
Abstract
Genome-wide association studies have identified thousands of single nucleotide polymorphisms in the human genome that are statistically associated with particular disease traits. In this Perspective, we review emerging data suggesting that most single nucleotide polymorphisms associated with immune-mediated diseases are found in regulatory regions of the DNA – parts of the genome that control expression of the protein encoding genes – rather than causing mutations in proteins. We discuss how the emerging understanding of particular gene regulatory regions, gene enhancers and the epigenetic mechanisms by which they are regulated is opening up new opportunities for the treatment of immune-mediated diseases, focusing particularly on the BET family of epigenetic reader proteins as potential therapeutic targets.
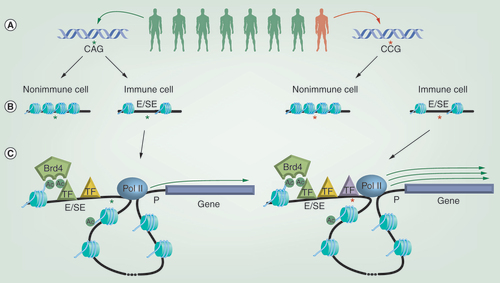
(A) Genome-wide association studies have shown that individuals harboring specific rare single nucleotide polymorphisms (SNPs) in their genetic sequence can show an increased incidence of developing particular immune-mediated diseases. (B) Rather than occurring in the protein-coding region of genes, most disease-associated SNPs sit within regions of the genome that regulate gene expression, such as enhancers (Es) and super enhancers (SEs), which are characterized by possessing relatively open and nucleosome free chromatin structures. SNPs associated with immune diseases are enriched within Es/SEs found selectively within relevant immune cell types. (C) Es/SEs are able to regulate the expression of distal genes through the ‘looping out’ of intervening DNA, bringing the E/SE in close proximity to the gene promoter (P). E/SE function is linked to the recruitment of transcription factors (TFs) and epigenetic regulators, such as Brd4, which binds to acetylated histones and TFs via its bromodomains. One mechanism by which SNPs may alter gene expression is by changing the binding sites for TFs within Es/SEs and ultimately the efficiency of recruiting components of the transcriptional machinery, such as RNA polymerase II. In the example depicted, the disease-associated SNP leads to increased TF recruitment and elevated expression of the gene controlled by that E/SE.
Dysregulation of gene expression as primary driver of human disease
Genome-wide association studies (GWAS) combine epidemiological data with molecular genetic analysis to identify statistical associations between single nucleotide polymorphisms (SNPs) (variance in the nucleotide at a particular location in the genome within the population) in the genome and particular disease or biochemical traits, but most commonly disease incidence. As hoped for, these studies have provided pivotal insight into the genetic basis of complex human diseases. However, the emerging picture is an unexpected one. Thus, unlike simple (Mendelian) genetic diseases such as cystic fibrosis that are caused by mutations in the protein coding region of a gene, the vast majority (>90%) of the thousands of GWAS disease-associated SNPs identified to date are located in noncoding regions, including the intergenic and intronic compartments of the genome [Citation1,Citation2]. While localization of disease-associated SNPs in these regions – dismissively referred to in the past as ‘gene deserts’ – initially dampened enthusiasm for GWAS results, interest has been renewed from recent advances in our understanding of the noncoding genome. It is now clear that the nonprotein coding DNA contains regions that play a key role in regulating the expression of protein coding genes, and moreover, that GWAS SNPs localize preferentially in these regulatory regions. Thus, rather than causing mutations in proteins, SNPs in noncoding regions commonly lead to quantitative changes in gene expression [Citation3], implicating dysregulation of gene expression as the key driver of complex human diseases. Recent advances in genome-wide ‘epigenomic’ approaches are beginning to elucidate the mechanisms by which these SNPs alter gene expression, and hence point to novel approaches to therapeutic intervention.
Frequent localization of disease-associated SNPs in gene enhancers
Enhancers are cis-regulatory elements, typically a few hundred base pairs in size, which function in concert with promoters to control gene expression [Citation4]. Like promoters, enhancers contain docking sites for transcription factors (TFs), and mediate recruitment of the components of the transcriptional machinery, including RNA polymerase II and the mediator complex. Unlike promoters, which are typically located less than 1 kb from the transcription start site, enhancers may be separated from their target gene by long stretches of DNA, in some cases more than 1 Mb [Citation5]. Enhancers are able to act over such long distances through the ‘looping’ of the intervening DNA, allowing for physical interaction with promoters [Citation6].
While historically identified on a one by one basis, predicated on the presence of TF-binding sites and experimentally demonstrated capacity to enhance gene expression, high-throughput methods for identifying putative enhancers on a whole genome scale have recently been developed and employed. One such approach involves the measurement of deoxyribonuclease I hypersensitive sites (DHSs). In this method, nuclear DNA is exposed to deoxyribonuclease I, which cuts preferentially at sites of nucleosome depletion, a characteristic of gene regulatory sites. While this approach on its own does not discriminate well between different types of regulatory elements (e.g., enhancers, promoters), the designation of DHSs as enhancers can be inferred from other complementary genome-wide measurements, such as the presence of specific histone modifications (as discussed further below). Strikingly, a very large number of DHSs and enhancers have been detected using this methodology – on average approximately 200,000 DHSs in a given cell type – which greatly exceeds the number of protein coding genes (∼20,000) [Citation7]. The implication is that individual genes are controlled by multiple regulatory elements. Indeed, it is now clear that multiple enhancers can function to control expression of individual genes. In some cases, these occur in clusters in the genome that can span large ranges of DNA (up to 50 kb), and have recently been referred to as super enhancers (SEs) or stretch enhancers [Citation8–11]. Additionally, enhancers located in distinct locations can also converge on the same gene promoter through long-range interactions. The presence of multiple enhancer elements, and particularly SEs is linked to high levels of gene expression, likely through the additive effects of the individual enhancers [Citation12].
