
Abstract
Early detection and characterization of molecular events associated with tumorgenesis remain high priorities. Genome-wide epigenetic assays are promising diagnostic tools, as aberrant epigenetic events are frequent and often cancer specific. The deposition and analysis of multiple patient-derived cancer epigenomic profiles contributes to our appreciation of the underlying biology; aiding the detection of novel identifiers for cancer subtypes. Modifying enzymes and co-factors regulating these epigenetic marks are frequently mutated in cancers, and as epigenetic modifications themselves are reversible, this makes their study very attractive with respect to pharmaceutical intervention. Here we focus on the novel modified base, 5-hydoxymethylcytosine, and discuss how genome-wide 5-hydoxymethylcytosine profiling expedites our molecular understanding of cancer, serves as a lineage tracer, classifies the mode of action of potentially carcinogenic agents and clarifies the roles of potential novel cancer drug targets; thus assisting the development of new diagnostic/prognostic tools.
(A) Overview of the DNA methylation/active demethylation cycle. (B) Schematic of the structures of the human TET enzymes. CXXC: domain which can bind directly to unmodified CpG dinucleotides, Cys: cysteine-rich domain, DSBH: double-stranded β-helix domain including binding sites for Fe(II) and 2-OG cofactors. (C) Activation of gene expression can occur through the active demethylation of regions around the transcription start sites (loss of 5mC through a 5hmC intermediate) with elevated promoter proximal and genic 5hmC levels allowing the binding and elongation of the RNAP-II complex, probably in concert with histone modification changes (not shown). Conversion of 5mC into 5hmC (and further derivatives) is mediated by the TET proteins (blue). TET proteins have been shown to interact with OGT as well as HCF1, component of the H3K4 methyltransferase SET1/COMPASS complex, resulting in altered chromatin environments (OGT, HCF1&SET1 complex = Yellow). Thymine DNA glycosylase, TDG, (not shown) is proposed to complete this promoter specific demethylation through base excision repair while regions lacking TDG lead to the accumulation of 5hmC (such as gene bodies and enhancers), perhaps by tracking the RNA Pol II complex in the case of gene bodies. Typical 5hmC (blue) and 5mC (red) profiles at an actively transcribing gene are shown below with patterns observed in cancer shown as an overlay. (D) Review of methods by which Tet enzyme levels or activity may be altered and how these may be related to perturbed epigenetic landscapes in cancer.
5hmC: 5-hydroxymethylcytosine; HCF1: Host Cell Factor 1; OGT: O-GlcNAc transferase.
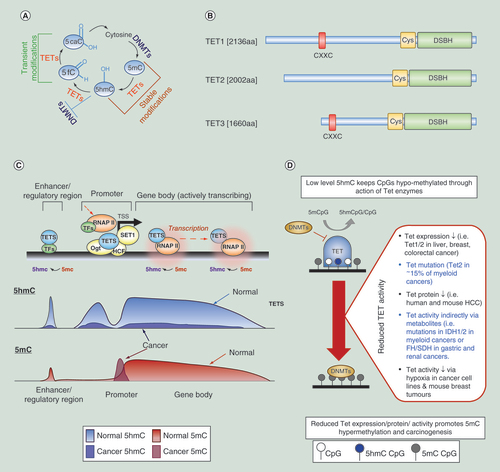
Genomic DNA (gDNA) is isolated from the host organ/tumor and fragmented to a desired size range. Following enrichment of 5hmC marked DNA fragments by hydroxymethyl-DNA-immunoprecipitation (hmeDIP), sequencing libraries can be prepared and genome-wide sequencing carried out. Following bioinformatic processing, regions of differential 5hmC modification can be identified which can lead to: (i) increased understanding of molecular mechanisms, (ii) identification of diagnostic, (iii) or cancer specific (patient stratification) biomarkers and (iv) identification of novel therapeutic targets.
5hmC: 5-hydroxymethylcytosine.
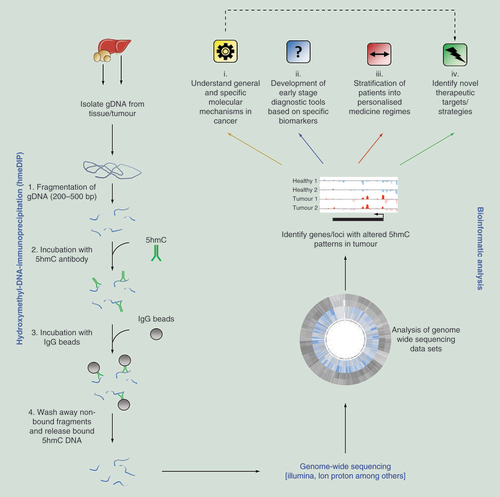
(A) Schematic examples for how epigenetic ‘barcoding’ based on genome-wide 5hmC patterns can aid in the stratification of novel drug compounds depending on potential carcinogenic outcome. (B) Schematic for how 5hmC ‘barcoding’ could be applied to identify a series of epigenetic changes which occur prior to the observation of a tumor. In doing so it may be possible to stratify patients into cohorts based on their specific cancer subtype which would aid in downstream personal medicine regimes.
5hmC: 5-hydroxymethylcytosine.
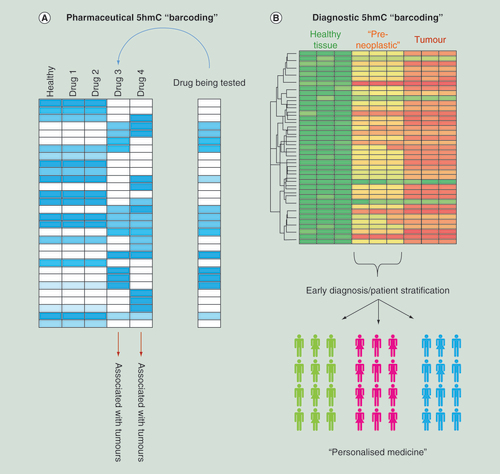
Examples for how 5hmC-based studies can aid in probing of molecular events that occur in metastasis, resulting in the identification of primary tumor sites distinct from metastatic samples or be adapted for novel early stage diagnostic tools through the study of circulating tumor cells in the bloodstream.
5hmC: 5-hydroxymethylcytosine.
5hmC-based analysis can aid both the understanding of the underlying mechanisms associated with cancer progression, identify novel diagnostic tools and result in the development of new therapeutic strategies for cancer treatment.
5hmC: 5-hydroxymethylcytosine.
