Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease with poor survival outcomes. Bosentan is an oral endothelin-1 receptor antagonist (ERA) that has been shown in a large randomized placebo-controlled trial (BREATHE-1) to be effective at improving exercise tolerance in patients with PAH in functional class III and IV. Further studies have been conducted showing: benefit in smaller subgroups of PAH, eg, congenital heart disease, efficacy in combination with other PAH therapies, eg, sildenafil, improved long-term survival compared with historical controls. More recently, controlled trials of new ERAs have included patients with milder symptoms; those in functional class II. Analysis of the functional class II data is often limited by small numbers. These trials have generally shown a similar treatment effect to bosentan, but there are no controlled trials directly comparing these new ERAs. The EARLY trial exclusively enrolled functional class II patients and assessed hemodynamics at 6 months. Though significant, the reduction in pulmonary vascular resistance is merely a surrogate marker for the intended aim of delaying disease progression. Significant adverse effects associated with bosentan include edema, anemia and transaminase elevation. These may preclude a long duration of treatment. Further studies are required to determine optimum treatment strategy in mild disease.
Introduction to the management of pulmonary arterial hypertension
Pulmonary hypertension is a broad term which refers to elevated pressure in the pulmonary arterial tree. There are several mechanisms by which this can occur and this is reflected in the Venice classification (see ).Citation1 Pulmonary arterial hypertension (PAH) is the term used in the presence of changes which directly affect the pulmonary vessels, ie, group 1 pulmonary hypertension which is the concern of this review. This includes a seemingly diverse group of diseases, but the underlying patho-physiology is thought to be similar: vasoconstriction, smooth muscle cell and endothelial proliferation, and intravascular thrombosis.Citation2 An updated classification is awaited from the expert meeting at Dana Point in 2008, but the make-up of groups 1–5 will not be significantly altered.
Table 1 The Venice classification of pulmonary hypertension 2003
The gold standard diagnostic test is a right heart catheter study and the criteria for diagnosis are: mean pulmonary artery pressure (mPAP) greater than 25 mmHg at rest or 30 mmHg with exercise, pulmonary capillary wedge pressure less than or equal to 15 mmHg and a pulmonary vascular resistance (PVR) of greater than or equal to 240 dynes/s/cm5.Citation1 The diagnosis of pulmonary hypertension is often delayed and requires a thorough assessment to exclude other pathologies and identify the probable cause of pulmonary hypertension. Vaso-reactivity challenge is important to identify those patients who will benefit from calcium channel blockers.Citation3 Cardio-pulmonary exercise testing is used at some centers and may be helpful to identify those patients with exercise-induced pulmonary hypertension.Citation4 It is important to exclude chronic thrombo-embolic disease with ventilation-perfusion scanning.Citation5 Screening programs using echocardiography are recommended for groups at high risk of developing PAH: first-degree relatives of patients with idiopathic PAH, people with known genetic mutations for PAH, scleroderma, congenital heart disease with systemic-to-pulmonary shunts and portal hypertension being considered for liver transplantation.Citation3 More information on further investigations and determining the type of pulmonary hypertension can be found in recognized guidelines.Citation3,Citation5,Citation6
Once a diagnosis of PAH is established there are several assessments widely used to monitor progress. The use of the World Health Organization (WHO) modified functional classification (FC) scale () allows for standardized grading, which is incorporated into treatment guidelines.Citation3 The six-minute walk distance (6MWD) is frequently utilized in trials of PAH therapy as the primary endpoint. It is an appealing measure because of its simplicity and replication of the dominant clinical feature of cardio-respiratory disease – reduced exercise ability – but also carries prognostic significance.Citation7 Additionally oxygen saturations can be measured at peak exercise, and those patients whose levels fall below 90% may benefit from ambulatory oxygen. The Borg dyspnea index is a visual analogue score which attempts to quantify effort during the 6MWD. General health-related questionnaires, such as short form survey 36 and specific ones, such as the Camphor questionnaire, are available tools.
Table 2 Functional classification of pulmonary hypertension modified after the NYHA functional classification according to the World Health Organization
Several general measures are recommended for PAH. Firstly, where there is an associated cause, eg, sickle cell anemia, optimization of this condition is recommended. It is noteworthy that pulmonary vasculopathy associated with systemic lupus erythematosis and mixed connective tissue disease may respond to immunosuppression.Citation8,Citation9 Lifestyle advice includes limiting exercise to avoid symptoms, family planning advice and advance planning if surgery or anesthesia are required.Citation6 Oxygen therapy is appropriate in the presence of hypoxemia and should also be considered for those undertaking air travel as the low cabin pressure may precipitate breathlessness. Because of the high risk of intravascular thrombosis in the small pulmonary arterioles, anticoagulation with warfarin is recommended. For patients in whom right heart failure has developed, diuretics are indicated to offload excess fluid. Digoxin is often prescribed for its inotropic effect. Calcium channel blockers are still used for those patients with evidence of a vasodilator response at right heart catheter.Citation6
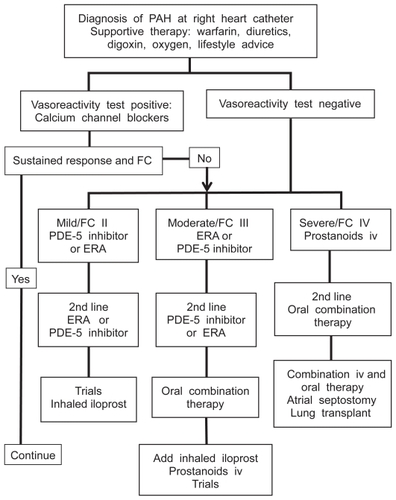
An understanding of the molecular pathways involved has resulted in the development of targeted therapies for PAH. There are three main molecules exploited by current therapies: prostacyclin, endothelin-1 and nitric oxide.Citation2 The first therapy developed was the prostacyclin analogue epoprostenol which requires a continuous intravenous infusion via a central tunneled line. The high maintenance requirement and potential complications of this delivery means that it is usually reserved for patients in functional class IV or who have failed to respond to other therapies.Citation6 The phosphodiesterase-5 inhibitor sildenafil is licensed for PAH in FC I–IV in the USA and FC II–III in Europe. Endothelin receptor antagonists (ERAs) including bosentan, sitaxentan and ambrisentan are oral therapies licensed for use in PAH. As no oral agent has shown superiority there remains debate as to which should be used first line. A treatment algorithm for patients with PAH is shown in to illustrate the place of ERA therapy.Citation6,Citation10
Figure 1 Outline treatment algorithm for pulmonary arterial hypertension (PAH) at a specialist center.