In addition to highlighting the large number of regulatory elements in the genome, DHS and other approaches have helped to elucidate other critical fundamental aspects of enhancer biology. First, enhancers are highly cell-type specific, reflecting cell-specific patterns of gene expression; SE’s appear to be particularly important as drivers of cell/tissue specific gene expression [Citation11]. Second, even in a given cell type, the enhancer landscape is highly flexible, as large numbers of new enhancers, including SEs can appear while others disappear when cells are exposed to stimuli that induce cell differentiation or activation [Citation13]. Most importantly, with the increasing understanding and cataloguing of enhancers has come the realization that a high proportion of GWAS SNPs fall within enhancer regions, consistent with the effects of these polymorphisms on gene expression [Citation7]. Moreover, as discussed further below, disease-associated SNPs are particularly enriched within cell-type specific enhancers in a manner that fits with known or predicted disease mechanisms, for example within immune cell-specific enhancers for autoimmune disease-associated SNPs. These findings not only provide a satisfying explanation for GWAS data, but also point to a commonality in mechanism – and potentially therapeutic strategy – for diverse diseases.
Epigenetics & enhancers
Enhancer function is linked to the presence of DNA-encoded recognition sequences for TF-binding motifs, which allow for recruitment of TFs and nucleation of the transcriptional machinery. Additionally, however, functional enhancer activity is highly dependent on the local structure of chromatin – the form in which DNA is packaged in the nucleus in close association with histone proteins. DNA in tightly packed ‘heterochromatin’ is inaccessible to DNA-binding proteins such as TFs; hence TF-binding motifs in such locations do not function as enhancers. The generation of accessible DNA regions and functional enhancers is mediated by a range of ‘epigenetic’ mechanisms capable of dynamically modifying chromatin structure.
The basic unit of chromatin is the nucleosome, which is comprised of approximately 146 bp of DNA wrapped around an octamer of core histone proteins (two copies each of histones H3, H4, H2A and H2B) [Citation14,Citation15]. Nucleosomes are further organized into higher order structures through interactions with additional histone and nonhistone proteins, allowing for compact packing of the genome. A fundamental mechanism underlying the regulation of chromatin structure is the chemical alteration of its constituents, with both DNA and histones being subject to modification. DNA is modified through methylation of cytosine residues via the activity of DNA methyltransferases to generate 5-methylcytosine (5mC), with 5mC being converted back to cytosine through the sequential activity of members of the ten-eleven translocation family of dioxygenases and thymine DNA glycosylase [Citation16–20]. Additionally, the amino acids within histones are subject to a wide variety of post-translational modifications, the most common being acetylation, methylation or ubiquitination of lysine residues, methylation of arginine residues and phosphorylation of serine, threonine or histidine residues [Citation21]. Families of proteins able to add and remove these modifications in the context of specific histone sequences have been identified, and have been termed epigenetic ‘writers’ and ‘erasers,’ respectively.
Chromatin modifications have been shown to change DNA accessibility in three ways. First, the extent of histone acetylation can directly affect the strength of the interaction between histones and DNA, since neutralization of positively charged lysine residues by acetylation weakens histone binding to negatively charged DNA [Citation22]. Second, DNA methylation can directly modify DNA recognition and binding by TFs. Thus, the binding of certain TFs has been shown to be disrupted by the conversion of cytosine to 5mC within TF-binding motifs [Citation23,Citation24]. Third, and most commonly, chromatin modifications serve as recognition signals for epigenetic ‘reader’ proteins, including 5mC-binding proteins as well as multiple families of proteins that recognize specific histone modifications. Reader proteins that bind to specific chromatin marks can bring with them a range of chromatin remodeling enzymatic activities, either as intrinsic constituents of the reader proteins themselves or through recruitment of interacting protein partners. Through such activity, reader protein recruitment can lead to the local removal of histones to create a nucleosome-free region of DNA, and hence an active regulatory region. Conversely, some readers recruit catalytic activities that promote the generation of closed heterochromatin, or transcriptional repressor complexes to shut down gene transcription.
In addition to possessing nucleosome-sparse TF-binding motifs, active enhancers are associated with a number of other characteristic epigenetic features. For example, the nucleosomes that flank TF-binding sites are marked by high levels of histone H3 lysine 27 acetylation (H3K27ac) and H3 lysine 4 mono- or di-methylation (H3K4me1/2), with extended stretches of these marks being commonly used to identify SEs [Citation25–27]. The histones flanking enhancers also show a high rate of turnover, indicating that the maintenance of the local structure is a dynamic and active process [Citation28,Citation29]. Consistent with this view, numerous chromatin regulating proteins are highly enriched within SEs [Citation10,Citation30]. Proteins that have been identified within SEs include epigenetic writer proteins, such as the histone acetyltransferases p300 and CBP, epigenetic readers such as CHD7, Brd4 and SMARCA4 (Brg1) and eraser proteins such as the histone demethlyase KDM1a and histone deacetylases HDAC1 and HDAC2 [Citation10]. Notably, several of these proteins contain intrinsic chromatin remodeling activity (e.g., CHD7, SMARCA4) and/or are components of multiprotein complexes with such activity (e.g., KDM1, HDA1/2) [Citation31–33]. Conversely, the Brd4 protein, which functions as an epigenetic reader based on possessing acetyl lysine-binding bromodomains, interacts with key regulators of gene transcription, including the Mediator and P-TEFb protein [Citation34]. Thus, in addition to regulating gene transcription indirectly through chromatin remodeling, epigenetic proteins can also interact directly with the transcriptional machinery. In this respect, it is worth noting that enhancer DNA is itself transcribed bi-directionally, generating enhancer RNAs (eRNAs) [Citation35]. These eRNAs are thought to play an important role in driving expression of enhancer-associated genes, although the precise mechanisms involved remain to be elucidated.
Targeting epigenetic mechanisms for treatment of disease
In view of the emerging link between GWAS SNPs, gene enhancers and altered gene expression in disease on one hand, and the increasing understanding of the epigenetic mechanisms involved in regulating enhancer function and gene expression on the other, a logical hypothesis would be that epigenetic regulatory proteins may be suitable targets for treatment of human complex diseases. A reasonable theoretical concern, however, would be that common epigenetic mechanisms may regulate expression of all genes, and hence such approaches would be overwhelmingly toxic. Most encouragingly, data generated with newly developed inhibitors of epigenetic proteins have shown that this is not the case. Rather, through targeting of specific epigenetic mediators, it is possible to have very selective effects on gene expression. Even more excitingly, certain targets appear to be intimately involved in regulating the very genes that are most commonly implicated in disease.
