts and their phenotypic conversion to myofibroblasts (CitationSappino et al 1990; CitationWalker et al 2004) and provokes “fetal” contractile protein gene expression in cardiomyocytes (CitationParker et al 1990). Additionally, TGF-β1 mediates the cardiac hypertrophy induced by Ang II (CitationSchultz Jel et al 2002), overexpression of TGF-β1 in transgenic mice results in hypertrophic growth of ventricular myocytes (CitationNakajima et al 2000), heterozygous TGF-β1 (±)-deficient mice exhibit decreased fibrosis of the aging heart (CitationBrooks and Conrad 2000) and functional blockage of TGF-β1 signaling in vivo by neutralizing antibodies prevents myocardial fibrosis and dysfunction in pressure overloaded hearts (CitationKuwahara et al 2002).
Structural remodeling of the atrium
Atrial remodeling, which stands for “any persistent change in atrial structure or function”, promotes the occurrence or maintenance of atrial fibrillation (AF), by acting on the fundamental mechanisms of the arrhythmia (CitationThompson et al 1988). Accordingly, AF can be induced by several physiological and artificial stimuli (), such as heart failure (CitationLi et al 1999; CitationLee et al 2006), senescence (CitationAnyukhovsky et al 2002; CitationHayashi et al 2002), atrial dilatation (CitationEckstein et al 2008), and rapid atrial rate of stimulation (CitationMorillo et al 1995). Structural remodeling and in particular interstitial fibrosis represents the major promoter of AF. This is because fibrosis provokes disruption of electrical conduction among adjacent myocytes, due to an increased deposition of extracellular matrix, which disrupts the normal myocardial ultrastructure. In fact, transgenic mice overexpressing cardiac TGF-β1 develop atrial fibrosis, heterogeneous conduction, and AF (CitationVerheule et al 2004). Additionally to atrial fibrosis, myocyte loss by either apoptosis or necrosis accompanies AF (Burstein and Nattel recently reviewed structural alterations associated to AF (CitationBurstein and Nattel 2008)).
Figure 3 Role of TGF-β on atrial remodeling. Certain physiological and pathophysiological conditions (ie, heart failure, rapid atrial rate of electrical stimulation, aging, and atrial dilatation) promotes the development of atrial fibrosis, which in turn provokes AF. Atrial fibrosis is developed in parallel to increased expression of TGF-β1, a well known profibrogenic agent. Interestingly, a compound named pirfenidone that exerts its biological actions by inhibiting the synthesis of TGF-β1, also prevents development of both atrial fibrosis and AF.41 This suggests a cause-effect relationship, by which TGF-β1 promotes development of atrial fibrosis, and thereby AF. Recent evidences obtained from cultured cells suggest thatTGF-β1 also provokes electrical remodeling (). These potentially in vivo electrophysiological effects by TGF-β1 could be attributed to a direct effect on ion channels expression and function (dotted arrow). Alternatively, they could be mediated by structural remolding (eg, fibrosis). Once established, electrical remodeling provokes AF and a vicious circle begins – termed atrial fibrillation begets atrial fibrillation, or AF begets AF.
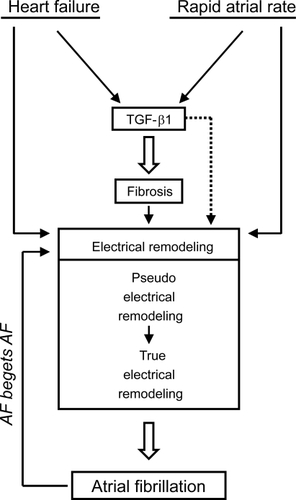
Table 1 Modulation of cardiac electrophysiology by TGF-β1
The cellular mechanisms that underlie cardiac fibrosis are similar to fibrosis in other epithelial organs. Accordingly, fibroblasts represent the principal cellular mediators of cardiac fibrosis. The increased number of these cells was initially thought to originate from proliferation of resident fibroblasts, bone marrow cells, and epithelial cells (derived through EMT). More recently, cardiac fibroblasts were also shown to be derived from endothelial cells, via endothelial–mesenchymal transformation (EndMT) (CitationKisseleva and Brenner 2008).
Role of TGF-β in structural remodeling of the atrium
A number of stimuli that promotes AF converge in increasing expression levels of TGF-β1, which in turn provokes interstitial fibrosis (). However, while upregulation of TGF-β1 represents a key event in inducing fibrosis, other growth factors are known to synergize with TGF-β1 for this effect. For example, connective tissue growth factor (CTGF), and platelet-derived growth factor (PDGF). Interestingly, while TGF-β1 promotes fibrosis and increases collagen gene expression by acting through Smad proteins signaling pathway (CitationPiek et al 1999; CitationHao et al 2000; CitationEvans et al 2003), inhibiting the activity of PI3K reduces TGF-β induced EMT, suggesting a role for Smad-independent pathways in fibrosis (CitationBakin et al 2000). In fact, the p38MAPK pathway is also believed to be required for TGF-mediated EMT (CitationBakin et al 2002).
