
Abstract
Aim: Obesity results from the interaction of genetic and environmental factors, which may involve epigenetic mechanisms such as DNA methylation (DNAm). Materials & methods: We have followed the PRISMA protocol to select studies that analyzed DNAm at baseline and end point of a weight loss intervention using either candidate-locus or genome-wide approaches. Results: Six genes displayed weight loss associated DNAm across four out of nine genome-wide studies. Weight loss is associated with significant but small changes in DNAm across the genome, and weight loss outcome is associated with individual differences in baseline DNAm at several genomic locations. Conclusion: The identified weight loss associated DNAm markers, especially those showing reproducibility across different studies, warrant validation by further studies with robust design and adequate power.
Worldwide, the prevalence of obesity has more than doubled since 1980. In 2014, 39% of adults older than 18 years were overweight, and 13% were obese. Most of the world’s population lives in countries where overweight and obesity are responsible for more deaths than underweight [Citation1]. Obesity is a major risk factor for Type-2 diabetes (T2D), cardiovascular disease and certain types of cancer such as endometrial, colon and breast cancer [Citation2]. The rising prevalence of these diseases is a top priority health concern and the combined medical costs associated with treatment of these preventable diseases are estimated to increase by $48–66 billion/year in the USA and by £1·9–2 billion/year in the UK by 2030 [Citation3]. Obesity is a multifactorial disease likely influenced by the interaction of genetic predisposition and exposure to obesogenic environmental factors. Among these factors are lifestyle choices including an excessive consumption of energy-dense foods and lack of physical activity [Citation4]. Nevertheless, people exposed to an obesogenic environment display a huge variability in body mass index (BMI), from normal weight (18.5–24.9 kg/m2) to overweight (25–29.9 kg/m2) or obese (30 kg/m2 and above) [Citation5]. Although recent studies indicate that 27–30% of this variability can be attributed to genetic variants including single nucleotide polymorphisms (SNPs) [Citation6,Citation7], these estimates may be inflated based on study-design factors and unaccounted shared conditions in family-based studies [Citation8].
Recent evidence indicates that not only genetic polymorphisms but also changes in gene expression underlie the development of obesity under exposure to an obesogenic environment [Citation9]. These changes in gene expression are regulated by a variety of mechanisms including epigenetic modifications, which interact with the genomic DNA or transcript without any change to the nucleotide sequence. Epigenetic modifications such as DNA methylation (DNAm) and histone modifications bring about alterations to chromatin structure, thereby modulating the accessibility of the transcriptional machinery to target genes [Citation10]. DNAm consists of methylation of the carbon 5 position within cytosine bases resulting in 5-methylcytosine, and occurs predominantly at cytosines within CG dinucleotides (‘CpG’ sites) in mammalian genomes. DNAm is typically associated with gene silencing when this occurs at gene promoters and enhancers, and with active gene expression when established within gene bodies [Citation11]. Regulatory noncoding RNAs represent a broad group of long (>200 bp) and short (<200 bp) RNA molecules that regulate gene expression pre- and post-transcriptionally, as reviewed elsewhere [Citation12]. Mechanistic studies in animal and plant model systems show that epigenetic modifications are modulated by environmental factors such as dietary components, chemical pollutants and temperature changes, as reviewed elsewhere [Citation13]. Epigenetics thus acts at the interface between environmental and genetic factors: environmental factors can induce epigenetic modifications, which can in turn affect gene activity. Through their effects on gene expression, epigenetic mechanisms can translate environmental stimuli into long-lasting alterations of cellular physiology. This is especially true when the target cells are stem or progenitor cells.
Epigenetic changes may play a causative role in obesity by inducing inappropriate expression or silencing of obesity-related genes and regulatory sequences, thus leading to metabolic imbalances [Citation14]. Alternatively, epigenetic changes may occur as a consequence of obesity, but once established, may predispose to obesity-related co-morbidities such as T2D mellitus (T2DM). In a recent population-based observational epigenome-wide association study (EWAS) by Wahl et al. BMI was found to be associated with changes in DNAm levels at 187 genetic loci. Furthermore, this study provided evidence to suggest that these changes were most likely caused by adiposity rather than a cause of it [Citation15]. The study by Wahl et al. is currently the most robust and significant EWAS in the field of obesity-associated DNAm in terms of population size, study design and methods used. However, since this was an observational study and not a weight loss intervention, the reversibility of obesity-associated DNAm could not be tested.