The potential for specific modulation of gene expression by targeting epigenetic mechanisms is rooted in the fact that disease-associated SNPs are enriched within cell-type specific enhancers, particularly within SEs, and include enhancers that are generated in response to signals associated with cellular differentiation, activation and stress [Citation8,Citation10,Citation11]. Regulation of these genes is quite distinct from that of constitutively expressed ‘housekeeping’ genes, which generally either do not require enhancer activity or contain an integrated enhancer [Citation36]. Hence epigenetic mechanisms involved more selectively in the generation or function of SEs represent more attractive potential targets for therapeutic intervention. One setting in which the potential for intervention at SE-driven genes has been noted is oncology, where high expression of oncogenes has been shown to be under the control of SEs [Citation9]. Here, we focus on the emerging opportunity in autoimmune and inflammatory diseases.
Epigenetic regulation of immune cell function
The immune system is comprised of a complex set of interacting cell types that are commonly classified into innate and adaptive subtypes based on the receptors utilized to recognize pathogens. Innate cells utilize germ-line encoded receptors to recognize relatively invariant features of infectious agents, such as lipopolysaccharide (LPS), a conserved component of the outer wall of Gram-negative bacteria. Conversely, adaptive immune cells express receptors generated through a process of somatic recombination of variable gene segments, which generates a vast array of ligand specificities; these are distributed in a clonal manner such that individual adaptive immune cells express receptors of a single specificity. Common to both innate and adaptive immune cells, however, is the capacity to undergo dramatic changes in gene expression as an integral and requisite mechanism in their function to defend the host against infection.
As an early line of defense, innate immune cells must respond rapidly following the detection of pathogens. This response is typically highly inflammatory in nature, with the production of large quantities of mediators involved in directly killing/containing the pathogen, recruiting additional innate cells to the site of infection and promoting the activation of adaptive immune cells. The magnitude of gene expression changes associated with this process can be illustrated by the response stimulated by LPS in macrophages, a key innate immune cell, wherein alterations in the expression of hundreds of genes is observed within a few hours [Citation37]. This response is associated with a marked remodeling of the enhancer landscape, including the appearance of thousands of de novo enhancers, while many pre-existing enhancers are lost (inferred from gain/loss of specific histone modifications and TF binding and increased/decreased expression of nearby genes) [Citation35,Citation38]. Moreover, many of the de novo enhancers induced by LPS fit the criteria of SEs [Citation13], and macrophage SE-driven genes appear to be most prominently affected by LPS stimulation [Citation39], or by exposure to inflammatory cytokines such as IFN-γ and TNF [Citation40]. Interestingly, similar de novo generation of SEs has been reported to occur in endothelial cells exposed to the inflammatory cytokine TNF [Citation13]. While production of inflammatory mediators by activated innate cells is necessarily transient, epigenetic modifications may persist and alter a subsequent response to stimulation, providing a form of innate cell ‘memory’ that can contribute to a relatively nonspecific resistance to re-infection [Citation41].
Adaptive immune cells – T and B lymphocytes – also undergo marked changes in gene expression during the course of a response to infection. Since the frequency of T and B cells expressing receptors specific for any particular infectious agent is very low in individuals who have not encountered that pathogen before (i.e., immunologically naive), the response initially involves extensive cell proliferation to expand the pathogen-specific cell population. During this process, cells also undergo differentiation into ‘effector’ cells that express molecules capable of combating infection. For B cells, this mainly involves their differentiation into antibody-secreting plasma cells. For T cells, effector cell differentiation is accompanied by acquisition of an ability to produce a range of cytokines that contribute directly or indirectly to pathogen clearance and/or a capacity to directly kill infected cells.
The precise function of effector T cells is determined by cell lineage (particularly CD4+ vs CD8+) as well as the presence of polarizing signals during T cell activation and differentiation. These polarizing signals are supplied predominantly by activated innate immune cells, and are linked to the type of pathogen encountered (e.g., Gram-negative bacteria, DNA virus, etc.). Polarized effector T cells include well characterized CD4+ T-helper (Th) subsets, typically categorized on the basis of the pattern of cytokines they produce (e.g., IFN-γ by Th1 cells, IL-4, IL-5 and IL-13 by Th2 cells and IL-17A/F by Th17 cells). T-cell proliferation and differentiation is accompanied by extensive changes in gene expression and associated epigenetic modifications at gene regulatory sites [Citation27]. Key TFs drive the polarization through the generation of new SEs [Citation42], resulting in hundreds of Th subset-specific genes, including Ifng in Th1, Il13 in Th2 and Il17a in Th17 cells being associated with SEs present selectively in those cells [Citation43]. Overall, cytokines and cytokine receptors appear to be the dominant class of SE-associated genes in T cells [Citation44].
Once the infection has been cleared, the majority of effector T cells disappear through a process of programmed cell death; this is necessary because of the highly inflammatory nature of the immune response and potential to cause collateral damage to host tissues. Notably, however, a proportion of cells survives and persists at an increased frequency relative to that present pre-infection. These cells no longer constitutively produce effector molecules, but retain epigenetic modifications that allow them to rapidly express these genes upon subsequent encounter with the same pathogen, providing immunological memory to re-infection [Citation45–47].
GWAS-insights into immune-mediated disease
Although inflammatory immune responses are essential for protection against infection, the damaging nature of inflammation necessitates that these responses are transient in nature and restricted to situations of genuine ‘danger.’ Indeed, failure in immune regulation results in a wide range of human diseases. This includes autoimmune diseases, caused by immune responses directed against self antigens, as well as inflammatory responses against foreign substances that are intrinsically harmless – for example the allergic response to pollen allergens. Both innate and adaptive cells typically contribute to immune-mediated diseases, with different cell types predominating in different diseases. Tissue damage and disease result when inflammation fails to resolve and becomes chronic. In addition, polarization of the immune response plays an important role in the nature of the pathology observed, or indeed whether disease occurs at all. For example, the symptoms of allergy or closely linked the generation of a Th2 response, whereas psoriasis is highly dependent on IL-17. The implication is that tight regulation of gene expression in immune cells, to prevent chronic or inappropriate expression of inflammatory mediators, is critical for the maintenance of tissue homeostasis and that dysregulation of gene expression is an important determinant in immune-mediated disease.