Ang II has been also implicated in promoting the activation of fibroblasts, as well as the synthesis of extracellular matrix proteins, such as collagen and proteoglycan. Additionally, Ang II regulates production of matrix metalloproteinases (eg, MMP-2), as well as the breakdown of collagen IV (CitationMehta and Griendling 2007). Interestingly, cardiac fibroblasts exposed to Ang II overexpress both fibronectin and TGF-β1 (CitationMoriguchi et al 1999). Actually, the profibrotic effects of Ang II are largely explained by its ability to increase expression levels of TGF-β1 (CitationKisseleva and Brenner 2008).
There are several differences between atrial and ventricular structural remodeling. For example, while at the ventricle TGF-β1 provokes hypertrophy, this is prevented by TGF-β1 at the atrium (CitationNakajima et al 2000). Additionally, compared to the ventricle, the atrial tissue is more susceptible to develop fibrosis (CitationNakajima et al 2000; CitationHanna et al 2004). Moreover, leukocyte infiltration, cell death, apoptosis and MAP kinase activation, are larger in atrium, occur earlier, and are more transient in atrium compared to ventricle (CitationHanna et al 2004). In a very interesting study, CitationBurstein and colleagues (2008) investigated the different behavior of atrial and ventricular fibroblasts. Basically, they found the atrial fibroblasts show enhanced reactivity, which contributes to greater atrial fibrotic responses. Apparently, a different behavior between atrial and ventricular fibroblasts is due to an increased expression of the PDGF receptor in the atrium (CitationBurstein et al 2008).
Electrical remodeling
Cardiac excitation-contraction (EC) coupling consists of intra-cellular Ca2+ release that occurs in response to activation of type 2 ryanodine receptors (RyR2s, located at the sarcoplasmic reticulum or SR). Intracellular Ca2+ release is triggered by Ca2+ entering through voltage dependent Ca2+ channels (L-type Ca2+ channels, or L-channels) located at the sarcolemma (termed Ca2+ induced Ca2+ release, or CICR) (CitationFabiato and Fabiato 1979). This combination of Ca2+ influx and release transiently increases intracellular free Ca2+ concentration (termed Ca2+ transient), which in turn activates the contractile machinery. The relaxation process occurs following termination of the Ca2+ transient, which depends mostly on the removal of cytosolic Ca2+ by the activities of: sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), sarcolemmal Na+/Ca2+ exchanger (NCX), sarcolemmal Ca2+-ATPase, and mitochondrial Ca2+ uniport (CitationBers 2002). Ca2+-dependent inactivation of L-channels, on the other hand, guarantees fast termination (in milliseconds) of the triggering Ca2+ influx (CitationBers and Perez-Reyes 1999; CitationCatterall 2000).
In addition to L-current (ICaL), cardiac myocytes express often a voltage-dependent T-type Ca2+ current (T-current, or ICaT) whose physiological relevance remains elusive (CitationBers and Perez-Reyes 1999). Voltage-dependent Na+ channels generate also quickly activating inward currents (INa), which are at least partially responsible for the fast depolarizing upstroke of the action potential (AP) (CitationBalser JR 1999; CitationClancy and Kass 2005). Repolarization, on the other hand, is favored by inactivation of inward currents, but depends primarily on the activation of the following K+ currents – which are often composed of two or more subcomponets: inward rectifier (IK1), transient outward (Ito), delayed rectifier (IKsus), and a time independent background current (IKP) (CitationSnyders 1999; CitationClancy and Kass 2005). Thus, as in other excitable cells APs of the cardiac myocytes depend on a delicate balance on the activity of several ion channels and transporters. Consequently, heart disease is commonly associated to several channelopathies, which are not only restricted to inherited disorders, but involve also acquired alterations in either the expression or post translational modification of ion channels (CitationClancy and Kass 2005; CitationTomaselli and Marbán 1999; CitationMarbán 2002). The observation that tachyarrhythmias modify electrophysiological properties in the same manner as cardiac disease has lead to the concept “electrical or electrophysiological remodeling” ( and ). However, the precise alterations involved in this process, regarding to ion channel function and expression remain controversial (CitationPandozi and Santini 2001).