More than 2000 studies have analyzed the association between DNAm and obesity traits (BMI, waist circumference, subcutaneous adiposity) in order to explore epigenetic mechanisms in obesity development, or to identify biomarkers of obesity risk and reversal by weight loss. Most of these studies are cross-sectional and do not enable the assessment of temporal associations between adiposity and methylation, and any inference regarding causality and directionality. In contrast, longitudinal studies like the one by Wahl et al. do allow one to explore causal relationships between epigenetic markers and obesity, and to evaluate the power of these markers to predict weight gain and onset of obesity-associated co-morbidities such as T2DM. In addition, longitudinal studies of DNAm and obesity that involve a weight loss intervention can also assess the potential reversibility of obesity-associated epigenetic markers by measuring them in the same individual at baseline and end point. Provided the assignment to the intervention group is done randomly and with a sufficiently large sample size, intervention studies represent the most effective way to control for confounding factors and avoid possible sources of bias. For the above reasons, in this review, we have focused on intervention studies that analyzed DNAm at baseline and end point of a weight loss intervention in obese/overweight human subjects using either candidate-locus or genome-wide approaches. We have also excluded studies of subjects with hereditary disease or cancer, as these diseases are associated with DNAm changes [Citation16] that could confound the association between DNAm and weight loss. Finally, we discuss the strengths and limitations of these studies, and provide a perspective on future horizons in the field of weight loss related epigenomics.
Materials & methods
Search strategy
Studies for this review were identified following the PRISMA protocol via extensive searches of three bibliographic databases of scientific literature published from 2008 to 2016: PubMed (includes MEDLINE); SCOPUS (a large, multidisciplinary database); and the Cochrane Library. Search terms related to DNAm included: DNAm, bisulfite treatment and bisulfite conversion. Terms related to obesity included: GBP, diet, BMI, obesity, adiposity, lean body weight, overweight, body weight, weight loss and weight reduction. The searches excluded, wherever feasible, animal-only studies, review articles and non-English language studies. Complete, specific search strategies for each database including the database’s native search syntax are provided below.
PubMed
(‘Diet, reducing’ [mesh] OR ‘Obesity’ [mesh] OR ‘Overweight’ [mesh] OR ‘Body mass index’ [mesh] OR ‘Body weight’ [mesh] OR ‘Weight loss’ [mesh] OR ‘weight loss’ [tw] OR ‘weight reduction’ [tw] OR obes* [tw] OR adipos* [ti] OR ‘body fat’ [ti] OR ‘Body Fat Distribution’ [mesh] OR ‘adipose tissue’ [mesh] OR ((weight [ti] OR bmi [ti) AND (dietary [ti] OR diet [ti] OR loss [ti] OR losing [ti]))) AND (epigenetic* [ti] OR ‘DNA methylation’ [mesh] OR methylat* [ti] OR ‘DNA methylation’ [tw] OR ‘DNA Methylations’ [tw] OR ‘bisulfite treatment’ [tw] OR ‘bisulfite conversion’ [tw]) AND English [lang] NOT (‘animals’ [mesh] NOT ‘humans’ [mesh]) AND (‘2008/01/01’ [PDAT]: ‘2016/08/01’ [PDAT]) NOT ‘review’ [pt]
SCOPUS
(KEY((obesity OR ‘body mass’ OR ‘lean body weight’ OR overweight OR ‘body weight’ OR ‘weight loss’ OR ‘weight reduction’ OR adipos* OR ‘bisulfite treatment’ OR ‘bisulfite conversion’) AND ‘DNA methylation’)) OR (TITLE((obesity OR ‘body mass’ OR ‘body weight’ OR ‘weight loss’ OR overweight OR adipos* OR ‘bisulfite treatment’ OR ‘bisulfite conversion’ OR epigenet* OR epigenom* OR ‘gastric bypass’) AND methylat*)) AND (LIMIT-TO(LANGUAGE,‘English’)) AND (LIMIT-TO(PUBYEAR, 2016) OR LIMIT-TO(PUBYEAR, 2015) OR LIMIT-TO(PUBYEAR, 2014) OR LIMIT-TO(PUBYEAR, 2013) OR LIMIT-TO(PUBYEAR, 2012) OR LIMIT-TO(PUBYEAR, 2011) OR LIMIT-TO(PUBYEAR, 2010) OR LIMIT-TO(PUBYEAR, 2009) OR LIMIT-TO(PUBYEAR, 2008)) AND (EXCLUDE(DOCTYPE, ‘re’))
Cochrane library
Title/Abstract/Keywords: Methylat* AND (obesity OR ‘body mass’ OR ‘body weight’ OR ‘weight loss’ OR overweight OR adipos* OR epigenet* OR epigenom* OR ‘gastric bypass’)
Screening
The results of the literature search were screened for duplicates, first using Endnote software and then by visual inspection for further confirmation. The remaining results were then screened based on title/abstract and full text in two subsequent steps by two individuals using the web-based systematic review tool Covidence. After each screening step, conflicting decisions were solved to ensure consensus on selected studies. Next, full articles were assessed for eligibility for inclusion using the inclusion and exclusion criteria outlined below.