First draft submitted: 15 September 2016, Accepted for publication: 14 October 2016, Published online: 12 December 2016
Cancer is now largely accepted as being both a genetic and epigenetic disease as specific tumor types are frequently found to harbor common sets of genetic mutations as well as recurrent changes to the epigenetic landscape [Citation1,Citation2]. Although epigenetic control of gene expression is related to the overall phenotypic output of a cell and dysregulation of epigenetic signals has been associated with cancer, whether or not these epigenetic changes are causal or simply reflective of signaling driven changes is still under debate [Citation3,Citation4]. Enzymes and protein complexes which are associated with the regulation of epigenetic patterns are among the most frequently mutated genes in many cancer types – indicating that genetic disruptions of these epigenetic landscapes may be associated either with the initiation and/or the progression of cancer [Citation1,Citation5–7]. Nevertheless these nonsequence based alterations are considered as viable cancer driver alternatives to chromosomal alterations and mutations [Citation8]. It is widely accepted that deregulation of normal DNA modification and gene expression patterns can aid cancer cells to evolve more rapidly, generating intratumoral heterogeneity and thus contribute to increased invasiveness, metastatic potential and potentially drug resistance [Citation9,Citation10]. These observations have been further strengthened by results from the International Cancer Genome Consortium (ICGC) whose genome-wide sequencing of a large cohort of cancers has provided a catalogue of common somatic mutations in the coding regions of numerous epigenetic regulators [Citation11].
In the mammalian genome the bulk of the epigenetic DNA modifications (70–80%) occur within a CpG dinucleotide (Cytosine-phosphate-Guanine), the majority of which include a methylated cytosine base (5-methylcytosine, [5mC]) and are found at relatively low densities throughout the genome [Citation12]. Methylated CpG dinucleotides are typically associated with heterochromatic portions of the genome and also have roles in phenomenon such as imprinting, X-inactivation and transcriptional silencing of genes and retrotransposons [Citation13,Citation14]. High density nonmethylated CpGs occur as CpG Islands (CGIs), which are typically associated with promoter regions of actively and potentially active transcribing genes. Unmodified CpGs can be converted to 5mC through the actions of a group of enzymes known as the DNA methyltransferases (the maintenance methyltransferase DNMT1, and the de novo methyltransferases DNMT3a&3b). These same 5mC marks can be converted back to an unmodified state, through inhibition of DNA methyltransferases and the action of a series of methylcytosine dioxygenase enzymes known as the Ten-Eleven-Translocases (TETs 1, 2&3) that can generate intermediates in a potential DNA demethylation pathway; 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5CaC) (A) [Citation15-Citation16]. The former of these of modified bases, 5hmC, has gathered much interest in recent years due to the fact that its stable relative abundance would predict that its roles may be more purposeful than simply acting as a DNA demethylation intermediate [Citation17]. 5fC and 5caC can be further excised by thymine-DNA glycosylase (Tdg) to allow the restoration of unmodified C by the base excision and repair machinery (A) [Citation18,Citation19]. Oocyte-specific Tdg conditional knockout gives rise to normal offspring which do not exhibit altered levels of zygotic 5hmC, which may indicate the existence of other demethylation mechanisms downstream of 5mC oxidation [Citation20,Citation21].
Recent advances in genome-wide sequencing strategies combined with reduced costs, the accessibility of equipment and growing bioinformatics expertise are opening up new avenues of research for academic, clinical and industrial research [Citation22], most evidently in the cancer field where studies are routinely carried out to define genetic mutations associated with particular cancer subtypes [Citation23]. However in many cancers, tumorigenesis is not always directly related to a clear set of genetic abnormalities but is also linked to changes in the epigenetic landscape [Citation24]. In this review we will focus on how recent genome-wide 5hmC studies have advanced our understanding of cancer at the molecular level and advocate how such studies could further aid in the identification of novel cancer drug targets and the development of new diagnostic tools.
Active DNA demethylation&5hmC
Active DNA demethylation pathways can be mediated through the activity of the TET enzymes (TET1, 2 and 3) in which 5mC bases are converted to 5-hydroxymethyl marked bases by oxidation in an iron and alpha-ketoglutarate dependent manner [Citation15,Citation25](B). Changes in TET activity has been suggested to be linked with altered 5mC patterns in many cancers; a hot topic is delineating how perturbation of this pathway may contribute to cancer initiation and progression [Citation26–30]. Although less abundant in absolute terms than 5mC (between 0.1 and 0.7% of all cytosines) the levels of 5-hydroxy-marked cytosines are far greater than the downstream DNA demethylation modifications; 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) representing approximately 20 × 10-6% cytosine for 5fC and 3 × 10-6% cytosine for 5caC in mouse embryonic stem (ES) cells, indicating that while 5fC and 5CaC may represent transient and ongoing DNA demethylation, the more abundant 5hmC may also have a functional role throughout the genome [Citation31,Citation32].
A myriad of work has revealed that both the global levels as well as the patterns of the modifications vary greatly between tissue and cell types – highlighting that fact that 5hmC can be used as an identifier of cell or tissue type [Citation33]. The consensus view is such that 5hmC modified CpGs are generally depleted over the majority of promoter elements but are enriched over the bodies of transcriptionally active genes and enhancer elements as well as a small number of transcriptional starts sites associated with silenced genes [Citation34,Citation35] (C). This contrasts with 5mC profiles which are present genome wide and enriched at satellite and repeat DNA sequences [Citation34]. Proximal enrichment of 5hmC at enhancers upstream of annotated transcriptional start sites suggests a role for these regions in the regulation of gene expression [Citation36]. The distribution of particular histone modifications around genes is strongly overlapping with peaks of 5hmC in normal tissues, for example active enhancer marks, H3K4me1/H3K27ac, are associated with 5hmC at regions flanking transcription start sites [Citation37,Citation38]. The fact that 5hmC profiles are related to the transcriptional landscape means that it is a far more dynamic modification than 5mC – which is typically thought of as a stable lock on inactive chromatin states. In addition, sequencing studies in ES cells correlates the presence of 5fC and 5caC with hypomethylated promoters of highly expressed genes, which is increased upon Tdg silencing, indicating the presence of an active DNA demethylation pathway on these promoters [Citation39]. These regions were found to be methylated by Dnmt1 and Dnmt3a, and demethylated by a Tet-dependent mechanism [Citation39]. Thus inhibition of Tet function may influence the CGI promoters of both active and repressed genes and this may also be relevant to downregulation of tumor suppressor genes in cancer.
DNA modification perturbations in cancer
Disruption of epigenetic landscapes, including 5hmC and 5mC patterns, is a hallmark of cancer [Citation1,Citation8,Citation10,Citation37,Citation40]. Although the underlying mechanisms of cancer-specific methylation changes are still largely unclear, it is apparent that they occur early in both cancer initiation and progression [Citation41]. In cancerous tissues, DNA methylation patterns are often drastically different from those found in the normal healthy tissue. Three major epigenetic alterations are frequently observed in cancer: global DNA hypomethylation in cancer across large domains and affecting repetitive DNA sequences, global hypo-hydroxymethylation across the majority of the genome including over promoters and gene bodies and discrete gene-specific hypermethylation of CGIs, CGI shores and enhancer elements affecting hundreds of loci [Citation26,Citation42–45]. Given the robust interplay that 5mC and 5hmC exhibit, the observed changes in each modification throughout cancer are dynamically linked [Citation46–48]. For the scope of this review we will focus mainly on the study of 5hmC patterns during carcinogenesis in the context of global hypo-hydroxymethylation and discrete hypermethylation.