This is partially due to the different experimental models that are commonly used in heart disease. Another source of variation is that the alterations change during disease progression or transition to failure. In fact, different times of exposure to the pathophysiologic substrate may result in either “pseudo” (acute) or “true” (chronic) electrical remodeling ( and ). Pseudo-remodeling have short onset and offset kinetics (minutes), and involve alterations in the activity (or functional properties) of a fixed number of ion channels, transporters, and pumps (CitationPandozi and Santini 2001). In contrast, true electrical remodeling is associated with alterations in the expression levels of functional ion channels, transporters, and pumps. Accordingly, alterations on mRNA levels encoding to these proteins are commonly observed. True electrical remodeling has therefore, long onset and offset kinetics, last for a long time (hours or days) and is not functional or metabolic (CitationPandozi and Santini 2001; CitationAllessie 1998).
Electrical remodeling of the ventricle
Shortenings of action potential duration (APD) and effective refractory period (ERP) are typical of atrial fibrillation (see below, Electrical remodeling of the atrium). In contrast, rapid rates of stimulation prolong both the APD and the ERP at the ventricle. Prolongation of the APD is characteristic of ventricular cells and tissues on which heart failure is induced by a number of mechanisms, including pressure and volume overload, genetic, metabolic, ischemia/infarction, and chronic pacing tachycardia. In certain models of ventricular failure prolongation of the APD is associated, as for the atrial myocytes, to reductions in ICaL, Ito, and expression levels of the corresponding genes. It is interesting thus that opposite alterations in APD can be related to changes in the same ionic currents (ICaL and Ito). However, the relative contribution of these channels to the AP differs substantially between atrium and ventricle, and this contributes to explaining the differences in APD between the two cavities.
Electrical remodeling of the ventricle also involves a parallel reduction in the amplitude and the rate of decay of Ca2+ transients, which provokes a reduced capability to is reduced in certain develop force. Interestingly, while ICaLmodels of HF, it does not necessarily contribute to explain the reduced Ca2+ transients. This is because many reports show in the face of a strong depression of both no change in ICaL contractions and Ca2+ transients (CitationKääb et al 1996; CitationGómez et al 1997). Instead, reduction of Ca2+ transients and the consequent contractile dysfunction, are both due to depletion of SR Ca2+, which may result from RyR2-dependent Ca2+ leak, an increased Ca2+ extrusion through the NCX, or a reduced function of SERCA (CitationBers 2001; CitationWehrens et al 2005).
Electrophysiological effects of TGF-β on the ventricle
The reported effects of TGF-β1 on cardiac electrophysiology are summarized on . Pioneer studies that investigated these effects were performed on confluent cultures of ventricular myocytes, forming a syncytium (CitationNeylon et al 1994; CitationKimura 1997; CitationCarrillo et al 1998). CitationRoberts and colleagues (1992) found that a chronic exposure of neonatal rat ventricular myocytes to TGF-β1 increases spontaneous beating rate of the syncytium. Conceivable, this effect could be at least partially explained by TGF-β1 provoking an increased SR Ca2+ content, and a higher frequency of spontaneous intracellular Ca2+ oscillations. Accordingly, both of these effects were subsequently reported by CitationNeylon and colleagues (1994). More recently, CitationCarrillo and colleagues (1998) reported that the increase in beating rate is also associated to a higher expression of the Na+/Ca2+ exchanger, which could prepare cells to deal better with the Ca2+ overload associated to high frequency of Ca2+ oscillations. While these studies support the notion that TGF-β1 increases beating rate of the syncytium, CitationKimura (1997) reported an opposite effect, which was associated to an increased IP3-sensitive intracellular Ca2+ store. Currently, molecular bases for this apparent contradiction remain unknown.
To our knowledge, only one study has investigated the electrophysiological effects of TGF-β1 in adult ventricular myocytes (CitationRoberts et al 1992). What these authors found is that ∼4 h of exposure to TGF-β1 provokes “contractile dysfunction”, characterized by a significant reduction on the maximum rate of myocyte shortening and re-lengthening. Additionally, TGF-β1 reduces the time to peak and the rate of decay of intracellular Ca2+ transients, in the absence of significant alterations on ICaL. Molecular mechanisms underlying these effects point to an enhanced reactive oxide species (ROS) production. However, downstream molecules other than ROS have yet to be elucidated.
For instance, the absence of alterations on ICaL could be interpreted to suggest that contractile dysfunction and effects on Ca2+ transients could both arise from a possible regulation on the Ca2+ handling proteins of the SR. However, neither the SR Ca2+ content nor spark properties (frequency and amplitude) were altered by TGF-β1, even thought this factor reduced the phosphorylation state of phospholamban, which in turn modulates SERCA. We should keep in mind that ICaL was recorded under whole-cell patch clamp conditions, whereas estimations of mechanical properties and intracellular Ca2+ were performed in intact (ie, nonpatched) myocytes (CitationLi et al 2008). This represents an important limitation, since misleading conclusions can be drawn from comparing results of different conditions. For example, intracellular Ca2+ can be strongly influenced by dialysis of the cytosol under whole-cell patch-clamp conditions. Finally, we should keep in mind that these data were obtained in cultured myocytes and the situation may be different in vivo.