Inclusion criteria
We have included longitudinal studies published between 2008 and 2016 that measured either candidate-locus or genome-wide DNAm at baseline and end point of a weight loss intervention in overweight/obese subjects free of hereditary disease and cancer.
Exclusion criteria
We excluded cross-sectional studies, noninterventional population-based longitudinal studies, studies providing only in vitro data, studies and data on global DNAm (i.e., total cytosine methylation content as measured by HPLC tandem mass spectrometry, luminometric methylation assay or pyrosequencing of Alu/LINE1 repeat elements as surrogates for total genomic DNAm), reviews or methodology studies, studies that had not been carried out in humans as well as studies focusing on obesity co-morbidities such as diabetes or cardiovascular disease as primary predictors of methylation outcome. We have also excluded studies of subjects with hereditary disease or cancer, as these diseases are associated with DNAm changes [Citation16] that could confound the association between DNAm and obesity.
Outcomes
We compared two primary outcomes: the difference in candidate-locus or genome-wide methylation before and after intervention; and the difference between high- and low-responders to the intervention, either at baseline or end point. The first outcome measures weight loss associated differences in DNAm within the same individuals to investigate mechanisms of epigenetic remodeling and potential reversal of obesity-associated methylation in response to weight loss. The second outcome measures differences of DNAm between subjects that may be potentially more stable and, therefore, serve as biomarkers of potential weight loss success when determined at baseline of a weight loss intervention.
To identify any overlap of weight loss associated genes displaying changes in DNAm across the 9 EWAS, all of the genes identified within each study as having a statistically significant change in DNAm levels were included. Analysis of the overlap of weight loss associated genes between studies was conducted using R version 3.3.1. Gene names were converted to HUGO gene symbols using the HGNChelper R package. The BioMaRt R package was used to convert differentially methylated mouse genes to human orthologs for the Multhaup study, as they reported mouse genes that were differentially methylated in both mice and humans. For studies that used Illumina 27 k and 450 k arrays, genes associated with cg probes were identified using the Illumina Human Methylation 27 k and 450 k annotation packages.