In accordance with this view, there is increasing evidence that GWAS SNPs associated with immune diseases localize preferentially within gene regulatory regions in immune cells. A seminal early study involving an unbiased analysis of 349 cell and tissue samples showed enrichment of GWAS SNPs for Crohn’s disease (a form of inflammatory bowel disease) in DHSs present in Th17 and Th1 cells, and for multiple sclerosis (MS) SNPs in DHSs in T and B cells [Citation7]. Subsequently, Farh and colleagues utilized a fine mapping algorithm to identify causal variants for 21 autoimmune diseases, and found that 60% of these mapped to immune cell enhancers, many of which gained histone acetylation and transcribed eRNA upon immune cell stimulation [Citation48]. These findings are supported by other studies focused on specific diseases, such the demonstration that asthma-associated SNPs are enriched in TH2-specific enhancers (in keeping with the known pathogenic role for TH2 cells in asthma) [Citation49], the report that Type 1 diabetes-associated SNPs localize to enhancer sequences active in thymus, T and B cells, and CD34+ stem cells [Citation50], and the identification of a Th1-specific enhancer as the location for an ankylosing spondylitis (a form of spinal arthritis) – associated SNP [Citation51].
Moreover, as the characteristic chromatin signatures of SEs have been elucidated, it has become evident that SEs are particular hotspots for GWAS SNPs. Thus, SNPs associated with a variety of autoimmune/inflammatory diseases, including rheumatoid arthritis, Type 1 diabetes, MS, celiac disease and ulcerative colitis were shown to be located in SEs present in Tfh cells, a subset of CD4+ T cells specialized in promoting B cell antibody production [Citation52]. Similarly a SNP associated with the autoimmune disease vitiligo, in which melanin-secreting cells are destroyed by self-reactive T cells, was shown to localize within an SE and promotes increased expression of major histocompatibility (MHC) genes [Citation53]. Since MHC proteins present peptides derived from pathogens and self proteins on the cell surface for recognition by peptide-specific T cells, dysregulation of MHC expression can have a major impact on T cell activation.
Targeting epigenetic enhancer regulation for treatment of immune-mediated disease
The accumulating evidence that dysregulation of gene expression is a key driver of immune-mediated diseases and that many disease-relevant genes are regulated by SEs raises the question of whether therapeutic strategies can be developed which target mechanisms involved in SE generation and function. Although much remains to be learned about the basic processes governing enhancer and SE biology, recent findings suggest that this may be a feasible approach. Optimism in this respect stems from a burgeoning understanding of the epigenetic mechanisms governing the functioning of enhancers, together with the recent development of small molecule inhibitors of key epigenetic mediators. Although not linked with differential effects on disease associated SNP variant containing SEs, we have chosen here to focus on one particular subfamily of epigenetic reader proteins where experiments with inhibitor compounds in a range of settings are beginning to provide a promising link between enhancers, epigenetic mechanisms and therapeutic opportunities.
As discussed above, numerous proteins involved in epigenetic mechanisms are found in high amounts within SEs. Although the potential value of most of these as targets for modulating SE function remains to be explored, a compelling story based on a specific subfamily of epigenetic readers, the BET family of proteins, is beginning to emerge. The BET family is comprised of four members, Brd2, Brd3, Brd4 and Brdt, each of which possesses two bromodomains, a protein module specialized in the recognition of acetylated lysine residues [Citation54]. BET bromodomains have been shown to bind to acetylated lysines in histone tails and TFs, both of which may contribute to BET protein localization to specific genomic sites. To date, the function of Brd4 has been explored most extensively among BET proteins, and its documented role in promoting transcriptional elongation has made it an enticing focus of studies on enhancers. However, it should be noted that Brd2 and Brd3 show broadly similar genome-wide distribution to Brd4, and there is evidence that these proteins may also be present at SEs [Citation55]. By contrast, Brdt is expressed exclusively in germ cells and hence has a much more specialized function [Citation56].
Elucidation of BET protein function has been greatly facilitated in recent years with the development of potent and selective small molecule inhibitors of BET bromodomains. Most of the molecules described to date are active against all eight BET bromodomains, owing to the high homology between the acetyl lysine binding sites present in these bromodomains. Hence, the data generated with these compounds cannot, for the most part, distinguish the contributions of specific BET family members. With that caveat, there is now a growing body of evidence that: BET inhibition mediates potent and selective inhibition of gene expression; selective effects on gene expression by BET inhibitors is linked to BET protein function at enhancers and SEs and; BET inhibition modulates expression of inflammatory genes in a therapeutically relevant manner.
The first demonstration that BET inhibition could selectively modulate inflammation-associated gene expression came from studies with the pan-BET inhibitor I-BET762, which was shown to reduce the expression of a range of cytokines and chemokines by LPS-stimulated mouse macrophages [Citation57]. Two key findings were noted in this study. First, the expression of housekeeping genes was not affected, indicating that their transcription was not BET dependent. Second, LPS-induced genes that were inhibited by I-BET762 corresponded to so-called secondary response genes, whereas expression of primary response genes was not modified. Beyond differences in the speed with which these classes of genes are induced (i.e., secondary response genes are expressed at a later time point after LPS stimulation than primary response genes), there is a fundamental difference at the chromatin level underlying these kinetic differences. Specifically, early-primary response genes have promoters/enhancers constitutively permissive for transcription, whereas secondary response genes require chromatin remodeling events before transcription can occur [Citation58].
Subsequent studies have linked the selective effects of BET inhibition on macrophage gene expression to BET protein function at enhancers, and SEs in particular. Thus, BET bromodomain inhibition has been shown to preferentially inhibit the expression of de novo generated SE-associated genes following LPS treatment of macrophages [Citation13]. Although the exact mechanism by which BET proteins function at enhancers/SEs remains to be fully understood, some insight in this respect was provided in a study showing that BET inhibition blocks eRNA expression from the large numbers of de novo enhancers generated following LPS stimulation of macrophages [Citation35]. These observations in macrophages also extend to inflammatory responses in other cells, as BET bromodomain inhibition has been shown to block TNF-induced de novo SE-driven gene expression in endothelial cells [Citation13], and to inhibit the induction of inflammatory gene eRNA and mRNA in a gastric adenocarcinoma cell line by Helicobactor pylori (the causative agent of stomach ulcers) [Citation59]. Moreover, the effects of BET inhibition observed in inflammation-associated SE function corroborate previous work in cancer cell lines showing that SE-associated oncogene expression is highly sensitive to BET inhibitors [Citation9]. Interestingly, recent work has extended this paradigm to virally induced SEs [Citation60]. In this study, BET inhibition was shown to greatly reduce the expression of viral oncogene expression driven by an SE generated upon genomic integration of human papillomavirus.