Idiopathic PAH patients make up the majority of study participants, with connective tissue disease associated pulmonary arterial hypertension (CTD-PAH) a significant minority in most trials. Guidance on treatment of specific subtypes is therefore limited and recommendations tend to follow idiopathic PAH guidelines. The use of ERAs is advocated in CTD-PAH as endothelin-1 is felt to play a role in the underlying pathogenesis of these diseases.Citation11 Specific studies using bosentan in HIV, congenital heart disease and pediatric patients have been performed.Citation12–Citation14 Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis are rare and pulmonary edema is a significant problem with vasodilator therapy.Citation6 Persistent pulmonary hypertension of the newborn is managed by neonatalologists. Patients with chronic thrombo-embolic disease not amenable to surgery can be considered for treatment with specific PAH therapies.Citation15 Detailed discussion of the differences between various subgroups and the formulation of treatment algorithms is beyond the scope of this review, and further information can be found in other articles.Citation1,Citation6,Citation16
As PAH is a progressive condition it would be an error to consider any form of the disease ‘mild’. It is possible with screening to detect it at an early stage, when symptoms may be mild. The easiest way to define such a group is to apply the WHO functional classification and accept those in group II as mildly symptomatic. This is far from perfect and is heavily based on patients’ subjective reporting and physicians’ interpretation. A more objective definition might use 6MWD or perhaps the percentage of predicted value, possibly with additional Borg dyspnea scores or quality of life measures. There are many potential confounders, eg, chronic adaptation, symptom exacerbation by co-morbid conditions like lung fibrosis and anemia. It is difficult to see how these could be fully accounted for and so any system, however complicated, is likely to remain flawed. Because of the high mortality in untreated cases there are much data on prognostic implications of various parameters, eg, right atrial pressure (RAP), cardiac index (CI), but these do not correlate with symptoms. Therapies are not licensed subject to hemodynamic parameters or 6MWD and consequently functional classification is the major determinant of initial therapy in PAH.
Bosentan has been licensed for the treatment of FC III/IV PAH since 2002 in North America and Europe. At that time treatment was either calcium channel blockers (for those with a positive vasodilator response) or intravenous prostanoids. Bosentan was therefore a practical first-line option for many patients, and though other oral agents are now available it is still a mainstay of PAH therapy either as monotherapy or, increasingly, as part of combination therapy.
Overview of pharmacology of bosentan in pulmonary arterial hypertension treatment
The mechanism of action of bosentan is competitive inhibition of endothelin-1 receptors.Citation17 Endothelin-1 is a potent vasoconstrictor, which also mediates cell proliferation, fibrosis and inflammation. It is mainly synthesized in endothelial cells and acts locally in autocrine and paracrine fashion.Citation18 Two subtypes of endothelin-1 receptor exist; when found on vascular smooth muscle cells they mediate vasoconstriction. Endothelin receptor subtype A (ETA) is predominantly found in smooth muscle and also on fibroblasts, whereas receptor subtype B (ETB) is expressed on smooth muscle and endothelial cells.Citation19 Endothelial ETB activation mediates clearance of endothelin-1 and vasodilatation by nitric oxide and prostacyclin release.Citation18 Because of these effects ETB activation is theoretically desirable in PAH. Selective agents with relative ETA:ETB affinity of greater than 100:1 have been developed, eg, sitaxentan, relative affinity 6500:1, which have also been shown to reduce endothelin-1 levels.Citation19 Bosentan exhibits a relative ETA:ETB affinity of 20:1 by in vitro assays and is therefore classed as a dual ERA.Citation20 Although ETA selectivity is attractive, these selective agents have not been shown to be superior to bosentan in clinical trials.