Results
Summary of reviewed studies
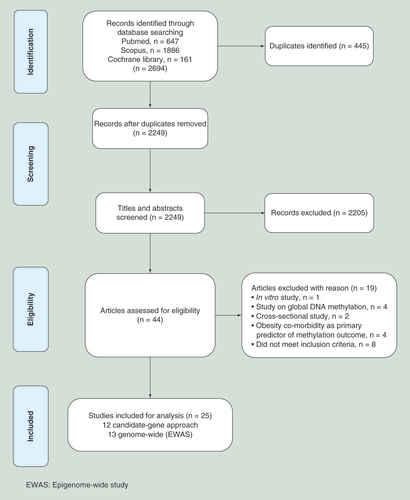
We searched three databases (PubMed, SCOPUS and Cochrane Library) following the PRISMA protocol to select studies that analyzed DNAm at baseline and end point of a weight loss intervention in overweight/obese subjects free from co-morbidities such as hereditary disease or cancer (). A total of 25 longitudinal intervention studies met our inclusion criteria and were included for detailed analysis in this review article. Each of these studies measured either pre- versus postintervention changes in DNAm, or differences in DNAm between high versus low responders to the intervention at baseline and/or end point. A wide range of tissues was used for these analyses, including adipocytes, muscle cells, blood and sperm cells. Of these studies, 16 used a candidate-gene approach, wherein the weight loss associated DNAm was measured at specific putative obesity susceptibility genes. Thirteen candidate-gene approach studies were diet and/or exercise interventions, whereas the other three studies were GBP surgery interventions. Details of each candidate-gene study, including a description of subjects, methodology employed and outcomes are summarized in & . The other nine studies were epigenome-wide association studies (EWAS) of weight loss associated DNAm, in which a genome-wide approach was used to identify differentially methylated individual CpG sites (DMS) or differentially methylated regions (DMRs). Three EWAS were diet and/or exercise interventions, and six were GBP surgery interventions. A summary of the subjects, methodology employed and outcomes of these studies is provided in & .
EWAS: Epigenome-wide study.
Candidate-gene approach studies
Sixteen studies investigated either pre- versus postintervention changes in DNAm, or differences in DNAm between high versus low responders to the weight loss intervention at genes implicated in the development of obesity based on evidence from previous studies. To quantify candidate-locus DNAm, these studies used a variety of quantitative methylation assays including pyrosequencing, methylation-sensitive single-nucleotide primer extension and MassARRAY EpiTyper.
Out of 16 candidate-gene approach studies, 13 were diet and/or exercise interventions, and 3 GBP surgery interventions ( & ). Seven diet studies [Citation17–18,Citation21–25], and all three GBP [Citation29–31] studies measured pre- versus postintervention changes in DNAm to explore mechanisms of epigenetic remodeling in response to weight loss (hereafter referred to as ‘mechanistic studies’). Overall, these mechanistic studies provide evidence that weight loss affects the methylation status of several genes, including obesity-associated genes (FTO, BDNF, SH2B1, PPARGC1A, PDK4, IL1B, SERPINE1 [Citation16,Citation21,Citation29,Citation31,Citation41]), metabolic genes (SCD1 [Citation22], PON1 [Citation25]), stroke-related genes (KCNQ1 and WT1 [Citation17]), genes regulated by diet (FADS1, FFAR3, CD14–36 [Citation18]) and circadian rhythm genes (CLOCK, PER2, BMAL1 and NR1D1 [Citation23–24]). The changes in the methylation of these genes in response to weight loss may reflect the reversal of epigenetic modifications that either cause or are caused by obesity. However, these changes may also be specifically associated with metabolic remodeling during weight loss rather than predisposing to, or be caused by, obesity.
Six diet intervention studies aimed at identifying DNAm biomarkers of weight loss response by analyzing baseline and/or end point methylation differences between high versus low responders to the diet, or in weight regainers versus nonregainers (hereafter referred to as ‘biomarker studies’) [Citation16,Citation19–20,Citation26–28]. These biomarker studies indicate that baseline methylation of TNF-α, LEP [Citation16,Citation19], POMC, NPY [Citation20], SERPINE-1 [Citation26], HTR2A [Citation28], WT1, ATP10A and CD44 [Citation27] can predict weight loss response at end point. Specifically, at baseline, high-responder dieters showed lower promoter methylation levels of LEP (−47%, p < 0.05) [Citation16] and TNF-α (-39%, p = 0.071 [Citation16], r = 0.74; p = 0.021 [Citation19]) than the nonresponder group, while obese men who regained weight after the diet showed higher methylation within POMC (+26%; p = 0.020) and lower methylation of NPY (-22%; p = 0.033) compared with nonregainers [Citation20]. In addition, lower HTR2A methylation (low ≤ 0.556; medium = 0.557–0.585) [Citation28] and higher methylation of SERPINE-1 (>7%) [Citation26], ATP10A (r = 0.42) and CD44 (r = 0.3) at baseline was associated with greater weight loss [Citation27], while end point methylation of the WT1 promoter was higher (+3–9%) in high versus low responders [Citation27].