Global hypo-hydroxymethylation
The 5hmC modification has been studied across a host of cancers both at the gross global level as well as in more detail by investigating genome-wide patterns [Citation37,Citation49–52]. These studies, most commonly using immunohistochemistry, dot blot assay and mass spectroscopy, consistently report a strong global loss of 5hmC in tumors, in a host of cancer cell lines [Citation33], human and mouse hepatocellular carcinomas [Citation45,Citation48,Citation53], human gastric cancers [Citation54], renal cell carcinomas [Citation55], acute myeloid leukemia [Citation56] and melanomas [Citation57]. Central to the reduction in global 5hmC levels is the fact that the enzymes responsible for generating 5hmC from 5mC – the TET enzymes – as well as several of their co-factors, are often mutated, transcriptionally downregulated or reduced at the protein level [Citation30,Citation48,Citation58]. Current data suggest that there exists substantial amount of 5hmC overlap deposited by the three members as evidenced by the finding that Tet-1, -2 or -3 null mice are viable and that loss of 5hmC is not absolute in Tet1 null mouse livers [Citation48,Citation59]. Short hairpin RNA (shRNA) reduction of each of the TET enzymes in human embryonic carcinoma cells has shown that loss of TET1 resulted in the greatest elevation of 5mC at promoter elements as well as widespread reduction of 5hmC, while depletion of TET2 and TET3 reduces 5hmC at a subset of TET1 targets suggesting some functional co-dependence [Citation60]. All TETs prevent hypermethylation throughout the genome, particularly at CpG island shores where loss of all three TETs was related to hypermethylation events [Citation37,Citation45,Citation61]. Loss of 5hmC at enhancers in Tet2-/- mouse ES cells resulted in their hypermethylation and impacted on gene expression during early stages of ES cell differentiation [Citation62]. This implies that alterations in enhancer function via epigenetic perturbation may affect gene expression during tumorigenesis.
The gene encoding TET1 was first identified as fusion partner of the mixed lineage leukemia (MLL1) gene in patients with acute myeloid leukemia [Citation63]. TET1 binds preferentially to CGIs via a CXXC domain and is frequently disrupted (either transcriptionally silenced or reduced in activity) during the progression of cancer, while re-expression in lung and colon cancer cells inhibits the proliferation and growth of tumor xenografts suggesting an important regulatory role during carcinogenesis [Citation27,Citation64]. Although specific mutations within the TET1 gene have not been directly associated with cancer progression, reduced transcriptional and/or protein levels of TET1 has been reported in colon, gastric, lung and liver cancers while TET2 transcription/protein levels are more typically reduced in leukemia and melanoma [Citation27,Citation46,Citation53,Citation57,Citation65–67] (D). Mutation in key cell surface receptors such as oncogenic epidermal growth factor receptor (EGFR) can result in the repression of TET1, but not TET2 or TET3, via the transcription factor C/EBPα which in turn induces silencing of a series of tumor suppressors in lung cancer and glioblastoma cells [Citation64]. TET1 downregulation has also been shown to promote malignancy in breast cancer and to act as a tumor suppressor that can inhibit colon cancer growth by de-repressing inhibitors of the WNT pathway [Citation27,Citation65,Citation68–70]. Interestingly TET1 is itself found both methylated and transcriptionally repressed in a series of cell lines and primary tumors of multiple carcinomas and lymphoma, although whether or not the methylation is itself causative or reflective of TET transcriptional inactivation is still to be fully elucidated [Citation29,Citation65]. However as mutations in TET1 are very infrequently found in cancer, particularly compared with TET2 in leukemia, signal induced epigenetic silencing may be an alternative method by which TET and 5hmC levels are reduced in cancer.
Aside from genetic or transcriptomic perturbation, the activity of TET enzymes can be inhibited or stimulated in the presence of particular co-factors, metabolites and post-translational modifications (D). This is most evident in a series of leukemias, glioblastomas and cholangiocarcinomas in which individuals harbor mutations in the genes IDH1 and IDH2 – the Krebs cycle enzymes isocitrate dehydrogenase 1 and 2 – responsible for the conversion of alpha ketoglutarate (αKG) into 2-hydroxyglutarate (2HG). These mutations result in overactive IDH1/2 enzymes which in turn results in elevated 2HG which inhibits TET catalytic activity [Citation46]. In addition to the IDH1 and 2 enzymes, mutations in two other Krebs cycle proteins, fumarate hydratase (FH) and succinate dehydrogenase (SDH), are relatively common in a subset of human cancers including Gastrointestinal stromal tumors (3–8% of SDH cases), renal cell carcinomas (1–4% of SDH and 71–93% of FH cases) and paraganglioma (12–15% of SDH cases) [Citation71–73]. Mutations in FH and SDH lead to an accumulation of fumarate and succinate which can inhibit multiple αKG-dependent dioxygenases, including the TET family of enzymes. In contrast it has been shown that increasing the levels of ascorbic acid (vitamin C) stimulates TET protein enzymatic activity in both cultured cells as well as mouse tissues [Citation74,Citation75]. Although the precise mechanism is currently unknown it is believed that vitamin C interacts with the catalytic domain of the TET enzymes resulting in a localized reduced environment favoring efficient binding of the Iron (Fe[II]) co-factor. The activity of TET enzymes can also be reduced by tumor hypoxia in human and mouse cells, which occurs independently of hypoxia-associated alterations in TET expression and depends directly on oxygen shortage [Citation30]. This can result in increased hypermethylation at gene promoters in vitro; patients exhibit markedly more methylation at selected promoters in hypoxic tumor tissue that is independent of proliferation, stromal cell infiltration and tumor characteristics. Increased hypoxia in mouse breast tumors results in hypermethylation, while restoration of tumor oxygenation abrogates this effect. Taken together these results highlight the complex relationship between 5hmC disruption and cancer progression that is not only reliant directly on the transcriptional state of the TET enzymes but the overall environment in the cancer cell (D).
In concert with a reduced global level during carcinogenesis, genome-wide patterns of 5hmC are also strongly altered between tumor samples and normal surrounding tissue [Citation37,Citation45,Citation48,Citation57,Citation61]. In melanoma, there is both loss and gain of genic 5hmC at a large number of genes – although the changes in 5mC were far more subtle than for 5hmC. These genes tended to be associated with melanoma related pathways, Wnt signaling components and not surprisingly general cancer progression. By reinstating normal levels of expression of the TET enzymes it was possible to somewhat restore the normal 5hmC landscape, resulting in increased tumor-free survival in a zebrafish model of melanoma [Citation57]. In lung and liver cancers the specific relationship between 5hmC and sets of chromatin marks present in normal tissue is largely absent in tumors, which may drive or reflect altered regulation of gene expression [Citation37].