Bosentan is an orally active nonpeptide compound with the chemical formula C27H29N5O6S-H2O.Citation17 It is usually started at a dose of 62.5 mg twice daily and uptitrated after 4 weeks to 125 mg twice daily. Bosentan reaches peak plasma concentration three to 5 hours after consumption with an absolute bioavailability of 50% that is not affected by food.Citation20 Bosentan is 98% bound to albumin with a volume of distribution of 30 L and a terminal half-life of 5.4 hours.Citation20 Steady-state plasma concentrations are achieved in 5 days with a multiple dose regimen.Citation20 The pharmacokinetics of bosentan are dose proportional up to 500 mg/day. Adult patients with PAH have a 2-fold increased exposure compared with pediatric patients and healthy subjects.Citation20 Severe renal impairment (creatinine clearance 15 to 30 mL/min) and mild hepatic impairment (Child-Pugh class A) do not have clinically relevant effects, but moderate to severe hepatic impairment are relative contraindications.Citation20
Metabolism of bosentan in the liver occurs by the cytochrome p450 enzymes CYP2C9 and CYP3A4, with excretion of metabolites in the bile. One of the three metabolites identified, Ro 48-5033, may be pharmacologically active.Citation20 Bosentan is an inducer of the CYP2C9 and CYP3A4 enzymes which may explain the increased clearance and reduced plasma levels of bosentan seen at steady-state.Citation20 This induction affects the plasma levels of other compounds metabolized by these enzymes: ciclosporin, glibenclamide, simvastatin and warfarin. Specifically, simvastatin levels may be reduced by 50%; warfarin concentrations are reduced but no relevant changes in INR have been experienced.Citation20 Glibenclamide and bosentan concentrations are both reduced when co-administered, and here is also an increased risk of transaminase elevation.Citation20 Concomitant ciclosporin use results in a reduced ciclosporin concentration and a tripled bosentan steady-state concentration via CYP3A4. The use of ciclosporin and glibenclamide with bosentan is contraindicated. Inhibitors of CYP2C9 (fluconazole, amiodarone) and CYP3A4 (ketoconazole, itraconazole) can be expected to increase bosentan concentration.Citation20 Another important interaction occurs with oral contraceptive agents: concomitant bosentan reduced norethisterone and ethinyl estradiol levels in a pharmacokinetic study and therefore hormonal contraception cannot be relied on.Citation21
Bosentan and Ro 48-5033 have been shown to be substrates of the human organic anion transporting polypeptides OATP1B1 and OATP1B3.Citation22 Ciclosporin A and rifampicin have been shown to inhibit these hepatic uptake transporter molecules which may explain increases in bosentan levels seen with co-administration.Citation22 The increase in bosentan trough levels seen initially with rifampicin turns into a decrease in steady-state conditions probably due to p450 enzyme induction.Citation23 Ritonavir also inhibits OATP-mediated uptake and pharmacological studies have shown increased bosentan levels when co-administered with lopinavir and ritonavir.Citation24 There was also a small reduction in lopinavir and ritonavir and concomitant use of bosentan and anti-retrovirals requires close monitoring. Both enzyme induction and OATP inhibition may be important in the interaction with sildenafil, another oral therapy used in PAH. In healthy subjects, co-administration of bosentan 125 mg twice daily and sildenafil 80 mg 3 times daily resulted in a 50% rise in bosentan levels and fall in sildenafil by nearly two thirds.Citation25 Tadalafil is a phosphodiesterase-5 inhibitor currently in phase III trials for PAH. It is metabolized by CYP3A4 and healthy volunteer studies showed similar results, with reduction in tadalafil levels and increased bosentan exposure when co-administered.Citation26 This may be clinically important but long-term data are awaited from combination studies in PAH.
Efficacy studies, including comparative studies
Bosentan has been studied in several clinical PAH trials with generally positive results. The first human study of bosentan treatment used an intravenous preparation which successfully reduced mPAP and PVR.Citation27 Unfortunately, it also provoked a drop in systemic arterial pressure and vascular resistance and subsequently an oral preparation has been favored.Citation27 After the benefit of oral bosentan in PAH was established in a randomized trial, subsequent papers have attempted to answer several outstanding questions. These include: long-term effect on survival, efficacy in subgroups of PAH and chronic thromboembolic disease, safety and benefit when used in combination with other PAH therapies, and the value in milder disease. A review of the relevant studies with consideration of these questions follows and a summary of bosentan trials is shown in .Citation12–Citation15,Citation28–Citation36
Table 3 PAH studies reporting effect of bosentan
In 2001, a small double-blind, randomized, placebo-controlled trial (study 351) was published. Trial subjects were all in FC III with a 6MWD of 150 to 500 m and had either idiopathic PAH or PAH associated with scleroderma. After 12 weeks improvement was seen in the primary endpoint, the 6MWD, of 70 m and 76 m compared with baseline and placebo respectively.Citation28 Repeated hemodynamic studies performed showed marked improvement in mPAP, PVR, RAP and CI compared with placebo as well.Citation28 Although all cases started in FC III, by 12 weeks nine of the bosentan treatment arm were in FC II.Citation28 The open-label extension showed the effect on 6MWD to be maintained at 6 months and after a year of therapy a significant number (41.4%) of patients had improved functional class.Citation37 To confirm the positive results of study 351 a multi-center, double-blind, randomized trial was undertaken. BREATHE-1 compared bosentan 125 mg and 250 mg twice daily with placebo over 16 weeks. 213 patients in WHO FC III and IV owing to idiopathic PAH or CTD-PAH with 6MWD of 150 to 450 m were enrolled. The observed treatment effect of bosentan on 6MWD was 35 m for the lower dose, licensed dose.Citation29 Other observations made were: improved FC in 42%, now and reduced Borg dyspnea index and prolonged time to clinical worsening (TTCW) compared with placebo.Citation29 One-year and 2-year survival data from the bosentan trial patients with idiopathic PAH (96% and 89%) compare favorably with National Institutes of Health registry predicted survival rates (69% and 57%).Citation38 These are not truly comparable data, but are important because trials in PAH have been unable to show survival benefit in the short controlled phases (a frequent criticism).