Electrical remodeling of the atrium
An important step toward understanding atrial fibrillation (AF) was the discovery that once initiated, AF alters atrial electrical properties, in a manner that favors the maintaining of the arrhythmia (ie, electrical remodeling, as a vicious circle (CitationWijffels et al 1995)). Initially, high frequencies of atrial activation (ie, rapid atrial rate; ) leads to what is known as a short-term APD (action potential duration) adaptation to rate, which is due to rapid (minutes) functional changes in ion channels and transporters (basically, what happens is that at high frequencies of stimulation, the atrium responds with a shortening of the APD). Thus, rapid atrial rate stimulation provokes “pseudo” electrical remodeling ().
This short-term adaptation can be at least partially explained by an increase in Ca2+-dependent inactivation of ICaL(CitationYagi et al 2002). Accordingly, cytosolic Ca2+ overload is also involved, which may activate intracellular signaling pathways that play a prominent role in the subsequent “true” electrical remodeling (CitationNattel 1999). Following this short-term adaptation, the atrial tachycardia begins to produce discrete changes in the expression levels of specific genes (in hours or days). Specifically, the proteins or mRNAs encoding to the principal subunits of ICaL(CitationYue et al 1999; CitationBrundel et al 2001), (but see Brundel and colleagues [CitationChrist et al 2004]), INa (CitationYue et al 1999), IKur (CitationLai et al 1999), and Ito (CitationYue et al 1999) are downregulated, whereas the corresponding subunits of IK1 and IK,Ach are upregulated (CitationDobrev et al 2001; CitationGaborit et al 2005) in a similar time-course and magnitude as the corresponding ionic currents (CitationPandozi and Santini 2001; CitationAllessie 1998; CitationNattel et al 2007; CitationNattel et al 2008).
True ionic remodeling (ie, changes in ion channel expression) permanently reduce the APD, the capability of APD to adapt to rate, and the effective refractory period (ERP). Altogether, these changes give rise to a vicious circle (). This is because decrease ERP promotes AF, by decreasing the wavelength (distance traveled by the electrical wave during the ERP, which determines the minimum path length that can support electrical reentry). This in turn allows the atrium to accommodate a larger number of reentry circuits, decreasing the chance of spontaneous termination. Thus, ionic remodeling can be seen as a protective mechanism against the initial Ca2+ overload. However, the protective effect occurs at the expense of promoting the maintenance of AF, which nonetheless eventually alters Ca2+ homeostasis (CitationNattel et al 2008).
Effects of TGF-β on electrical remodeling of the atrium
An interesting effect of TGF-β1 is that it can provoke an increased susceptibility to develop atrial fibrillation. This was discovered in 2004 by CitationVerheule and colleagues (2004), using transgenic mice overexpressing cardiac TGF-β1. These animals also present a decreased epicardial conduction velocity on the right atria, as well as a more heterogeneous conduction on the left atria. Since no differences in action potential properties were found, CitationVerheule and colleagues (2004) concluded that atrial fibrosis induced by TGF-β1 is sufficient to enhance the inducibility of AF (as opposed to electrical remodeling) (Citation2004). Unfortunately, they did not investigate ICaL or a potential loss of the capability of APs to adapt to high rate of stimulation, which would have suggested alterations on ICaL. In keeping with this possibility, we have previously reported that TGF-β1 reduces expression levels of ICaL, in neonatal rat atrial myocytes (CitationAvila et al 2007). Moreover, in these cells TGF-β1 also reduces current densities associated to INa, IKsus, and IK1 (CitationRamos-Mondragón 2008). Thus, at least in cardiomyocytes from neonatal rat TGF-β1 reproduces electrophysiological alterations that are commonly seen in AF.
Molecular bases for downregulation of ICaL by TGF-β1 point to a decreased expression of mRNA encoding to CaV1.2, the principal subunit of L-type Ca2+ channels. Accordingly, TGF-β1 also provokes reduction in the amount of immobilization-resistant charge movement, which reflects voltage sensor’s activity of CaV1.2. Moreover, inhibition of ICaL by TGF-β1 cannot be reverted by okadaic acid, an inhibitor of protein phosphatases, supporting the notion that this effect is not due to a possible reduction in the permanent phosphorylation state of the channels (CitationAvila et al 2007).