The original report showing selective inhibition of LPS-induced inflammatory gene expression in macrophages by I-BET762 has been confirmed and extended to demonstrate that several chemically distinct pan-BET inhibitor compounds are able to similarly affect this response in various innate immune cells, including macrophages, monocytes and dendritic cells, and in response to multiple innate-cell activating stimuli [Citation61–68]. In addition, BET bromodomain inhibitors have been shown to modulate the pro-inflammatory activity of adaptive immune cells. Pan-BET inhibitors I-BET762 and JQ1 were shown to greatly diminish the differentiation of CD4+ Th cells into polarized effector subsets, and also to inhibit the ability of already polarized Th cells to produce effector cytokines [Citation69,Citation70]. As for innate cell responses, BET inhibition was shown to result in a highly selective modulation of gene expression [Citation69]. Similarly, B-cell function is also modulated by BET inhibitors, as JQ1 has been reported to inhibit B-cell proliferation and expansion, as well as the ability of these cells to switch the isotype of antibody they produce [Citation71,Citation72]. Although the direct impact of BET inhibition on enhancer activity was not investigated in these studies, such a mechanism seems likely given the emerging recognition that enhancers and SEs play a key role in driving these adaptive immune cell functions, and the demonstrated importance of BET proteins at SEs in other contexts. Moreover, direct evidence that BET inhibition can affect T cell SE-driven gene expression in a disease relevant manner was provided in a recent study by Peeters et al. [Citation73]. Here, investigating CD4+ memory/effector T cells isolated from inflamed joint synovial fluid of patients with juvenile idiopathic arthritis, the authors demonstrated that JQ1 preferentially inhibited the expression of SE-regulated disease-associated genes.
Importantly, in addition to mediating selective, SE-biased modulation of immune cell gene expression in vitro, BET inhibitors have shown striking anti-inflammatory effects in vivo, associated with protection from disease in a variety of animal models. To date, this includes models of sepsis, MS, atherosclerosis, periodontitis, HIV-induced kidney injury, psoriasis, type I diabetes and arthritis [Citation13,Citation57,Citation61,Citation62,Citation64,Citation69,Citation70,Citation74–79]. Note that as well as effects on immune cells, selective modulation of gene expression in other tissue cells that contribute to disease, such as synovial fibroblasts in rheumatoid arthritis and pancreatic islet β-cells in type I diabetes, contributes to BET inhibitor-mediated protection from pathology in these models [Citation77,Citation80]. These findings support the potential for BET proteins specifically, and SE-related mechanisms more generally as therapeutic targets for immune-mediated diseases.
Future perspective
Great advances in the understanding of enhancer biology and epigenetic mechanisms have recently combined to provide a new interpretation of GWAS data, and in doing so offer a fresh paradigm for the genetic basis of complex human diseases. These advances have been aided by the demonstration that key epigenetic regulators are tractable to inhibition with small molecules and the development of potent and selective tool compounds with which to evaluate this paradigm. The remarkable emerging link between BET bromodomain inhibitor actions, SE biology and disease genetics is associated with their promising therapeutic potential; BET inhibitors have already entered the clinic for oncology indications and may soon be doing so for immune-mediated diseases.
In coming years, further important insight into the functional significance of GWAS disease-associated genetic polymorphisms will be needed, as numerous challenges remain. In particular, direct evidence that disease-associated SNPs falling within regulatory regions alter gene expression is sparse. Such functional effects can be evaluated on a SNP by SNP basis using conventional methods for evaluating enhancer function, but might be conducted on a much larger scale, for example, using approaches that measure eRNA expression on a genome-wide basis. Such data would be of great value in defining disease-causative SNPs in the context of a phenomenon known as linkage disequilibrium, in which groups of SNPs that occur in close proximity in the genome are inherited together, with essentially no recombination occurring between them (and hence making it impossible to assign one of these SNPs as causative). In addition to showing effects of disease-associated SNPs on enhancer function, much remains to be learned about the genes that are regulated by such SNPs. Thus, because enhancers can be separated from their targets genes by long stretches of DNA, it is difficult, if not impossible to ascribe an enhancer to its target gene(s) based on knowledge of the linear sequence of DNA alone. However, a number of techniques have been developed that can detect interactions between distinct regions of chromatin, such as those that occur between enhancers and gene promoters. These approaches are now beginning to be widely applied, and should help to identify the true target genes regulated by disease-associated enhancer SNPs, pointing to new potential therapeutic targets. Finally, there remains the question of ‘missing heritability,’ which is the conundrum that the additive genetic risk derived by combining GWAS associations often confers far less disease risk than expected from empirical estimates of heritability [Citation81]. For example, Crohn’s disease shows about 80% heritability, while the genetic components identified to date only explain 20% of this heritability [Citation82]. Whether such discrepancies reflect an overestimation of the true disease heritability, as yet unidentified genetic contributions or ‘transgenerational’ epigenetic causes remains to be determined [Citation81].
Continuing rapid advances in our understanding of the epigenetic mechanisms involved in enhancer/SE generation and function can also be expected, bringing with it new opportunities for therapeutic intervention. Thus, although numerous proteins and chromatin modifications have been shown to be associated with SEs, their functional significance remains unknown in most cases. As was the case for BET proteins, progress in this area will be greatly aided by the development of new chemical tools to elucidate the role of specific proteins. With respect to BET proteins, compounds with greater selectivity than the current pan-BET inhibitors may help to delineate the function of individual BET proteins or bromodomains.
Insight gained from such tools into the function of BET family members as well as other SE-associated proteins may point to opportunities for more selective modulation of SE genes if, for example, different proteins control SE output for subsets of genes or in a cell type specific manner. In this respect, it will also be important to investigate further the role of epigenetic proteins in the specific regulation of GWAS-identified disease-associated genes. Epigenetic inhibitors that preferentially target disease-associated SEs may have better safety profiles than those that affect SE-regulated genes more generally. As mentioned above, progress in this area will be greatly aided by new methods to identify the specific target genes for enhancers and SEs, the results of which can then be integrated with GWAS data. Moreover, linking the ‘directionality’ of effects will be critical, in other words, inhibitors of epigenetic regulators involved in enhancing or reducing gene expression should be matched with genes for which GWAS SNPs increase or decrease expression respectively.
In conclusion, the emerging understanding of GWAS data, enhancer biology and epigenetic mechanisms, together with the development of potent and selective inhibitors of BET bromodomains have highlighted dysregulation of gene expression as a key driver of immune-mediated diseases and the potential for therapeutic intervention via SE-targeting drugs. Continued progress in these rapidly advancing areas is expected, providing new opportunities for the treatment of human disease ().