The majority of study participants in BREATHE-1 were idiopathic PAH patients. Subgroup analysis of BREATHE-1 by diagnosis suggests a difference in response. Bosentan improved 6WMD in idiopathic PAH, whereas it prevented deterioration in CTD-PAH: a 3 m improvement was seen compared to a 40 m decline in placebo-treated CTD-PAH patients.Citation29 Combined analysis of the CTD-PAH patients in study 351 and BREATHE-1 suggests a trend favoring bosentan with a 22 m improvement in 6MWD.Citation39 This was despite less favorable baseline 6MWD and PVR. Survival estimates of 86% and 73% at 1 and 2 years, respectively, although better than historical data, were not as good as for idiopathic PAH.Citation39 The TRUST study sought to assess survival and quality of life in CTD-PAH patients. At 48 weeks, the survival was 92% with absence of clinical worsening in 68% by Kaplan-Meier estimation.Citation34 The broader implication of these data is that benefit seen in one type of PAH may not be manifest in another. The BREATHE-4 study demonstrated benefit in 16 patients with HIV associated PAH. Sixteen weeks of bosentan therapy improved 6MWD to 424 m from 333 m at baseline, with hemodynamic improvement and 13 patients attaining FC II status.Citation12 A placebo-controlled study (BREATHE-5) looked at the effects of bosentan in 52 subjects with Eisenmenger syndrome with an added safety endpoint of systemic oxygen saturation. 6MWD, mPAP and PVR were significantly improved with only a 1% drop in oxygen saturations.Citation14 Data from the open-label extension reported an overall improvement in 6MWD at 24 weeks with a drop of only 0.5% in systemic arterial saturations.Citation40 A multi-center trial of bosentan in patients with inoperable chronic thromboembolic pulmonary hypertension showed hemodynamic improvement but not benefit in exercise capacity.Citation15
As PAH is a progressive condition it seems reasonable to suppose that early treatment can prevent clinical deterioration in mildly symptomatic patients. Most trials have reported subjects improving from FC III to II before the open-label extension. Therefore many patients in FC II have received treatment, albeit they had to deteriorate to FC III status first. Unfortunately this reflects what happens to many FC II patients in practice. The combined open-label extension studies do not contain enough evidence to justify treating those in FC II. Although patients in FC II have higher mortality and event rates than healthy subjects, these rates are lower than seen in FC III and IV patients. An event-driven outcome would therefore require a longer placebo-controlled period than was ethically acceptable for the early PAH trials. BREATHE-1 reported a diminished 6MWD treatment effect in patients with more favorable hemodynamics: mPAP of < 50 mmHg and CI ≥2.3 L/min/m2.Citation29 The limited effect on 6MWD with sitaxentan in STRIDE-1 raised concerns of a ceiling effect in 6MWD improvement, as no upper limit of 6MWD was specified.Citation41 Trials with disappointing results may point to the inclusion of those with milder PAH as a potential confounder, but several trials with a minority of FC II patients have posted significant results (see ).Citation32,Citation36,Citation42–Citation50 Enough FC I and II patients were treated in the SUPER-1 and ARIES trials to show effect on 6MWD in these subgroups.Citation42,Citation44 Long-term follow-up data from patients treated with the ERA ambrisentan show improved 6MWD at 2 years, relative to baseline, and estimated survival of 95%.Citation51
Table 4 PAH studies including (more than 10) patients in FC II
The EARLY trial remains the only PAH trial designed specifically for FC II patients. It was prospective, double-blinded, randomized and placebo-controlled. Patients with idiopathic, familial, HIV, anorexigen, connective tissue disease and congenital heart disease (with some restrictions) associated PAH were recruited.Citation36 Rather than fixing an absolute upper limit for the baseline 6MWD a value of less than 80% predicted or 500 m with a Borg score ≥ 2 and a PVR ≥ 320 dynes/s/cm5 were specified. Co-primary endpoints of change in PVR and 6MWD at 6 months were used. Secondary endpoints included TTCW and change from baseline to 6 months in: WHO FC, Borg score, RAP, mPAP, CI and mixed venous oxygen saturation (mVO2). Definitions of TTCW vary between trials. EARLY used death, hospitalization due to PAH complications and symptomatic progression (new or worsening right heart failure or a 10% reduction in 6MWD from baseline assessed twice more than 2 weeks apart or 5% reduction in 6MWD from baseline assessed more than 2 weeks apart with an increase in Borg score of at least 2 points). Using this definition the clinical worsening event rate was 14% in the placebo group over 6 months.Citation36
Of the 476 patients screened 139 were not felt to be in functional class II and 76 met exclusion criteria.Citation36 Due to trial rules only 81 patients taking bosentan and 82 assigned placebo completed the study. Mean 6MWD data suggested a trend towards improvement with bosentan: +11.2 m versus −7.9 m for placebo.Citation36 Analysis of PVR values using the Wilcoxon rank-sum test showed a significant decrease in PVR for the bosentan group (83.2% versus placebo 107.5%).Citation36 Other hemodynamic variables that showed improvement were mPAP, CI and mVO2.Citation36 TTCW was delayed with bosentan therapy (p < 0.02) compared with placebo with a hazard ratio of 0.23.Citation36 Twelve patients taking placebo progressed to FC III compared with only 3 patients on bosentan (p < 0.05). N-terminal-pro-BNP levels were only assessed in certain pre-specified centers; the treatment effect was −471 ng/L (p < 0.0005) comparing bosentan with placebo.Citation36 Overall the results were therefore positive, even if some of the selection criteria were unusual.