As previously discussed (see Electrical remodeling of the atrium), molecular mechanisms underlying reduction of ICaL in AF include an altered function of CaV1.2. For example, ICaL is functionally reduced in AF by at the least the following mechanisms: 1) Ca2+-dependent inactivation (CitationYagi et al 2002). 2) Reduced phosphorylation state due to either increased activity of protein phosphatases (CitationChrist et al 2004), or impaired regulation by src kinase (CitationGreiser et al 2007). 3) Increased S-nitrosylation, due to decreased expression of the antioxidant glutation (CitationCarnes et al 2007). Remarkably, ICaL reduction has been also attributed to reduced expression levels of CaV1.2.(CitationYue et al 1999; CitationBrundel et al 2001).
The question of whether TGF-β1 actually regulates ICaL in adult atrial myocytes still remains to be solved. A recent study suggests increased in vivo levels of TGF-βs (in AF induced by rapid atrial pacing) do not significantly alter expression levels of CaV1.2 (CitationChen et al 2007). In contrast, the following evidences suggest possible functional effects. TGF-β1 increases intracellular levels of reactive oxide species (ROS) in adult ventricular myocytes (CitationLi et al 2008). On the other hand, increased ROS levels can provoke a reduced density of ICaL in adult atrial myocytes (CitationCarnes et al 2007). Thus, conceivable, in adult atrial myocytes TGF-β1 could decrease ICaL, by means of increasing levels of ROS. Undoubtedly, this possibility deserves to be elucidated.
Perspectives
Recently, arrythmogenic right ventricular dysplasia type 1 (ARVD1) was associated to mutations in the gene encoding TGF-β3 (CitationBeffagna et al 2005). ARVD1 is a progressive myocardial disease characterized by ventricular arrhythmias that lead to sudden unexpected death. The reported mutations increase expression levels of TGF-β3, which might in turn provoke the arrhythmia by promoting fibrosis and electrophysiological effects. In 2000, Brooks and Conrad (CitationBrooks and Conrad 2000) reported that senescent animals from TGF-β1 heterozygous (±) mice exhibit a decreased amount of myocardial fibrosis compared to controls. Additionally, these mice develop increased left ventricular compliance, live longer, and do not present a normal increase in diastolic pressure. This suggests decreased levels of the growth factor by loss of one TGF-β1 allele prevents development of myocardial stiffness induced by fibrosis. Alternatively, decreased levels of TGF-β1 in heterozygous (±) mice could also prevent development of a possible normal “contractile dysfunction” in senescent animals, similar to the electrophysiological effects that TGF-β1 exerts on cultured adult ventricular myocytes (CitationRoberts et al 1992), see also .
On the other hand, CitationKubin and colleagues (2005) reported that in the absence of serum TGF-β1 reduces in 6 days the beating rate of adult rat ventricular myocytes, but does not alter the corresponding morphological properties. Thus, it will be interesting to investigate a possible relationship between the acute contractile dysfunction (CitationRoberts et al 1992), and the chronic decrease in beating rate (CitationKubin et al 2005).
There are a number of unanswered questions regarding electrophysiological effects by TGF-β. To this date only the type 1 TGFβ has been used to perform electrophysiological studies (see ). However, there are numerous of noncompensated functions among the three isoforms of the TGF-β subfamily (see the section role of TGF-β in cardiac development). An interesting prediction would be that different TGF-βs preferentially regulate specific regions of the heart. Support to this view comes from the fact that TGF-β1 exerts opposite effects on atrial and ventricular hypertrophy (inhibition and stimulation, respectively (CitationNakajima et al 2000)). Moreover, while transgenic mice overexpressing TGF-β1 develop increased vulnerability to atrial fibrillation (CitationVerheule et al 2004), mutations in the gene encoding TGF-β3 are linked to a genetically determined myocardial dystrophy (ie, arrythmogenic right ventricular dysplasia type 1 or ARVD1 (CitationBeffagna et al 2005)). TGF-β2, on the other hand, is preferentially involved in mitral valve disease (CitationNg et al 2004) and certain cardiovascular anomalies (CitationSandford et al 1997; CitationBartram 2001). Thus, we anticipate elucidating effects of the two other isoforms will be of paramount relevance.
The beneficial effects of certain neurohumoral blockers on cardiac disease can be at least partially explained by their ability to inhibit electrical remodeling (CitationKatz 2002; CitationNattel 2002; CitationShinagawa et al 2003). Nevertheless, while certain drugs can be successfully used for the treatment of atrial fibrillation, they often result in potentially lethal alterations on ventricular electrophysiology. These opposite effects could be potentially explained by the aforementioned differences in electrical remodeling of the atrium and the ventricle. Thus, the identification of factors that selectively control ion channel expression and function in atrial vs ventricular myocytes, could be advantageous to find a more effective treatment of cardiac disease.