Dysregulation of gene expression as primary driver of human disease
Most single nucleotide polymorphisms (SNPs) associated with common human diseases are found within nonprotein coding regions of the genome.
Most disease-associated SNPs lead to alterations in gene expression rather than mutation of proteins.
Frequent localization of disease-associated SNPs in gene enhancers
Enhancers are nonprotein coding regions of DNA that function to regulate gene expression.
Single genes can be controlled by multiple enhancers, including larger clusters of enhancers termed super enhancers (SEs) or stretch enhancers.
Enhancers are dynamic structures that are generated in a cell-type and stimulus-specific manner.
Disease-associated SNPs are frequently found within cell-type specific enhancers and SEs.
Epigenetics & enhancers
Enhancers are regions of accessible DNA that possess recognition sequences for transcription factors.
Dynamic generation of transcription factor-accessible DNA and enhancers is dependent on epigenetic mechanisms that modify interactions between DNA and associated histones.
Multiple epigenetic regulatory proteins are associated with active enhancers and SEs.
Targeting epigenetic mechanisms for treatment of disease
Epigenetic mechanisms involved in the control of SE-regulated genes represent potential targets for therapeutic intervention.
Epigenetic regulation of immune cell function
Activation of the immune system in response to infection results in marked changes in gene expression patterns, accompanied by epigenetic changes to the enhancer and SE landscape.
Generation of inflammatory immune responses must be tightly controlled to avoid immune-mediated pathology and disease.
GWAS-insights into immune-mediated disease
GWAS SNPs associated with immune diseases localize preferentially within gene regulatory regions in immune cells.
SEs are particular hotspots for immune disease-associated GWAS SNPs.
Targeting epigenetic enhancer regulation for treatment of immune-mediated disease
Bromodomain and extra-terminal (BET) family of proteins localize in enhancers and SEs.
Inhibitors of BET bromodomains preferentially reduce expression of SE-driven genes.
BET bromodomain inhibitors reduce inflammatory gene expression in vitro and immune-mediated disease pathology in vivo.
Future perspective
BET bromodomain inhibitors have entered the clinic for oncology and may soon do so for immune-mediated diseases.
New approaches to identify the genes controlled by enhancers harboring GWAS SNPs will lead to new therapeutic targets.
New opportunities for therapeutic intervention will come with increasing understanding of the mechanisms controlling enhancers/SEs.
Financial & competing interests disclosure
DF Tough and RK Prinjha are employees and shareholders of GlaxoSmithKline, which is involved in research and development of epigenetic inhibitors for the treatment of disease. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Hindorff LA , SethupathyP , JunkinsHAet al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits . Proc. Natl Acad. Sci. USA106 ( 23 ), 9362 – 9367 ( 2009 ).
- Manolio TA . Genomewide association studies and assessment of the risk of disease . N. Engl. J. Med.363 ( 2 ), 166 – 176 ( 2010 ).
- Nicolae DL , GamazonE , ZhangW , DuanS , DolanME , CoxNJ . Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS . PLoS Genet.6 ( 4 ), e1000888 ( 2010 ).
- Bulger M , GroudineM . Functional and mechanistic diversity of distal transcription enhancers . Cell144 ( 3 ), 327 – 339 ( 2011 ).
- Symmons O , UsluVV , TsujimuraTet al. Functional and topological characteristics of mammalian regulatory domains . Genome Res.24 ( 3 ), 390 – 400 ( 2014 ).
- De Laat W , DubouleD . Topology of mammalian developmental enhancers and their regulatory landscapes . Nature502 ( 7472 ), 499 – 506 ( 2013 ).
- Maurano MT , HumbertR , RynesEet al. Systematic localization of common disease-associated variation in regulatory DNA . Science337 ( 6099 ), 1190 – 1195 ( 2012 ).
- Whyte W-A , OrlandoD-A , HniszDet al. Master transcription factors and mediator establish super-enhancers at key cell identity genes . Cell153 ( 2 ), 307 – 319 ( 2013 ).
- Loven J , HokeH-A , LinC-Yet al. Selective inhibition of tumor oncogenes by disruption of super-enhancers . Cell153 ( 2 ), 320 – 334 ( 2013 ).
- Hnisz D , AbrahamB-J , LeeT-Iet al. Super-enhancers in the control of cell identity and disease . Cell155 ( 4 ), 934 – 947 ( 2013 ).
- Parker SCJ , StitzelML , TaylorDLet al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants . Proc. Natl Acad. Sci. USA110 ( 44 ), 17921 – 17926 ( 2013 ).
- Hay D , HughesJR , BabbsCet al. Genetic dissection of the [alpha]-globin super-enhancer in vivo . Nat. Genet.48 ( 8 ), 895 – 903 ( 2016 ).
- Brown J-D , LinC-Y , DuanQet al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis . Mol. Cell56 ( 0 ), 1 – 13 ( 2014 ).
- Davey CA , SargentDF , LugerK , MaederAW , RichmondTJ . Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution . J. Mol. Biol.319 ( 5 ), 1097 – 1113 ( 2002 ).
- Luger K , MaderAW , RichmondRK , SargentDF , RichmondTJ . Crystal structure of the nucleosome core particle at 2.8 Å resolution . Nature389 ( 6648 ), 251 – 260 ( 1997 ).
- Tahiliani M , KohKP , ShenYet al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1 . Science324 ( 5929 ), 930 – 935 ( 2009 ).
- He YF , LiBZ , LiZet al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA . Science333 ( 6047 ), 1303 – 1307 ( 2011 ).
- Ito S , ShenL , DaiQet al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine . Science333 ( 6047 ), 1300 – 1303 ( 2011 ).
- Cortellino S , XuJ , SannaiMet al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair . Cell146 ( 1 ), 67 – 79 ( 2011 ).
- Maiti A , DrohatAC . Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implicatons for active demethylation of CpG sites . J. Biol. Chem.286 ( 41 ), 35334 – 35338 ( 2011 ).
- Kouzarides T . Chromatin modifications and their function . Cell128 ( 4 ), 693 – 705 ( 2007 ).
- Hong L , SchrothGP , MatthewsHR , YauP , BradburyEM . Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA . J. Biol. Chem.268 ( 1 ), 305 – 314 ( 1993 ).