With specific therapies for PAH acting on three different pathways and their efficacy as monotherapy proven, a logical step forward has been to look at combining agents. In BREATHE-2, 33 patients starting intravenous epoprostenol were randomized to either additional placebo or bosentan. Assessment at 16 weeks showed 6MWD and functional class improvement in both groups with a trend towards better hemodynamics in the bosentan-treated patients.Citation30 A small study conducted in Japan also showed improvement with the addition of bosentan to epoprostenol in idiopathic PAH. Eight patients on stable doses of epoprostenol (average 100 ng/kg/min) had continuous hemodynamic monitoring for 2 days during initiation with 62.5 mg bosentan twice daily.Citation35 Even while reducing epoprostenol to maintain SvO2 at baseline value, systolic pulmonary artery pressure and PVR fell; these changes were maintained in six of the patients at 1 year.Citation35
Several investigators have looked at the combination of bosentan and sildenafil. In an observational cohort of 9 patients with idiopathic PAH adjunctive therapy was deemed necessary after several months due to deterioration.Citation52 Initial response to bosentan was significant (+57 m) to an average of 403 m, but this fell to just 277 m at the time of adding sildenafil between 6 and 16 months later. The resulting increase in 6MWD (+122 m) was sustained for 6 months. Citation52 In a group including 13 idiopathic PAH and twelve scleroderma-associated PAH patients started on bosentan monotherapy, sildenafil was offered instead of intravenous therapy, after clinical deterioration. Improvement in FC and 6MWD was only seen in the idiopathic PAH group however.Citation33 Sildenafil received a license for use in PAH while the EARLY trial was ongoing. Because of this development an amendment was made to the study protocol allowing concomitant sildenafil use; provided it was started more than one month before enrolment and the dose remained steady during the trial. The FC status of these 28 patients before commencing sildenafil is not known. The reduction in PVR in this group was similar to that seen in the main analysis but the confidence interval ranged from −44% to 13% because of the small numbers.Citation36 A trend towards reduction in 6MWD was also seen in this subgroup.Citation36
The effects of bosentan have also been compared with other specific therapies for PAH. The SERAPH investigators randomized 26 patients with PAH in FC III to either standard regimen bosentan or sildenafil (50 mg twice daily for 4 weeks, then 50 mg 3 times daily).Citation31 At 16 weeks there were no significant differences in effect between the two drugs: bosentan improved 6MWD by 59 m compared with 75 m in the sildenafil group.Citation31 Both therapies improved CI, but only sildenafil reduced plasma BNP levels.Citation31 A survival comparison with a historical cohort of 346 IPAH patients treated with epoprostenol favored bosentan. One hundred and thirty-nine patients in FC III at commencement of bosentan had a survival of 97% and 91% at 1 and 2 years compared with 91% and 84% with epoprostenol.Citation53 The STRIDE-2 trial included an open-label bosentan arm in addition to the placebo and sitaxentan treatment arms. After 18 weeks, sitaxentan 100 mg and bosentan both increased 6MWD; the difference in treatment effect was less than two metres.Citation32 Sitaxentan also improved functional class but neither treatment delayed time to clinical worsening.Citation32 The extension study converted all patients to 100 mg dose and followed up for 1 year. No difference was seen in 6MWD or change in functional class.Citation54 For the CTD-PAH subgroup; the difference in TTCW reached significance favoring sitaxentan (hazard ratio 0.20).Citation55 There are no data comparing bosentan and ambrisentan but efficacy data from trials with the three available ERAs do not suggest superiority of any one agent.
Safety and tolerability
The ERA class of drugs are associated with many side effects and even though significant safety problems are rare, there are many cautions necessary for their use. As discussed earlier, the pharmacology of bosentan and ERAs can induce hepatic enzymes making hormonal contraception unreliable. Of course they are contraindicated in pregnancy as well due to teratogenicity. Breast-feeding is not recommended either because it is unknown whether ERAs pass into breast milk. The safety of bosentan use in children with PAH has been explored in an open label study which confirmed safe dose reduction in the pediatric population. Concurrent use of ciclosporin A with any ERA is contraindicated and glibenclamide with bosentan due to drug-drug interactions. Experience with bosentan in HIV associated PAH is limited and interaction with anti-retroviral drugs requires careful monitoring for the same reasons.Citation12 Clinical studies report very few patients stopping bosentan, but many experiencing adverse effects.
The principal problem with bosentan is hepatotoxicity initially manifest as elevated levels of alanine aminotransferase and aspartate aminotransferase. The bosentan information insert quotes an incidence of 11% for elevation of these liver enzymes to levels more than 3 times the upper limit of normal (ULN).Citation56 These changes are dose-dependent; in BREATHE-1 there was a 14% incidence of hepatic enzyme elevation in the 250 mg twice daily group compared with 4% in the 125 mg arm and 3% in the placebo arm.Citation29 Levels above 8 times the ULN occurred in 2 patients on the 125 mg twice daily and 5 patients taking 250 mg twice daily.Citation29 In BREATHE-5, 2 of the 37 patients developed elevated transaminases; this led to discontinuation in one case.Citation14 In TRUST, 17% of CTD-PAH patients developed elevated liver enzymes, requiring discontinuation of bosentan in three.Citation34 Postmarketing surveillance reports led to a strengthening of the warning on monthly liver enzyme monitoring. This includes one case of biopsy confirmed cirrhosis in a patient taking bosentan.Citation57 Although the patient was suffering from an intravenous catheter related infection and subsequently recovered; a contribution of bosentan to the liver damage could not be excluded.Citation57 A more recent case of hepatotoxicity that developed with the addition of methotrexate, resolved after bosentan was stopped and did not recur with reintroduction of methotrexate.Citation58 Currently, product guidance recommends stopping bosentan if levels are more than 5 times ULN, and re-introduction can only be considered if levels were less than 8 times ULN. For levels between 3 and 5 times ULN dose reduction may be adequate, but fortnightly monitoring is recommended.Citation56 Although lasting damage has occurred rarely, hepatic enzyme elevation is the most frequently cited reason for bosentan discontinuation. The reported incidence of elevated transaminases is lower with sitaxentan (7%) and ambrisentan (<1%) therapy. It should be noted that the incidence in the placebo groups in these trials is also lower.Citation19
Hemoglobin levels fall by approximately 0.9 g/dL with bosentan therapy, predominantly in the first few weeks of therapy with stabilization beyond 12 weeks.Citation20 A decrease of >1 g/dL was seen in 68% of patients treated with bosentan compared with 29% on placebo.Citation56 Three patients in study 351 had significant falls in hemoglobin recorded; levels did not drop below 10.4 g/dL though.Citation28 One patient in BREATHE-5 developed a significant anemia with a hemoglobin of <10 g/dL.Citation14,Citation40 The effect of chronic oral therapy on blood pressure does not seem to be dramatic; in contrast to the intravenous preparation. Mean systemic arterial pressure fell by 3 mmHg in BREATHE-1 and 4 mmHg in BREATHE-5. Citation14,Citation29 A safety endpoint was included in BREATHE-5 because of concerns that saturations may be reduced by bosentan. There was a nonsignificant, small reduction in mixed venous saturations in the bosentan group with no adverse events reported as a result.Citation14
When bosentan was trialled as therapy for chronic heart failure it provoked increased peripheral edema within weeks.Citation18 This effect has been documented in PAH with an incidence of 10% in study 351 and many patients anecdotally requiring diuretics or hospital treatment.Citation28 Rates in other PAH trials are higher, eg, BREATHE-2 27%, BREATHE-5 19% and TRUST 17%.Citation14,Citation30,Citation34 The mechanism of edema is not entirely clear; in some patients it may be sequelae of worsening right heart failure (and some trials have considered it so). It may be the result of vasodilatation caused by ERAs, or from effects upon renal tubular function.Citation18 In the open-label extension of study 351, a large number of adverse symptoms were reported: headache, dizziness, cough, dyspnea, syncope, flushing, respiratory tract infection, chest pain, palpitation, fatigue, arthralgia, dyspepsia, epistaxis and nausea.Citation28 Many of these might be attributable to progression of PAH and were not seen with significantly greater frequency in the bosentan groups compared with placebo in BREATHE-1.Citation29 Headache, dizziness, palpitations and chest pain were seen more frequently in the bosentan group in BREATHE-5.Citation14 Diarrhea, exacerbated dyspnea and nausea were reported in more than 10% of TRUST participants.Citation34 These differences in reported side effects may reflect the different study populations.