In conclusion, it has been recognized during the last 20 years that TGF-β plays a critical role in cardiac disease. Accordingly, significant efforts are been currently made to discover potential therapeutic roles for TGF-β signaling in cardiac pathology.(CitationLi et al 2005; CitationNarine et al 2004; CitationNg et al 2004; CitationLiao 2005; CitationOkada et al 2005). Thus, we believe that unraveling the corresponding electrophysiological effects may contribute to improve the clinical use of TGF-β-related agents, by providing the grounds for more rational and specific therapies.
Acknowledgements
This work was supported by CONACyT.
References
- AllessieMA1998Atrial electrophysiologic remodeling: another vicious circle?J Cardiovasc Electrophysiol91378939869538
- AnyukhovskyEPSosunovEAPlotnikovA2002Cellular electrophysiologic properties of old canine atria provide a substrate for arrhythmogenesisCardiovasc Res54462912062351
- AvilaGMedinaIMJimenezE2007Transforming growth factor-β1 decreases cardiac muscle L-type Ca2+ current and charge movement by acting on the Cav1.2 mRNAAm J Physiol Heart Circ Physiol292H6223116980347
- BakinAVTomlinsonAKBhowmickNA2000Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migrationJ Biol Chem275368031010969078
- BakinAVRinehartCTomlinsonAK2002p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migrationJ Cell Sci115319320612118074
- BalserJR1999Structure and function of the cardiac sodium channelsCardiovasc Res423273810533571
- BartramUMolinDGWisseLJ2001Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout miceCirculation10327455211390347
- BeffagnaGOcchiGNavaA2005Regulatory mutations in transforming growth factor-beta3 gene cause arrhytmogenic right ventricular cardiomyopathy type 1Cardiovasc Res653667315639475
- BersDMPerez-ReyesE1999Ca channels in cardiac myocytes: structure and function in Ca influx and intracellular Ca releaseCardiovasc Res423396010533572
- BersDM2001Excitation-contraction coupling and cardiac contractile force. Series: Developments in cardiovascular medicine2542nd EdLondon, UKSpringer Publishing Inc
- BersDM2002Cardiac excitation-contraction couplingNature41519820511805843
- BhambiBEghbaliM1991Effect of norepinephrine on myocardial collagen gene expression and response of cardiac fibroblast after nor-epinephrine treatmentAm J Pathol1391131421951630
- BöttingerEP2007TGF-beta in renal injury and diseaseSemin Nephrol273092017533008
- BoyerASAyerinskasIIVincentEB1999TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heartDev Biol2085304510191064
- BreitkopfKHaasSWiercinskaE2005Anti-TGF-beta strategies for the treatment of chronic liver diseaseAlcohol Clin Exp Res29121S131S16344596
- BrooksWWConradCH2000Myocardial fibrosis in transforming growth factor β1 heterozygous miceJ Mol Cell Cardiol321879510722796
- BrownCBBoyerASRunyanRB1999Requirement of type III TGF-beta receptor for endocardial cell transformation in the heartScience2832080210092230
- BrundelBJVan GelderICHenningRH2001Ion channel remodeling is related to intraoperative atrial effective refractory periods in patients with paroxysmal and persistent atrial fibrillationCirculation1036849011156880
- BujakMFrangogiannisNG2007The role of TGF-beta signaling in myocardial infarction and cardiac remodelingCardiovasc Res741849517109837
- BursteinBNattelS2008Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillationJ Am Coll Cardiol51802918294563
- BursteinBLibbyECalderoneA2008Differential behaviors of atrial versus ventricular fibroblasts: a potential role for platelet-derived growth factor in atrial-ventricular remodeling differencesCirculation11716304118347210
- CarnesCAJanssenPMRuehrML2007Atrial glutathione content, calcium current, and contractilityJ Biol Chem282280637317656369
- CarrilloCCafferataEGGenoveseJ1998TGF-beta1 up-regulates the mRNA for the Na+/Ca2+ exchanger in neonatal rat cardiac myocytesCell Mol Biol44543519620452
- CatterallWA2000Structure and regulation of voltage-gated Ca2+ channelsAnnu Rev Cell Dev Biol165215511031246
- ChangHBrownCWMatzukMM2002Genetic analysis of the mammalian transforming growth factor-beta superfamilyEndocr Rev2378782312466190
- ChenCLLinJLLaiLP2007Altered expression of FHL1, CARP, TSC-22 and P311 provide insights into complex transcriptional regulation in pacing-induced atrial fibrillationBiochim Biophys Acta17723172917174532
- ChristTBoknikPWöhrlS2004L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatasesCirculation1102651715492323
- ClancyCEKassRS2005Inherited and acquired vulnerability to ventricular arrhythmias: Cardiac Na+ and K+ channelsPhysiol Rev85334715618477