- Campanero MR , ArmstrongMI , FlemingtonEK . CpG methylation as a mechanism for the regulation of E2F activity . Proc. Natl Acad. Sci. USA97 ( 12 ), 6481 – 6486 ( 2000 ).
- Iguchi-Ariga SM , SchaffnerW . CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation . Genes Dev.3 ( 5 ), 612 – 619 ( 1989 ).
- Heintzman ND , HonGC , HawkinsRDet al. Histone modifications at human enhancers reflect global cell-type-specific gene expression . Nature459 ( 7243 ), 108 – 112 ( 2009 ).
- Creyghton MP , ChengAW , WelsteadGGet al. Histone H3K27ac separates active from poised enhancers and predicts developmental state . Proc. Natl Acad. Sci. USA107 ( 50 ), 21931 – 21936 ( 2010 ).
- Roh T-Y , CuddapahS , ZhaoK . Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping . Genes Dev.19 ( 5 ), 542 – 552 ( 2005 ).
- Kraushaar DC , JinW , MaunakeaA , AbrahamB , HaM , ZhaoK . Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3 . Genome Biol.14 ( 10 ), 1 – 15 ( 2013 ).
- Deaton AM , Gómez-RodríguezM , MieczkowskiJet al. Enhancer regions show high histone H3.3 turnover that changes during differentiation . eLife5 , e15316 ( 2016 ).
- Rada-Iglesias A , BajpaiR , SwigutT , BrugmannSA , FlynnRA , WysockaJ . A unique chromatin signature uncovers early developmental enhancers in humans . Nature470 ( 7333 ), 279 – 283 ( 2011 ).
- Schnetz MP , HandokoL , Akhtar-ZaidiBet al. CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression . PLoS Genet.6 ( 7 ), e1001023 ( 2010 ).
- Kadoch C , CopelandRA , KeilhackH . PRC2 and SWI/SNF chromatin remodeling complexes in health and disease . Biochemistry55 ( 11 ), 1600 – 1614 ( 2016 ).
- Torchy MP , HamicheA , KlaholzBP . Structure and function insights into the NuRD chromatin remodeling complex . Cell. Mol. Life Sci.72 ( 13 ), 2491 – 2507 ( 2015 ).
- Jang MK , MochizukiK , ZhouM , JeongHS , BradyJN , OzatoK . The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription . Mol. Cell19 ( 4 ), 523 – 534 ( 2005 ).
- Kaikkonen M-U , SpannN-J , HeinzSet al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription . Mol. Cell51 ( 3 ), 310 – 325 ( 2013 ).
- Zabidi MA , ArnoldCD , SchernhuberKet al. Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation . Nature518 ( 7540 ), 556 – 559 ( 2015 ).
- Ramsey SA , KlemmSL , ZakDEet al. Uncovering a macrophage transcriptional program by integrating evidence from motif scanning and expression dynamics . PLoS Comput. Biol.4 ( 3 ), e1000021 ( 2008 ).
- Ostuni R , PiccoloV , BarozziIet al. Latent enhancers activated by stimulation in differentiated cells . Cell152 ( 1–2 ), 157 – 171 ( 2013 ).
- Hah N , BennerC , ChongL-W , YuRT , DownesM , EvansRM . Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs . Proc. Natl Acad. Sci. USA112 ( 3 ), E297 – E302 ( 2015 ).
- Schmidt SV , KrebsW , UlasTet al. The transcriptional regulator network of human inflammatory macrophages is defined by open chromatin . Cell Res.26 ( 2 ), 151 – 170 ( 2016 ).
- Netea MG , QuintinJ , Van Der MeerJW . Trained immunity: a memory for innate host defense . Cell Host Micr.9 ( 5 ), 355 – 361 ( 2011 ).
- Hertweck A , EvansC-Á , EskandarpourMet al. T-bet activates Th1 genes through mediator and the super elongation complex . Cell Rep.15 ( 12 ), 2756 – 2770 ( 2016 ).
- Fang Z , HecklauK , GrossFet al. Transcription factor co-occupied regions in the murine genome constitute T-helper-cell subtype-specific enhancers . Eur. J. Immunol.45 ( 11 ), 3150 – 3157 ( 2015 ).
- Vahedi G , KannoY , FurumotoYet al. Super-enhancers delineate disease-associated regulatory nodes in T cells . Nature520 ( 7548 ), 558 – 562 ( 2015 ).
- Northrop JK , WellsAD , ShenH . Cutting edge: chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T Cells . J. Immunol.181 ( 2 ), 865 – 868 ( 2008 ).
- Zediak VP , JohnnidisJB , WherryEJ , BergerSL . Cutting edge: persistently open chromatin at effector gene loci in resting memory CD8+ T cells independent of transcriptional status . J. Immunol.186 ( 5 ), 2705 – 2709 ( 2011 ).
- Messi M , GiacchettoI , NagataK , LanzavecchiaA , NatoliG , SallustoF . Memory and flexibility of cytokine gene expression as separable properties of human TH1 and TH2 lymphocytes . Nat. Immunol.4 ( 1 ), 78 – 86 ( 2003 ).
- Farh KK-H , MarsonA , ZhuJet al. Genetic and epigenetic fine mapping of causal autoimmune disease variants . Nature518 ( 7539 ), 337 – 343 ( 2015 ).
- Seumois G , ChavezL , GerasimovaAet al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility . Nat. Immunol.15 ( 8 ), 777 – 788 ( 2014 ).
- Onengut-Gumuscu S , ChenWM , BurrenOet al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers . Nat. Genet.47 ( 4 ), 381 – 386 ( 2015 ).
- Roberts AR , VecellioM , ChenLet al. An ankylosing spondylitis-associated genetic variant in the IL23R-IL12RB2 intergenic region modulates enhancer activity and is associated with increased Th1-cell differentiation . Ann. Rheum. Dis.75 ( 12 ), 2150 – 2156 ( 2016 ).
- Weinstein JS , Lezon-GeydaK , MaksimovaYet al. Global transcriptome analysis and enhancer landscape of human primary T follicular helper and T effector lymphocytes . Blood124 ( 25 ), 3719 – 3729 ( 2014 ).
- Cavalli G , HayashiM , JinYet al. MHC class II super-enhancer increases surface expression of HLA-DR and HLA-DQ and affects cytokine production in autoimmune vitiligo . Proc. Natl Acad. Sci. USA113 ( 5 ), 1363 – 1368 ( 2016 ).