Discrete hyper-methylation events in cancer
Typically, hyper-methylation events occur at either normally un-methylated CGI promoters, CGI ‘shores’ (the regions up to 2kb away from a CGI) or gene regulatory enhancer elements [Citation37,Citation43–44,Citation48,Citation76]. The mechanisms by which these cancer-associated epigenetic patterns arise are still debated however recent evidence suggests that disruption of the normal DNA methylation/demethylation cycle during carcinogenesis may be responsible for aberrant CGI hyper-methylation events [Citation10]. Inactivation of TET1 is associated with hematopoietic malignancy in which loss of 5hmC and/or gain of 5mC on promoters in TET1−/− cells may result in downregulation of expression and derailment of the differentiation process [Citation77]. Of interest is the observation that oncogenic KRAS can inhibit TET1 expression via the ERK-signaling pathway; restoration of TET1 expression by ERK pathway inhibition or ectopic TET1 reintroduction in KRAS-transformed cells reactivates a select number of target genes [Citation29]. This indicates a dichotomy between signaling-induced regulation of methylation of a discrete number of CGI target genes and generalized tissue determined CpG island methylator profiles that are variable within tumor types [Citation10,Citation78]. In the latter case, de novo methylation occurs predominately at already silenced genes (passenger genes) and there does not affect their expression status while in the former, silencing by DNA methylation of select target genes is dependent on an active signaling pathway and therefore is not epigenetic in character [Citation29,Citation40]. Oncogenic RAS- or BRAF-directed transcriptional silencing of tumor suppressor genes is not truly epigenetic because the activated oncogene is required for both initiation of the pathway and maintenance of repression [Citation3–4,Citation79]. This consideration leads to two questions: how do tumor suppressor genes become methylated and how is DNA methylation of tumor suppressor genes inherited through multiple generations [Citation78]? One suggestion would be that DNA methylation at tumor suppressor genes is not epigenetically inherited, but is maintained by an instructive transcriptional mechanism that can potentially repress multiple genes. In contrast, we have recently shown that the aberrant CGI hyper-methylation in several mouse models of liver cancer occurs at sites marked by a unique chromatin state in the healthy liver [Citation48]. The promoter proximal sites destined to become hyper-methylated in liver cancer were found to be rich in 5hmC and associated with ‘bivalently’ marked histone tail modifications (H3K27me3 and H3K4me3), which are typically associated with a transcriptionally poised but not active states. We observe loss of 5hmC occurs at these sites prior to accumulation of 5mC and this is related to a reduction in the levels of the TET1 enzyme, which has previously been shown to bind preferentially to CGIs. Loss or reduced binding of TET1 from these CGIs would ultimately result in a loss of active, ‘protective’ DNA demethylation and acquisition of 5mC.
Although aberrant hyper-methylation at CGIs is often invoked as a mechanism causing the transcriptional inactivation of tumor suppressor genes that directly drives the carcinogenic process, many of the genes associated with hypermethylated CGIs in cancer are already silent in the host tissue to begin with [Citation40,Citation48]. Emerging data reveal that changes in 5mC over enhancer elements may instead be more related to the phenotypic and transcriptomic changes observed during cancer progression [Citation37]. In recent studies enhancers are consistently the most differentially methylated regions during the progression from normal tissue to primary tumors and then to metastases, compared with other genomic features, and changes in the 5mC levels at these loci were linked to cancer type as well as the overall patient outcome [Citation37,Citation44]. Future focus on such loci will be key to unravelling the role of DNA methylation and hydroxymethylation disruption during cancer progression [Citation36]. In addition, the anticancer effects of DNA methyltransferase inhibitors has been linked with upregulation of immune signaling in cancer through the viral defense pathway, independently of CpG island methylator profiles [Citation80,Citation81]. Upregulation of hypermethylated endogenous retrovirus genes accompanies the response and endogenous retrovirus overexpression activates the response. Interferon pathway genes were also upregulated by the DNMT inhibitor, 5-Azacytidine (AZA), and this was correlated with increased expression of endogenous retroviral transcripts rather than de-repression of interferon pathway transcription factors [80,81].
Applications of genome-wide 5hmC studies in cancer research
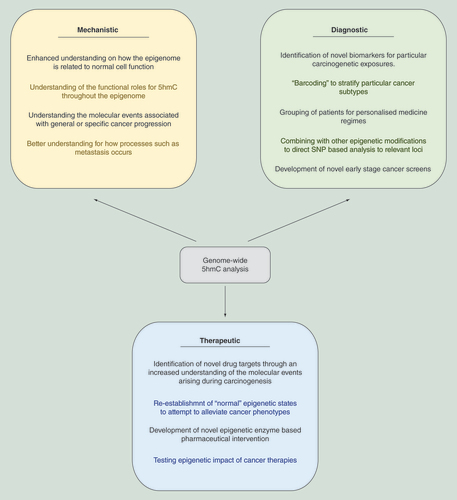
The rapid development of genome-wide sequencing-based technologies is resulting in an ever growing number of studies across a broad range of research areas [Citation22]. Currently the bulk of genome-wide 5hmC datasets are either generated through affinity-based methods (antibody or chemical capture – see [Citation35]) or emerging single base resolution-based strategies which can differentiate 5mC from 5hmC [Citation37,Citation82–83]. Although incredibly informative, these latter approaches require between 50 and 100× coverage to accurately call 5hmC content and as such are both costly and data heavy. Bioinformatic pipelines are in development to extract usable information based on 15X coverable using single nucleotide resolution methods [Citation37]. Here we highlight the utility of 5hmC-based assays – particularly those centered on antibody-based 5-hydroxmethyl DNA immunoprecipitation or hmeDIP – to both aid in the understanding of how cancer progresses at the molecular level as well as how such research can impact on the development of diagnostic tools and therapeutic strategies ().