From the limited amount of data available, most of which is not placebo-controlled, it seems combination therapy is well tolerated. In one report of bosentan and sildenafil combination no greater number of liver enzyme elevations were seen compared with bosentan monotherapy.Citation10 In another trial several patients had to stop sildenafil for severe dyspepsia (mostly CTD-PAH). Two patients developed LFT abnormalities after addition of sildenafil – one resolved with discontinuation of sildenafil, the other with reduction of bosentan. One patient developed intractable headaches which required discontinuation of sildenafil. In BREATHE-2 peripheral edema was significantly more common in the bosentan/epoprostenol group, although liver enzyme elevation was seen more often in the placebo/epoprostenol group.Citation30
Obviously, when using a drug in situations where there may be less positive clinical effect, safety must come under closer scrutiny to ensure that the risk-benefit ratio is still favorable. The commonest adverse events in the bosentan group in the EARLY trial were nasopharyngitis and elevated liver enzymes.Citation36 Twelve patients on bosentan developed transaminase levels over 3 times ULN, 10 within 20 weeks. In all cases transaminase levels returned towards baseline levels with appropriate management (at least 6 discontinued altogether). In the open-label extension seven serious adverse events occurred: cardiac failure and anemia, progression of PAH, abnormal liver enzymes (n = 3), 1 death after seizures probably caused by vasculitis reactivation, and an episode of toxic epidermal necrolysis and acute hepatitis.Citation36 Postmarketing surveillance reports have flagged hypersensitivity, rash, thrombocytopenia, jaundice, anemia requiring transfusion and hepatic cirrhosis/failure as serious adverse events. These potentially grave consequences bear thought when considering therapy in those with mild symptoms.
Patient-focused outcomes such as quality of life
Several different scores can be used to quantify the patient’s perception of improvement in PAH. The Borg dyspnea index or score is the most widely used patient-reported measure in PAH trials; a fall in the score suggests less exertional breathlessness. This was seen in study 351 and BREATHE-1 with a net treatment effect of −0.6 (CI −1.2 to −0.1) in the latter.Citation28,Citation29 BREATHE-4 found bosentan improved Borg score in HIV associated PAH (1.5 vs 3.4 at baseline, p < 0.02).Citation12 Perhaps it should have been expected that there was no change in Borg score was seen in the EARLY trial.Citation36 BREATHE-2 used a dyspnea-fatigue index which did not change with the addition of bosentan.Citation30
PAH trials have not consistently used any one symptom scoring system. Although the camphor score was specifically designed for PAH the different aims of trials means it is often not suited for this purpose. Quality of life assessed by the EQ-5D visual analog scale improved in BREATHE-4 (p < 0.001), as did 6 of the 8 items in the SF-36 health survey form: physical functioning, role-physical, general health perception, vitality, social functioning, mental health.Citation12 The TRUST study analyzed a broader set of items looking for additional benefits in connective tissue disease patients. There were minimal improvements at 48 weeks in SF-36 scores; more patients felt they had improved than deteriorated on the health transition item (mean −0.83, 95% confidence intervals −1.27 to −0.39).Citation34 The health assessment questionnaire and visual analogue scores used tended to increase suggesting a lack of perceived benefit.Citation34 In EARLY the SF-36 health questionnaire scores suggested that 57% of bosentan patients and 38% of placebo patients felt their condition had improved (p < 0.05).Citation36 Breakdown of domains did not show significance for any individual part, the most marked trend was for general health perceptions followed by role-physical and mental health and vitality.
Conclusions, place in therapy
The progressive natural history of PAH gives us a clear rationale for treating the disease early. However, the criteria we use to decide who to treat and how we treat them are still open to debate. The combined complexities of ethics and study design for this patient group mean the optimal trial endpoints are not clear. The EARLY trial shows that unless they are treated we can expect patients in FC II to progress to a more symptomatic state. Recent meta-analysis of PAH trials has demonstrated improved survival with targeted therapy; resulting in a number needed to treat of 20 to prevent one death at 1 year.Citation59 While survival is perhaps the dominant concern in patients with more severe disease, ideally those at the milder end of the spectrum would not progress that far. Those therapies which provide mortality benefit in severe disease may not delay progression in the earlier stages as effectively as other agents. Advocates of targets in PAH therapy would include FC II status as a clear goal. Thus, many PAH patients who are in FC II are treated, if only because they have shown clinical evidence of deterioration previously. Waiting for such evidence before commencing therapy is not a logical approach. The EARLY trial provides clear evidence for treating the FC II patient; this is supported by sub-group data from other trials, eg, ARIES.Citation36,Citation44
Bosentan has been shown to be effective at improving exercise tolerance in PAH patients in FC III and IV in randomized controlled trials. Of the currently available oral therapies for PAH there is more long-term data relating to bosentan use. The existence of small studies which have addressed specific problems and subgroups provides reassurance when confronted with the more unusual cases. In the absence of comparative data showing superiority of any oral therapy in terms of clinical effect, prescribing decisions may be based on other factors. Given the likelihood that a treatment for FC II patients may be needed for several years the ideal drug would be one without significant side effects or drug interactions. No oral therapy is free of adverse effects but bosentan has perhaps a worse side effect profile than the other ERAs and phosphodiesterase-5 inhibitors. Long-term and post-marketing data are awaited to see if the lower incidence of adverse events reported in trials of other agents persists. In conclusion, bosentan seems to offer benefit to those patients with PAH in FC II, but the relatively high incidence of adverse events may be prohibitive for some patients and clinicians.