- Zeng L , ZhouMM . Bromodomain: an acetyl-lysine binding domain . FEBS Lett.513 ( 1 ), 124 – 128 ( 2002 ).
- Pinz S , UnserS , RascleA . Signal transducer and activator of transcription STAT5 is recruited to c-Myc super-enhancer . BMC Mol. Biol.17 ( 1 ), 1 – 11 ( 2016 ).
- Barda S , PazG , YogevLet al. Expression of BET genes in testis of men with different spermatogenic impairments . Fert. Ster.97 ( 1 ), 46 – 52 ( 2012 ).
- Nicodeme E , JeffreyKL , SchaeferUet al. Suppression of inflammation by a synthetic histone mimic . Nature468 ( 7327 ), 1119 – 1123 ( 2010 ).
- Ramirez-Carrozzi VR , NazarianAA , LiCCet al. Selective and antagonistic functions of SWI/SNF and Mi-2β nucleosome remodeling complexes during an inflammatory response . Genes Devel.20 ( 3 ), 282 – 296 ( 2006 ).
- Chen J , WangZ , HuXet al. BET inhibition attenuates Helicobacter pylori–induced inflammatory response by suppressing inflammatory gene transcription and enhancer activation . J. Immunol.196 ( 10 ), 4132 – 4142 ( 2016 ).
- Dooley KE , WarburtonA , McbrideAA . Tandemly integrated HPV16 can form a Brd4-dependent super-enhancer-like element that drives transcription of viral oncogenes . mBio7 ( 5 ), doi:10.1128/mBio.01446-16 ( 2016 ).
- Belkina AC , NikolajczykBS , DenisGV . BET protein function Is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1impair mouse macrophageinflammatory responses . J. Immunol.190 ( 7 ), 3670 – 3678 ( 2013 ).
- Barrett E , BrothersS , WahlestedtC , BeurelEO . I-BET151 selectively regulates IL-6 production . Bioch. Biophy. Acta1842 ( 9 ), 1549 – 1555 ( 2014 ).
- Wienerroither S , RauchI , RosebrockFet al. Regulation of NO synthesis, local inflammation, and innate immunity to pathogens by BET family proteins . Mol. Cell. Biol.34 ( 3 ), 415 – 427 ( 2014 ).
- Meng S , ZhangL , TangYet al. BET Inhibitor JQ1 blocks inflammation and bone destruction . J. Dent. Res.93 ( 7 ), 657 – 662 ( 2014 ).
- Qiao Y , GiannopoulouE-G , ChanC-Het al. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and toll-like receptor signaling . Immunity39 ( 3 ), 454 – 469 ( 2013 ).
- Chan CH , FangC , QiaoY , YarilinaA , PrinjhaRK , IvashkivLB . BET bromodomain inhibition suppresses transcriptional responses to cytokine-Jak-STAT signaling in a gene-specific manner in human monocytes . Eur. J. Immunol.45 ( 1 ), 287 – 297 ( 2015 ).
- Toniolo PA , LiuS , YehJEet al. Inhibiting STAT5 by the BET bromodomain inhibitor JQ1 disrupts human dendritic cell maturation . J. Immunol.194 ( 7 ), 3180 – 3190 ( 2015 ).
- Schilderink R , BellM , ReginatoEet al. BET bromodomain inhibition reduces maturation and enhances tolerogenic properties of human and mouse dendritic cells . Mol. Immunol.79 , 66 – 76 ( 2016 ).
- Bandukwala HS , GagnonJ , TogherSet al. Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors . Proc. Natl Acad. Sci. USA109 ( 36 ), 14532 – 14537 ( 2012 ).
- Mele DA , SalmeronA , GhoshS , HuangHR , BryantBM , LoraJM . BET bromodomain inhibition suppresses TH17-mediated pathology . J. Exp. Med.210 ( 11 ), 2181 – 2190 ( 2013 ).
- Belkina AC , BlantonWP , NikolajczykBS , DenisGV . The double bromodomain protein Brd2 promotes B cell expansion and mitogenesis . J. Leuk. Biol.95 ( 3 ), 451 – 460 ( 2014 ).
- Stanlie A , YousifAS , AkiyamaH , HonjoT , Begum NasimA . Chromatin reader Brd4 functions in Ig class switching as a repair complex adaptor of nonhomologous end-joining . Mol. Cell55 ( 1 ), 97 – 110 ( 2014 ).
- Peeters JGC , VervoortSJ , TanSCet al. Inhibition of super-enhancer activity in autoinflammatory site-derived T cells reduces disease-associated gene expression . Cell Rep.12 ( 12 ), 1986 – 1996 ( 2015 ).
- Michaeloudes C , MercadoN , ClarkeCet al. Bromodomain and extraterminal proteins suppress NF-E2-related factor 2–mediated antioxidant gene expression . J. Immunol.192 ( 10 ), 4913 – 4920 ( 2014 ).
- Zhang G , LiuR , ZhongYet al. Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition . J. Biol. Chem.287 ( 34 ), 28840 – 28851 ( 2012 ).
- Nadeem A , Al-HarbiNO , Al-HarbiMMet al. Imiquimod-induced psoriasis-like skin inflammation is suppressed by BET bromodomain inhibitor in mice through RORC/IL-17A pathway modulation . Pharmacol. Res.99 ( 0 ), 248 – 257 ( 2015 ).
- Fu W , FaracheJ , ClardySMet al. Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and β cells . eLife3 , e04631 ( 2014 ).
- Park-Min KH , LimE , LeeMJet al. Inhibition of osteoclastogenesis and inflammatory bone resorption by targeting BET proteins and epigenetic regulation . Nat. Commun.5 , 5418 ( 2014 ).
- Zhang QG , QianJ , ZhuYC . Targeting bromodomain-containing protein 4 (BRD4) benefits rheumatoid arthritis . Immunol. Lett.166 ( 2 ), 103 – 108 ( 2015 ).
- Klein K , KabalaPA , GrabiecAMet al. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts . Ann. Rheum. Dis.75 ( 2 ), 422 – 429 ( 2014 ).
- Trerotola M , RelliV , SimeoneP , AlbertiS . Epigenetic inheritance and the missing heritability . Hum. Genomics9 , 17 ( 2015 ).
- Park J-H , WacholderS , GailMHet al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries . Nat. Genet.42 ( 7 ), 570 – 575 ( 2010 ).