5hmC ‘barcoding’ to identify carcinogenic compounds
As discussed above, 5hmC patterns are dynamic, tissue specific and strongly perturbed during disease progression [Citation84]. We have previously reported that in the mouse liver that promoter 5hmC patterns can reflect both acute and chronic exposure to carcinogenic agents in a model of nongenotoxic carcinogenesis [Citation84–86]. In addition, a global decrease in the level of 5hmC was noted following subchronic exposures to the genotoxic carcinogens riddelliine and aristolochic acid in rat liver [Citation87]. Thus, 5hmC can be regarded as an ‘identifier of cell type or disease state,’ with specific 5hmC patterns dependant on the length of time of exposure to a carcinogenic agent. Through administration of the anticonvulsant Phenobarbital (PB), we noted a number of time dependant gene body and regulatory region changes in the liver hydroxymethylome with only modest changes observed in the methylome. Importantly, these time dependant changes were reproducible across individual mice – highlighting the potential robustness of 5hmC analysis for industrial and clinical-based assays using isogenic animal models. Changes in genome-wide 5hmC patterns were far more efficient at stratifying exposed animals from the control cohort than analysis of 5mC modifications [Citation85,Citation86]. A series of genes exhibited a strong elevation in their genic 5hmC level, some of which have well defined roles in the metabolism of drugs such as PB; the cytochrome P450s Cyp2b10 and Cyp2c55 (B). Others were related to key signaling pathways associated with cancer progression such as Wisp1 (WNT1 inducible signaling pathway protein 1) and the noncoding RNA cluster Dlk1/Dio3 [Citation85,Citation88–89]. As such, analysis of 5hmC perturbations in the drug exposed animal can result in the identification of novel drug/disease specific biomarkers as well as giving an insight into the molecular mechanisms for how particular compounds promote tumorigenesis. Ultimately, using genome-wide based approaches it may be possible to generate 5hmC ‘barcodes’ (digital outputs unique to a given drug exposure/disease state) in which loci exhibiting reproducible 5hmC change can be compared with healthy tissues (normal 5hmC patterns), as well as to a catalogue of barcodes generated through exposure to different compounds, some of which may be carcinogenic (A). Through the comparative analysis of 5hmC barcodes it will be possible to delineate the response of novel drug compounds and through analysis of 5hmC changes predict the likelihood that a given drug will result in a carcinogenic effect in the host. Such approaches may be particularly useful in satisfying the 3Rs (replace, reduce and refine) in both general scientific studies and industrial toxicity testing screens.
5hmC profiling to aid in the identification of novel therapeutic targets & strategies
As the epigenome is often dramatically altered in cancer there has been much interest in linking these perturbations with the identification of novel drug targets. It is critical to learn if the reinstatement of normal epigenetic patterns is coincident with disease reversal. Identification of genes exhibiting strong 5hmC changes has the potential to identify novel therapeutic targets, while the study of 5hmC patterns using in vitro or in vivo models in which potential tumor suppressor genes or oncogenes are mutated or removed can give a novel insight into their function. For example, the enzyme TET1 has been defined as a tumor suppressor that inhibits both colon cancer and hematopoietic malignancies [Citation27,Citation77] and, as described above, is along with TET2 and TET3 responsible for the conversion of 5mC to 5hmC in mammalian systems. TET1 activity is lost in several cancer types, including liver cancer, and has been linked to increased proliferation and metastatic potential in human gastric cancer [Citation66]. We have previously shown through genome-wide sequencing in Tet1 deficient mice that a global reduction in the levels of 5hmC occurs in mutant liver compared with its healthy wild-type counterpart [Citation48]. As the re-expression of TET1 in colorectal cancer cells results in a reduction in the number of tumors in mouse xenografts it will be important to study if such recovery is directly related to a restoration of epigenetic state. One potential therapeutic strategy is centered on the TET enzyme co-factor vitamin C, which has been found to increase the activity of the TET enzymes [Citation74,Citation75]. In a recent study, Gustafson et al. found that treatment of melanoma cell lines with vitamin C at relatively physiological doses increased the level of 5hmC and decreased tumor-cell invasiveness [Citation90].
Application of diagnostic 5hmC-based studies in personalized cancer treatment regimes
The identification of novel biomarkers that can define specific cancer subtypes at an early stage remains a high priority in oncology. The dynamic nature of 5hmC during cancer progression reveals its utility in understanding how cancers develop as well as identifying different cancer types. 5hmC holds both prognostic and diagnostic value – as shown in recent studies for both prostate cancer and gliomas where a global reduction in the mark was associated with poor prognostic outcomes [Citation53,Citation91–93]. Application of genome-wide sequencing assays to such samples would give a valuable insight into the epigenetic changes occurring in particular cancer subtypes and aid in the identification of novel early stage diagnostic biomarkers. We have previously shown that during the progression of three independent mouse liver cancers (either PB drug exposed, Ha-Ras mutated or nonalcoholic fatty liver disease-associated hepatocellular carcinomas) that although global promoter core hypo-hydroxymethylation/hyper-methylation is a hallmark of cancer progression, there are a number of unique epigenetic perturbations linked to specific tumor pathologies. Such a result reinforces the possibility that combined epigenetic analysis may allow the stratification of particular tumor subtypes [Citation40,Citation94–95] and may be a useful diagnostic tool in humans [Citation45]. Epigenetic ‘barcoding’ may be able to identify particular subtypes of a given cancer, which will allow for stratification and assessment toward downstream clinical treatment regimens (B). Application of similar studies to test the 5hmC patterns in circulating tumor cells in the serum of blood samples may provide novel early stage diagnostic tools while investigating 5hmC landscapes at isolated metastatic samples may be able to identify an unknown primary tumor site based on epigenetic perturbations common to particular tumor types ().
The potential for epigenetic-based studies to refine clinical decision-making holds great promise. Indeed in a recent small study of oncogenic EGFR lung cancers it was reported that individuals who exhibited resistance to EGFR tyrosine kinase inhibitors did not express the TET1, unlike the tyrosine kinase inhibitors responsive samples, which instead exhibited elevated TET1 levels [Citation64]. Such a study highlights the importance for further tests establishing the utility of TET1 expression – and expansion to investigate 5hmC patterns in resistant individuals – to define biomarkers for predicting responses to cancer therapeutic agents.
Conclusion
The discovery of the dynamic DNA modification, 5hmC, has provided mechanistic insights into the process of DNA methylation reprogramming that accompanies cellular transformation of multiple cancer types. At the same time, using genetically similar mouse models, it has proved to be a potential read out of physiological and environmental induced changes in tissue status. Whether this is applicable to genetically diverse human populations, experiencing multiple exposure environments remains to be assessed. However 5hmC profiling has proved to be efficacious in tracking and describing the epigenetic dysregulation associated with human tumor samples. It provides additional data for integrative pathway analysis (IPA) that may correlate with documented phenotypic, pathological, genetic and molecular outcomes to better describe disease states [Citation96]. Is it possible that there are characteristic epigenetic disruption signatures that remain after exposure to a disruptive agent? Resolution of these questions will aid in the development of improved biomarkers and eventual therapies.
Future perspective
Genome-wide epigenetic-based assays continue to hold tremendous potential toward both the enhancement of our understanding of cancer progression at the molecular level as well as in the development of new diagnostic screens with direct clinical utility. Not only are these epigenetic patterns strongly altered during the early stages of cancer progression, emerging evidence suggests that these are specific to particular cancer types – highlighting the potential utility in stratification of patients into optimal downstream treatment regimes. Linking these stratifications with genetic studies will be important. There may be SNPs associated with specific epigenomic responses that link to enhancer or topological chromatin organization [Citation38]. Unlike genetic mutations which are encoded in the DNA sequence – epigenetic patterns are potentially reversible. As such the development of refined drugs that restore normal epigenetic patterns are of great interest in the field of cancer research, as they hold great promise as effective treatments in combined therapies [Citation80,Citation81]. Combining genome-wide epigenetic-based assay with cutting edge technologies – such as recent advances in CRISPR–Cas9 technologies which allow researcher to manipulate the epigenetic states of specific loci [Citation97] – will be pivotal in driving future research into cancer progression and in the identification of novel cancer therapies.