- de CaesteckerMPParksWTFrankCJ1998Smad2 transduces common signals from receptor serine-threonine and tyrosine kinasesGenes Dev121587929620846
- DerynckRZhangYE2003Smad-dependent and Smad-independent pathways in TGF-beta family signallingNature4255778414534577
- DesmouliéreAGeinozAGabbianiF1993Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblastsJ Cell Biol122103118314838
- DobrevDGrafEWettwerE2001Molecular basis of downregulation of G-protein-coupled inward rectifying K(+) current (I(K,ACh) in chronic human atrial fibrillation: decrease in GIRK4 mRNA correlates with reduced I(K,ACh) and muscarinic receptor-mediated shortening of action potentialsCirculation1042551711714649
- EcksteinJVerheuleSde GrootN2008Mechanisms of perpetuation of atrial fibrillation in chronically dilated atriaProg Biophys Mol Biol974355118378284
- EvansRATianYCSteadmanR2003TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteinsExp Cell Res2829010012531695
- FabiatoAFabiatoF1979Use of chlorotetracycline fluorescence to demonstrate Ca2+-induced release of Ca2+ from the sarcoplasmic reticulum of skinned cardiac cellsNature2811468471060
- GaboritNSteenmanMLamiraultG2005Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillationCirculation1124718116027256
- GómezAMValdiviaHHChengH1997Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failureScience27680069115206
- GreiserMHalaszovichCRFrechenD2007Pharmacological evidence for altered src kinase regulation of I (Ca,L) in patients with chronic atrial fibrillationArch Pharmacol37538392
- HannaNCardinSLeungTK2004Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failureCardiovasc Res632364415249181
- HaoJWangBJonesSC2000Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitroAm J Physiol Heart Circ Physiol279H30203011087260
- HayashiHWangCMiyauchiY2002Aging-related increase to inducible atrial fibrillation in the rat modelJ Cardiovasc Electrophysiol13801812212701
- HillJA2003Electrical remodeling in cardiac hypertrophyTrends Cardiovasc Med133162214596946
- KääbSNussHBChiamvimonvatN1996Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failureCirc Res78262738575070
- KatzAM2002Maladaptive growth in the failing heart: the cardiomyopathy of overloadCardiovasc Drugs Ther16245912374903
- KimuraHTakemuraHImotoK1997Upregulation of expression of sarcoplasmic reticulum by TGF-β1 in cultured rat cardiac myocytesAm J Physiol41H2639449227541
- KisselevaTBrennerDA2008Mechanisms of fibrogenesisExp Biol Med23310922
- KubinTTomarsMFachC2005Transforming growth factor β-1 downregulates beating frequency and remodeling of cultured rat adult cardiomyocytesCell Tissue Res321576615902494
- KuwaharaFKaiHTokudaK2002Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded ratsCirculation106130512093782
- LaiLPSuMJLinJL1999Changes in the mRNA levels of delayed rectifier potassium channels in human atrial fibrillationCardiology922485510844385
- LeeKWEverettTH4thRahmutulaD2006Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation11417031217030685
- LiDFarehSLeungTK1999Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sortCirculation100879510393686
- LiRKLiGMickleDA1997Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic cardiomyopathyCirculation96874819264495
- LiSLiXZhengH2008Pro-oxidant effect of transforming growth factor-β1 mediates contractile dysfunction in rat ventricular myocytesCardiovasc Res771071718006470
- LiTSHayashiMItoH2005Regeneration of infarcted myocardium by intramyocardial implantation of ex vivo transforming growth factor-beta- preprogrammed bone marrow stem cellsCirculation11124384515883211
- LiaoR2005Yin and Yang of myocardial transforming growth factor-beta1: timing is everythingCirculation1112416715897356
- LongCS1996Autocrine and paracrine regulation of myocardial cell growth in vitro. The TGFβ paradigmTrends Cardiovasc Med621726
- MarbánE2002Cardiac channelopathiesNature4152131811805845
- MasliahEHoGWyss-CorayT2001Functional role of TGF beta in Alzheimer’s disease microvascular injury: lessons from transgenic miceNeurochem Int3939340011578774
- MehtaPKGriendlingKK2007Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular systemAm J Physiol Cell Physiol292C829716870827
- MoriguchiYMatsubaraHMoriY1999Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-beta synthesis via transcriptional and posttranscriptional mechanismsCirc Res8410738410325245
- MorilloCAKleinGJJonesDL1995Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillationCirculation911588957867201
- NakajimaHNakajimaHOSalcherO2000Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heartCirc Res86571910720419
- NakajimaYYamagishiTHokariS2000Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP)Anat Rec2581192710645959