Disclosure
CJ Valerio had no conflicts of interest to declare. JG Coghlan has received consultancy and lecture fees from Actelion, GlaxoSmithKline, Pfizer and Gilead. His department has benefited from research grants from Actelion and Pfizer.
References
- SimonneauGGalièNRubinLJClinical classification of pulmonary hypertensionJ Am Coll Cardiol200443Suppl S5S12S15194173
- FarberHWLoscalzoJMechanisms of disease: pulmonary hypertensionNew Engl J Med20043511655166515483284
- BarstRMcGoonMTorbickiADiagnosis and differential assessment of pulmonary arterial hypertensionJ Am Coll Cardiol200443SupplS4047
- SunXGHansenJEOudizRJExercise pathophysiology in patients with primary pulmonary hypertensionCirculation200110442943511468205
- McGoonMGuttermanDSteenVScreening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP Evidence-Based Clinical Practice GuidelinesChest2004126SupplS1434
- GalièNTorbickiABarstRGuidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of CardiologyEur Heart J2004252243227815589643
- MiyamotoSNagayaNSatohTClinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension: comparison with cardiopulmonary exercise testingAm J Resp Crit Care Med200016148749210673190
- SanchezOSitbonOJaïsXSimonneauGHumbertGImmunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertensionChest200613018218916840400
- JaisXLaunayDYaiciAImmunosuppressive therapy in lupus- and mixed connective tissue disease–associated pulmonary arterial hypertension a retrospective analysis of twenty-three casesArthritis Rheum20085852153118240255
- HoeperMMMarkevychISpiekerkoetterEWelteTNiedermeyerJGoal-oriented treatment and combination therapy for pulmonary arterial hypertensionEur Respir J20052685886316264047
- GibbsSCorrisPfor National pulmonary hypertension centres of the UK and IrelandConsensus statement on the management of pulmonary hypertension in clinical practice in the UK and IrelandHeart200894Suppl Ii1i4118276826
- SitbonOGressinVSpeichRBosentan for the treatment of human immunodeficiency virus–associated pulmonary arterial hypertensionAm J Respir Crit Care Med20041701212121715317666
- BarstRJIvyDDingemanseJPharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertensionClin Pharmacol Ther20037337238212709727
- GalièNBeghettiMGatzoulisMABosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled studyCirculation2006114485416801459
- JaïsXD’ArminiAMJansaPBosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronic Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trialJ Am Coll Cardiol2008522127213419095129
- McLaughlinVVArcherSLBadeschDBACCF/AHA 2009 Expert Consensus Document on Pulmonary Hypertension: A Report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart AssociationJ Am Coll Cardiol2009531573161919389575
- KenyonKWNappiJMBosentan for the treatment of pulmonary arterial hypertensionAnn Pharmacother2003371055106212841819
- MotteSMcEnteeKNaeijeREndothelin receptor antagonistsPharmacol Ther200611038641416219361
- OpitzCFEwertRKirchWPittrowDInhibition of endothelin receptors in the treatment of pulmonary arterial hypertension: does selectivity matterEur Heart J2008291936194818562303
- DingemanseJvan GiersbergenPLClinical pharmacology of bosentan, a dual endothelin receptor antagonistClin Pharmacokinet2004431089111515568889
- van GiersbergenPLHalabiADingemanseJPharmacokinetic interaction between bosentan and the oral contraceptives norethisterone and ethinyl estradiolInt J Clin Pharmacol Ther20064411311816550733
- TreiberASchneiterRHäuslerSStiegerBBosentan is a substrate of human OATP1B1 and OATP1B3: inhibition of hepatic uptake as the common mechanism of its interactions with cyclosporin A, rifampicin, and sildenafilDrug Metab Dispos2007351400140717496208
- van GiersbergenPLTreiberASchneiterRDietrichHDingemanseJInhibitory and inductive effects of rifampin on the pharmacokinetics of bosentan in healthy subjectsClin Pharmacol Ther20078141441917251982
- McRaeMPLoweCMTianXRitonavir, saquinavir, and efavirenz, but not nevirapine, inhibit bile acid transport in human and rat hepatocytesJ Pharmacol Exp Ther20063181068107516720753
- BurgessGHoogkamerHCollingsLDingemanseJMutual pharmacokinetic interactions between steady-state bosentan and sildenafilEur J Clin Pharmacol200864435018040672
- WrishkoREDingemanseJYuADarsteinCPhillipsDLMitchellMIPharmacokinetic interaction between tadalafil and bosentan in healthy male subjectsJ Clin Pharmacol20084861061818305126
- WilliamsonDJWallmanLLJonesRHemodynamic effects of bosentan, an endothelin receptor antagonist, in patients with pulmonary hypertensionCirculation200010241141810908213
- ChannickRNSimmoneauGSitbonOEffects of the dual endothelin-receptor antagonist Bosentan in patients with pulmonary hypertension: a randomised placebo-controlled studyLancet20013581119112311597664
- RubinLJBadeschDBBarstRJBosentan therapy for pulmonary arterial hypertensionNew Eng J Med200234689690311907289
- HumbertMBarstRJRobbinsIMCombination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2Eur Respir J20042435335915358690
- WilkinsMRPaulGAStrangeJWSildenafil versus Endothelin receptor Antagonist for Pulmonary Hypertension (SERAPH) studyAm J Respir Crit Care Med20051711292129715750042