Genome-wide sequencing technologies aimed at mapping epigenetic DNA modifications have shed light on the origins of epigenetic abnormalities in cancer ().
5-hydroxymethylcytosine (5hmC) based analysis lends itself well to the field of cancer research due to its conserved tissue/cell type specific genome-wide distribution, relationship to transcriptional state, association with enhancers and relatively dynamic behavior during carcinogenesis.
Layering of 5hmC datasets against other epigenetic modifications can further refine analytical approaches, as has been recently shown in T-cell specific disease such as Crohn’s disease and multiple sclerosis Citation38. Here combined regions of differential 5hmC signal were overlapped to histone marks denoting enhancer elements that were linked with disease associated genetic variants.
As the number of studies employing genome-wide epigenetic-based assays increases, so too will our overall understanding of how cancer progresses at the molecular level, our identification of novel diagnostic tools and development of novel therapeutic interventions.
Author contributions
J Thomson and R Meehan wrote manuscript and drew the figures. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank M Drake (QMRI) and D Sproul (IGMM) at the University of Edinburgh for helpful comments and insight.
Financial & competing interests disclosure
J Thomson was a recipient of IMI-MARCAR funded career development fellowships at the MRC HGU. Part of this work was supported by the Metastasis prize from the The Beug Foundation to J Thomson. R Meehan is supported by the Medical Research Council (Project Ref: MC_PC_U127574433). Work in R Meehan’s lab is supported by Cefic-LRI, the BBSRC and the MRC. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Feinberg AP , KoldobskiyMA , GondorA . Epigenetic modulators, modifiers and mediators in cancer aetiology and progression . Nat. Rev. Genet.17 ( 5 ), 284 – 299 ( 2016 ).
- Liu F , WangL , PernaF , NimerSD . Beyond transcription factors: how oncogenic signalling reshapes the epigenetic landscape . Nat. Rev. Cancer16 ( 6 ), 359 – 372 ( 2016 ).
- Fang M , OuJ , HutchinsonL , GreenMR . The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG Island Methylator phenotype . Mol. Cell55 ( 6 ), 904 – 915 ( 2014 ).
- Serra RW , FangM , ParkSM , HutchinsonL , GreenMR . A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype . Elife3 , e02313 ( 2014 ).
- Shah MA , DentonEL , ArrowsmithCH , LupienM , SchapiraM . A global assessment of cancer genomic alterations in epigenetic mechanisms . Epigenetics Chromatin7 ( 1 ), 29 ( 2014 ).
- Yang Z , JonesA , WidschwendterM , TeschendorffAE . An integrative pan-cancer-wide analysis of epigenetic enzymes reveals universal patterns of epigenomic deregulation in cancer . Genome Biol.16 , 140 ( 2015 ).
- Dalgliesh GL , FurgeK , GreenmanCet al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes . Nature463 ( 7279 ), 360 – 363 ( 2010 ).
- Hanahan D , WeinbergRA . Hallmarks of cancer: the next generation . Cell144 ( 5 ), 646 – 674 ( 2011 ).
- Timp W , FeinbergAP . Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host . Nat. Rev. Cancer13 ( 7 ), 497 – 510 ( 2013 ).
- Sproul D , MeehanRR . Genomic insights into cancer-associated aberrant CpG island hypermethylation . Brief Funct. Genomics12 ( 3 ), 174 – 190 ( 2013 ).
- Forbes SA , BindalN , BamfordSet al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer . Nucleic Acids Res.39 , D945 – D950 ( 2011 ).
- Bird A . DNA methylation patterns and epigenetic memory . Genes Dev.16 ( 1 ), 6 – 21 ( 2002 ).
- Dunican DS , CruickshanksHA , SuzukiMet al. Lsh regulates LTR retrotransposon repression independently of Dnmt3b function . Genome Biol.14 ( 12 ), R146 ( 2013 ).
- Hackett JA , ReddingtonJP , NestorCEet al. Promoter DNA methylation couples genome-defence mechanisms to epigenetic reprogramming in the mouse germline . Development139 ( 19 ), 3623 – 3632 ( 2012 ).
- Tahiliani M , KohKP , ShenYet al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1 . Science324 ( 5929 ), 930 – 935 ( 2009 ).
- Thomson JP , HunterJM , MeehanRR . Deep C diving: mapping the low-abundance modifications of the DNA demethylation pathway . Genome Biol.14 ( 5 ), 118 ( 2013 ).
- Bachman M , Uribe-LewisS , YangX , WilliamsM , MurrellA , BalasubramanianS . 5-Hydroxymethylcytosine is a predominantly stable DNA modification . Nat. Chem.6 ( 12 ), 1049 – 1055 ( 2014 ).
- He YF , LiBZ , LiZet al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA . Science333 ( 6047 ), 1303 – 1307 ( 2011 ).
- Xu X , WattDS , LiuC . Multifaceted roles for thymine DNA glycosylase in embryonic development and human carcinogenesis . Acta Biochim. Biophys. Sin.48 ( 1 ), 82 – 89 ( 2016 ).
- Guo F , LiX , LiangDet al. Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote . Cell Stem Cell15 ( 4 ), 447 – 458 ( 2014 ).
- Xu G-L , WongJ . Oxidative DNA demethylation mediated by Tet enzymes . Natl Sci. Rev.2 , 318 – 328 ( 2015 ).
- Goodwin S , McphersonJD , MccombieWR . Coming of age: ten years of next-generation sequencing technologies . Nat. Rev. Genet.17 ( 6 ), 333 – 351 ( 2016 ).
- Nik-Zainal S , DaviesH , StaafJet al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences . Nature534 ( 7605 ), 47 – 54 ( 2016 ).
- Gu L , FrommelSC , OakesCCet al. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence . Nat. Genet.47 ( 1 ), 22 – 30 ( 2015 ).
- Kriaucionis S , HeintzN . The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain . Science324 ( 5929 ), 929 – 930 ( 2009 ).
- Ficz G , GribbenJG . Loss of 5-hydroxymethylcytosine in cancer: cause or consequence?Genomics104 ( 5 ), 352 – 357 ( 2014 ).
- Neri F , DettoriD , IncarnatoDet al. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway . Oncogene34 ( 32 ), 4168 – 4176 ( 2015 ).