- NarineKDeWeverOCathenisK2004Transforming growth factor-beta-induced transition of fibroblasts: a model for myofibroblast procurement in tissue valve engineeringJ Heart Valve Dis13281915086268
- NattelSBursteinBDobrevD2008Atrial remodeling and atrial fibrillation, mechanisms and implicationsCirc Arrhythmia Electrophysiol16273
- NattelSMaguyALe BouterS2007Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillationPhysiol Rev874255617429037
- NattelS1999Atrial electrophysiological remodeling caused by rapid atrial activation: underlying mechanisms and clinical relevance to atrial fibrillationCardiovasc Res4229830810533568
- NattelS2002Therapeutic implications of atrial fibrillation mechanisms: can mechanistic insights be used to improve AF management?Cardiovasc Res543476012062340
- NeylonCBBryantSMLittlePJ1994Transforming growth factor-β1 regulates the expression of ryanodine-sensitive Ca2+ oscillations in cardiac myocytesBiochem Biophys Res Commun204678847980529
- NgCMChengAMyersLA2004TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndromeJ Clin Invest11415869215546004
- OkadaHTakemuraGKosaiK2005Postinfarction gene therapy against transformig growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failureCirculation1112430715867170
- PandoziCSantiniM2001Update on atrial remodelling owing to rate; does atrial fibrillation always ‘beget’ atrial fibrillation?Eur Heart J225415311259142
- ParkerTGPackerSESchneiderMD1990Peptide growth factors can provoke “fetal” contractile protein gene expression in rat cardiac myocytesJ Clin Invest85507141688886
- PauschingerMKnopfDPetschauerS1999Dilated cardiomyopathy is associated with significant changes in collagen type I/III ratioCirculation992750610351968
- PiekEHeldinCHTen DijkeP1999Specificity, diversity, and regulation in TGF-beta superfamily signalingFASEB J1321052410593858
- PottsJDRunyanRB1989Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor betaDev Biol1343924012744239
- Ramos-MondragónRJiménezEAvilaG2008TGF-β1 chronically regulates voltage-gated sodium and potassium channels in neonatal rat atrial myocytesLong Beach, CAAnnual Meeting of the Biophysical Society
- RobertsABWakefieldLM2003The two faces of transforming growth factor beta in carcinogenesisProc Natl Acad Sci U S A1008621312861075
- RobertsABRocheNSWinokurTS1992Role of transforming growth factor-β in maintenance of function of cultured neonatal cardiac myocytes. Autocrine action and reversal of myocytesJ Clin Invest902056621430228
- RosenkranzS2004TGF-β1 and angiotensin networking in cardiac remodelingCardiovasc Res634233215276467
- SandfordLPOrmsbyIGittenberger-de GrootAC1997TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypesDevelopment1242659709217007
- SappinoAPSchürchWGabbianiG1990Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulationsLab Invest63144612116562
- Schultz JelJWittSAGlascockBJ2002TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin IIJ Clin Invest1097879611901187
- ShinagawaKDerakhchanKNattelS2003Pharmacological prevention of atrial tachycardia induced atrial remodeling as a potential therapeutic strategyPacing Clin Electrophysiol267526412698678
- SnydersDJ1999Structure and function of cardiac potassium channelsCardiovasc Res423779010533574
- ThompsonNLBazoberryFSpeirEH1988Transforming growth factor beta-1 in acute myocardial infarction in ratsGrowth Factors19193078566
- TomaselliGFMarbánE1999Electrophysiological remodeling in hypertrophy and heart failureCardiovasc Res422708310533566
- VerheuleSSatoTEverettT4th2004Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta 1Circ Res9414586515117823
- VillarrealFJDillmannWH1992Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagenAm J Physiol262H186161535758
- WalkerGAMastersKSShahDN2004Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve diseaseCirc Res952536015217906
- WehrensXHLehnartSEMarksAR2005Intracellular calcium release and cardiac diseaseAnnu Rev Physiol67699815709953
- WijffelsMCKirchhofCJDorlandR1995Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goatsCirculation921954687671380
- WillisBCBorokZ2007TGF-beta-induced EMT: mechanisms and implications for fibrotic lung diseaseAm J Physiol Lung Cell Mol Physiol293L5253417631612
- WünschMSharmaHSMarkertT1991In situ localization of transforming growth factor beta 1 in porcine heart: enhanced expression after chronic coronary artery constrictionJ Mol Cell Cardiol231051621942095
- YagiTPuJChandraP2002Density and function of inward currents in right atrial cells from chronically fibrillating canine atriaCardiovasc Res544051512062345
- YueLMelnykPGaspoR1999Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillationCirc Res847768410205145