- BarstRJLanglebenDBadeschDfor STRIDE-2 Study GroupTreatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentanJ Am Coll Cardiol2006472049205616697324
- MathaiSCGirgisREFisherMRAddition of sildenafil to bosentan monotherapy in pulmonary arterial hypertensionEur Respir J20072946947517079256
- DentonCPPopeJEPeterHHLong-term effects of bosentan on quality of life, survival, safety and tolerability in pulmonary arterial hypertension related to connective tissue diseasesAnn Rheum Dis2008671222122818055477
- AkagiSMatsubaraHMiyajiKAdditional effects of bosentan in patients with idiopathic pulmonary arterial hypertension already treated with high-dose epoprostenolCirc J2008721142114618577825
- GalièNRubinLJHoeperMMTreatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trialLancet20083712093210018572079
- SitbonOBadeschDChannickRNEffects of the dual endothelin receptor antagonist bosentan in patients with pulmonary arterial hypertension. A 1-year follow-up studyChest200312424725412853530
- McLaughlinVVSurvival in patients with pulmonary arterial hypertension treated with first-line bosentanEur J Clin Invest200636Suppl 3101516919005
- DentonCPHumbertMRubinLBlackCMBosentan treatment for pulmonary arterial hypertension related to connective tissue disease: a subgroup analysis of the pivotal clinical trials and their open-label extensionsAnn Rheum Dis2006651336134016793845
- GatzoulisMABeghettiMGalièNLonger-term bosentan therapy improves functional capacity in Eisenmenger syndrome: Results of the BREATHE-5 open-label extension studyInt J Cardiol2008127273217658633
- FrostAELanglebenDOudizRThe 6-min walk test (6MW) as an efficacy endpoint in pulmonary arterial hypertension clinical trials: demonstration of a ceiling effectVascul Pharmacol200543363915890561
- GalièNGhofraniHATorbickiAfor the Sildena fil Use in Pulmonary Arterial Hypertension (SUPER) Study GroupSildenafil citrate therapy for pulmonary arterial hypertensionN Engl J Med20053532148215716291984
- GalièNBadeschDOudizRAmbrisentan therapy for pulmonary arterial hypertensionJ Am Coll Cardiol20054652953516053970
- GalièNOlschewskiHOudizRJAmbrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2Circulation20081173010301918506008
- SimonneauGBarstRJGalièNContinuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension. A double-blind, randomized, placebo-controlled trialAm J Respir Crit Care Med200216580080411897647
- GalièNHumbertMVachiéryJLfor Arterial Pulmonary Hypertension and Beraprost European (ALPHABET) Study GroupEffects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trialJ Am Coll Cardiol2002391496150211985913
- BarstRJMcGoonMMcLaughlinVfor Beraprost Study GroupBeraprost therapy for pulmonary arterial hypertensionJ Am Coll Cardiol2003412119212512821234
- BarstRJLanglebenDFrostAfor STRIDE-1 Study GroupSitaxsentan therapy for pulmonary arterial hypertensionAm J Respir Crit Care Med200416944144714630619
- SastryBKSNarasimhanCReddyNKRajuBSClinical efficacy of sildenafil in primary pulmonary hypertension: A randomized, placebo-controlled, double- blind, crossover studyJ Am Coll Cardiol2004431149115315063421
- SimonneauGRubinLGalièNfor the Pulmonary Arterial Hypertension combination Study of Epoprostenol Sildenafil (PACES) Study GroupAddition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertensionAnn Intern Med200814952153018936500
- TorresFLong-term ambrisentan therapy in patients with pulmonary arterial hypertension: an analysis by WHO Functional Class [abstract]Am J Respir Crit Care Med2009179A3370
- HoeperMMFaulenbachCGolponHWinklerJWelteTNiedermeyerJCombination therapy with bosentan and sildenafil in idiopathic pulmonary arterial hypertensionEur Respir J2004241007101015572546
- SitbonOMcLaughlinVVBadeschDBSurvival in patients with class III idiopathic pulmonary arterial hypertension treated with first line oral bosentan compared with an historical cohort of patients started on intravenous epoprostenolThorax2005601025103016055621
- BenzaRLFrostAGirgisRLanglebenDLawrenceECNaeijeRChronic treatment of pulmonary arterial hypertension (PAH) with sitax-entan and bosentan [abstract]Proc Am Thorac Soc20063A729
- HighlandKBStrangeCGirgisREBlackCComparison of sitaxentan and bosentan in pulmonary arterial hypertension associated with connective tissue disease [abstract]Ann Rheum Dis200665Suppl 2393
- No author listedTracleer (bosentan tablets) 62.5 and 125 mg film-coated tablets http://www.tracleer.com/pdf/01 001 01 09 0309_Tra_PromoPI_SinglePages_TR4385_030409_FINAL.pdfAccessed April 2009
- SegalSTravcleer_dhcp_final.pdf [Letter dated 2006 March 1]United States Food and Drug Administration [safety information pages]Silver Spring [updated 2009 June 19; cited 2009 June 19]. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm150762.htmAccessed April 2009
- DwyerNJonesGKilpatrickDSevere hepatotoxicity in a patient on bosentan upon addition of methotrexate: reversible with resumption of methotrexate without bosentanJ Clin Rheumatol200915888919265355
- GalièNManesANegroLPalazziniMBacchi-ReggianiMLBranziAA meta-analysis of randomized controlled trials in pulmonary arterial hypertensionEur Heart J20093039440319155250