- Ichimura N , ShinjoK , AnBet al. Aberrant TET1 methylation closely associated with CpG island methylator phenotype in colorectal cancer . Cancer Prev. Res. (Phila.)8 ( 8 ), 702 – 711 ( 2015 ).
- Wu BK , BrennerC . Suppression of TET1-dependent DNA demethylation is essential for KRAS-mediated transformation . Cell Rep.9 ( 5 ), 1827 – 1840 ( 2014 ).
- Thienpont B , SteinbacherJ , ZhaoHet al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity . Nature537 ( 7618 ), 63 – 68 ( 2016 ).
- Laird A , ThomsonJP , HarrisonDJ , MeehanRR . 5-hydroxymethylcytosine profiling as an indicator of cellular state . Epigenomics5 ( 6 ), 655 – 669 ( 2013 ).
- Shen L , WuH , DiepDet al. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics . Cell153 ( 3 ), 692 – 706 ( 2013 ).
- Nestor CE , OttavianoR , ReddingtonJet al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes . Genome Res.22 ( 3 ), 467 – 477 ( 2012 ).
- Song CX , YiC , HeC . Mapping recently identified nucleotide variants in the genome and transcriptome . Nat. Biotechnol.30 ( 11 ), 1107 – 1116 ( 2012 ).
- Thomson JP , HunterJM , NestorCEet al. Comparative analysis of affinity-based 5-hydroxymethylation enrichment techniques . Nucleic Acids Res.41 ( 22 ), e206 ( 2013 ).
- Bogdanovic O , SmitsAH , De La Calle MustienesEet al. Active DNA demethylation at enhancers during the vertebrate phylotypic period . Nat. Genet.48 ( 4 ), 417 – 426 ( 2016 ).
- Li X , LiuY , SalzT , HansenKD , FeinbergAP . Whole genome analysis of the methylome and hydroxymethylome in normal and malignant lung and liver . Genome Res.26 , 1730 – 1741 ( 2016 ).
- Nestor CE , LentiniA , Hägg-NilssonCet al. 5-hydroxymethylcytosine remodeling precedes lineage specification during differentiation of human CD4+ T-cells . Cell Reports16 ( 2 ), 559 – 570 ( 2016 ).
- Neri F , IncarnatoD , KrepelovaAet al. Single-base resolution analysis of 5-formyl and 5-carboxyl cytosine reveals promoter DNA methylation dynamics . Cell Rep.10 , 674 – 683 ( 2015 ).
- Sproul D , KitchenRR , NestorCEet al. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns . Genome Biol.13 ( 10 ), R84 ( 2012 ).
- Feinberg AP , OhlssonR , HenikoffS . The epigenetic progenitor origin of human cancer . Nat. Rev. Genet.7 ( 1 ), 21 – 33 ( 2006 ).
- Baylin SB , JonesPA . A decade of exploring the cancer epigenome – biological and translational implications . Nat. Rev. Cancer11 ( 10 ), 726 – 734 ( 2011 ).
- Irizarry RA , Ladd-AcostaC , WenBet al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores . Nat. Genet.41 ( 2 ), 178 – 186 ( 2009 ).
- Bell RE , GolanT , SheinboimDet al. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality . Genome Res.26 ( 5 ), 601 – 611 ( 2016 ).
- Gao F , XiaY , WangJet al. Integrated analyses of DNA methylation and hydroxymethylation reveal tumor suppressive roles of ECM1, ATF5, and EOMES in human hepatocellular carcinoma . Genome Biol.15 ( 12 ), 533 ( 2014 ).
- Figueroa ME , Abdel-WahabO , LuCet al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation . Cancer Cell18 ( 6 ), 553 – 567 ( 2010 ).
- Rampal R , AlkalinA , MadzoJet al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia . Cell Rep.9 ( 5 ), 1841 – 1855 ( 2014 ).
- Thomson JP , OttavianoR , UnterbergerEBet al. Loss of Tet1-associated 5-hydroxymethylcytosine is concomitant with aberrant promoter hypermethylation in liver cancer . Cancer Res.76 ( 10 ), 3097 – 3108 ( 2016 ).
- Gambichler T , SandM , SkryganM . Loss of 5-hydroxymethylcytosine and ten-eleven translocation 2 protein expression in malignant melanoma . Melanoma Res.23 ( 3 ), 218 – 220 ( 2013 ).
- Jin SG , JiangY , QiuRet al. 5-hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations . Cancer Res.71 ( 24 ), 7360 – 7365 ( 2011 ).
- Kudo Y , TateishiK , YamamotoKet al. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation . Cancer Sci.103 ( 4 ), 670 – 676 ( 2012 ).
- Orr BA , HaffnerMC , NelsonWG , YegnasubramanianS , EberhartCG . Decreased 5-hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma . PLoS ONE7 ( 7 ), e41036 ( 2012 ).
- Liu C , LiuL , ChenXet al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1 . PLoS ONE8 ( 5 ), e62828 ( 2013 ).
- Park JL , KimHJ , SeoEHet al. Decrease of 5hmC in gastric cancers is associated with TET1 silencing due to with DNA methylation and bivalent histone marks at TET1 CpG island 3′-shore . Oncotarget6 ( 35 ), 37647 – 37662 ( 2015 ).
- Chen K , ZhangJ , GuoZet al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer . Cell Res.26 ( 1 ), 103 – 118 ( 2015 ).
- Kroeze LI , AslanyanMG , Van RooijAet al. Characterization of acute myeloid leukemia based on levels of global hydroxymethylation . Blood124 ( 7 ), 1110 – 1118 ( 2014 ).
- Lian CG , XuY , CeolCet al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma . Cell150 ( 6 ), 1135 – 1146 ( 2012 ).
- Rasmussen KD , HelinK . Role of TET enzymes in DNA methylation, development, and cancer . Genes Dev.30 ( 7 ), 733 – 750 ( 2016 ).
- Dawlaty MM , GanzK , PowellBEet al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development . Cell Stem Cell9 ( 2 ), 166 – 175 ( 2011 ).
- Putiri EL , TiedemannRL , ThompsonJJet al. Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells . Genome Biol.15 ( 6 ), R81 ( 2014 ).
- Ye C , TaoR , CaoQet al. Whole-genome DNA methylation and hydroxymethylation profiling for HBV-related hepatocellular carcinoma . Int. J. Oncol.49 ( 2 ), 589 – 602 ( 2016 ).
- Hon GC , SongCX , DuTet al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation . Mol. Cell56 ( 2 ), 286 – 297 ( 2014 ).
- Ono R , TakiT , TaketaniT , TaniwakiM , KobayashiH , HayashiY . LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23) . Cancer Res.62 ( 14 ), 4075 – 4080 ( 2002 ).
- Forloni M , GuptaR , NagarajanAet al. Oncogenic EGFR represses the TET1 DNA demethylase to induce silencing of tumor suppressors in cancer cells . Cell Rep.16 ( 2 ), 457 – 471 ( 2016 ).
- Li L , LiC , MaoHet al. Epigenetic inactivation of the CpG demethylase TET1 as a DNA methylation feedback loop in human cancers . Sci. Rep.6 , 26591 ( 2016 ).
- Pei YF , TaoR , LiJFet al. TET1 inhibits gastric cancer growth and metastasis by PTEN demethylation and re-expression . Oncotarget7 ( 21 ), 31322 – 31335 ( 2016 ).
- Kroeze LI , Van Der ReijdenBA , JansenJH . 5-hydroxymethylcytosine: an epigenetic mark frequently deregulated in cancer . Biochim. Biophys. Acta1855 ( 2 ), 144 – 154 ( 2015 ).
- Pei YF , LeiY , LiuXQ . MiR-29a promotes cell proliferation and EMT in breast cancer by targeting ten eleven translocation 1 . Biochim. Biophys. Acta11 ( 10 ), 16 ( 2016 ).
- Wu MZ , ChenSF , NiehSet al. Hypoxia drives breast tumor malignancy through a TET–TNFalpha–p38–MAPK signaling axis . Cancer Res.75 ( 18 ), 3912 – 3924 ( 2015 ).
- Sang Y , ChengC , TangXF , ZhangMF , LvXB . Hypermethylation of TET1 promoter is a new diagnosic marker for breast cancer metastasis . Asian Pac. J. Cancer Prev.16 ( 3 ), 1197 – 1200 ( 2015 ).
- Letouze E , MartinelliC , LoriotCet al. SDH mutations establish a hypermethylator phenotype in paraganglioma . Cancer Cell23 ( 6 ), 739 – 752 ( 2013 ).
- Oermann EK , WuJ , GuanKL , XiongY . Alterations of metabolic genes and metabolites in cancer . Semin. Cell Dev. Biol.23 ( 4 ), 370 – 380 ( 2012 ).
- Toro JR , NickersonML , WeiMHet al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America . Am. J. Hum. Genet.73 ( 1 ), 95 – 106 ( 2003 ).
- Yin R , MaoSQ , ZhaoBet al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals . J. Am. Chem. Soc.135 ( 28 ), 10396 – 10403 ( 2013 ).
- Nestor CE , OttavianoR , ReinhardtDet al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems . Genome Biol.16 ( 1 ), 11 ( 2015 ).
- Baylin SB , HoppenerJW , De BustrosA , SteenberghPH , LipsCJ , NelkinBD . DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas . Cancer Res.46 ( 6 ), 2917 – 2922 ( 1986 ).
- Cimmino L , DawlatyMM , Ndiaye-LobryDet al. TET1 is a tumor suppressor of hematopoietic malignancy . Nat. Immunol.16 ( 6 ), 653 – 662 ( 2015 ).
- Struhl K . Is DNA methylation of tumour suppressor genes epigenetic?Elife3 , e02475 ( 2014 ).
- Gu J , StevensM , XingXet al. Mapping of variable DNA methylation across multiple cell types defines a dynamic regulatory landscape of the human genome . G3 (Bethesda)6 ( 4 ), 973 – 986 ( 2016 ).
- Chiappinelli KB , StrisselPL , DesrichardAet al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses . Cell162 ( 5 ), 974 – 986 ( 2015 ).
- Roulois D , Loo YauH , SinghaniaRet al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts . Cell162 ( 5 ), 961 – 973 ( 2015 ).
- Booth MJ , OstTW , BeraldiDet al. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine . Nat. Protoc.8 ( 10 ), 1841 – 1851 ( 2013 ).
- Yu M , HonGC , SzulwachKEet al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome . Cell149 ( 6 ), 1368 – 1380 ( 2012 ).
- Thomson JP , MoggsJG , WolfCR , MeehanRR . Epigenetic profiles as defined signatures of xenobiotic exposure . Mutat. Res.764–765 , 3 – 9 ( 2014 ).
- Thomson JP , HunterJM , LempiainenHet al. Dynamic changes in 5-hydroxymethylation signatures underpin early and late events in drug exposed liver . Nucleic Acids Res.41 ( 11 ), 5639 – 5654 ( 2013 ).
- Thomson JP , LempiainenH , HackettJAet al. Non-genotoxic carcinogen exposure induces defined changes in the 5-hydroxymethylome . Genome Biol.13 ( 10 ), R93 ( 2012 ).
- Lian CG , XuS , GuoWet al. Decrease of 5-hydroxymethylcytosine in rat liver with subchronic exposure to genotoxic carcinogens riddelliine and aristolochic acid . Mol. Carcinog.54 ( 11 ), 1503 – 1507 ( 2014 ).
- Gurbuz I , Chiquet-EhrismannR . CCN4/WISP1 (WNT1 inducible signaling pathway protein 1): a focus on its role in cancer . Int. J. Biochem. Cell. Biol.62 , 142 – 146 ( 2015 ).
- Lempiainen H , CouttetP , BolognaniFet al. Identification of Dlk1-Dio3 imprinted gene cluster noncoding RNAs as novel candidate biomarkers for liver tumor promotion . Toxicol. Sci.131 ( 2 ), 375 – 386 ( 2013 ).
- Gustafson CB , YangC , DicksonKMet al. Epigenetic reprogramming of melanoma cells by vitamin C treatment . Clin. Epigenetics7 ( 1 ), 51 ( 2015 ).
- Strand SH , HoyerS , LynnerupASet al. High levels of 5-hydroxymethylcytosine (5hmC) is an adverse predictor of biochemical recurrence after prostatectomy in ERG-negative prostate cancer . Clin. Epigenetics7 , 111 ( 2015 ).
- Zhang F , LiuY , ZhangZet al. 5-hydroxymethylcytosine loss is associated with poor prognosis for patients with WHO grade II diffuse astrocytomas . Sci. Rep.6 , 20882 ( 2016 ).
- Yang Q , WuK , JiMet al. Decreased 5-hydroxymethylcytosine (5-hmC) is an independent poor prognostic factor in gastric cancer patients . J. Biomed. Nanotechnol.9 ( 9 ), 1607 – 1616 ( 2013 ).
- Sproul D , NestorC , CulleyJet al. Transcriptionally repressed genes become aberrantly methylated and distinguish tumors of different lineages in breast cancer . Proc. Natl Acad. Sci. USA108 ( 11 ), 4364 – 4369 ( 2011 ).
- Moarii M , ReyalF , VertJP . Integrative DNA methylation and gene expression analysis to assess the universality of the CpG island methylator phenotype . Hum. Genomics9 , 26 ( 2015 ).
- Gustafsson M et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis . Genome Med.6 , 82 ( 2014 ).
- Amabile A , MigliaraA , CapassoPet al. Inheritable silencing of endogenous genes by hit-and-run targeted epigenetic editing . Cell167 ( 1 ), 219 – 232 ( 2016 ).