Most of the candidate genes studies reviewed herein investigated different loci, therefore reproducibility between studies could not be formally assessed. Nevertheless, one obesity-associated gene, TNF-α, that was investigated in two independent diet intervention studies did show reproducibility between them [Citation16,Citation19]. Both studies found an association between lower baseline methylation of the TNF-α promoter and greater weight loss in response to a calorie-restricted diet. Given that these two studies used different methodological approaches (bisulfite-specific PCR or methyl-specific PCR) and different tissues (peripheral blood mononuclear cells[PBMCs] or adipose tissue) this represents a promising weight loss associated biomarker. Other potential biomarkers for weight loss include the circadian rhythm genes CLOCK and PER2. Baseline methylation of these two genes in blood was associated with magnitude of weight loss in two diet intervention studies reported by the same group [Citation23–24]. However, this association reached statistical significance only in the pilot study (CLOCK: r = 0.337, PER2: p = 0.01, r = 0.318, p = 0.016) [Citation23] and not in the follow-up one [Citation24], likely due to differences in anthropometric and phenotypic features (i.e., baseline BMI, co-morbidities) of the subjects compared with the previous study [Citation23]. Two of the biomarker studies reported that baseline methylation levels of SERPINE-1 could predict weight loss success in response to a diet [Citation26] or a GBP intervention [Citation31]. However, higher levels of baseline methylation of SERPINE-1 were associated with opposite weight loss outcomes in the two studies: with a greater weight loss in response to a calorie-restricted diet, and with lower weight loss after GBP surgery. Finally, discrepant results were found for the gene PDK4, whose methylation levels decreased after a diet intervention (-0.5 to -2%) [Citation18], and increased upon GBP surgery (11.3% at baseline vs 6.4% 2-days post-GBP, and 19% 1-year post-GBP) [Citation29].
Epigenome-wide association studies
In the past decade, the emergence of affordable high-throughput genome-wide methylation profiling techniques has enabled the first EWAS. These studies have measured DNAm at single CpGs or regions containing CpGs across thousands of genes using a genome-wide approach, and analyzed associations with lifestyle factors and disease phenotype. Of the 25 intervention studies included in this review, 9 were EWAS that measured genome-wide DNAm before and after a weight loss intervention ( & ). The majority of the EWAS used array-based platforms, most frequently the Infinium 27 k or 450 k arrays (Illumina), to interrogate specific CpG sites to identify DMSs. The 27 k array represents less than 0.1% of the 28 million CpG sites in the human genome, covering on average 2 CpG sites in the promoter regions of approximately 14,000 genes with CpG island promoters. The 450 k array, represents 2% of the CpG sites in the genome, covering 99% of RefSeq genes with probes located within promoters, gene bodies, 3′UTRs and other intergenic sequences as well, but remains promoter-centric. Other studies used a fractionation method for the selective enrichment of either hypermethylated or unmethylated DNA sequences, followed by relative hybridization to tiling arrays or bisulfite sequencing, to identify DMRs [Citation32,Citation35,Citation39]. One study used methylated DNA immunoprecipitation [Citation35], in which hypermethylated DNA regions were enriched via capture on magnetic beads labeled with methyl binding domain proteins. Another study used McrBC digestion to cleave methylated DNA, followed by comprehensive high-throughput arrays-based relative methylation analysis [Citation39], which interrogates 7.5 million of the 28 million CpGs in the human genome [Citation42]. Another study employed reduced representation bisulfite sequencing, whereby genomic DNA is systematically cleaved with the MspI restriction enzyme (recognition site C’CGG) and size-fractionated to enrich for fragments containing CpG sites, then subjected to bisulfite conversion and sequencing to determine the methylation status at individual CpG resolution [Citation43]. Most of the EWAS included in this review subsequently validated a subset of the DMS or DMR initially identified by a genome-wide approach via an independent, targeted quantitative method, such as EpiTYPER or pyrosequencing. This is especially important for EWAS, as unlike genotype which is a categorical variable, DNAm shows a bimodal pattern of methylation at any individual CpG site on a single allele (is either methylated or unmethylated), but requires evaluation as a continuous variable when applied across the numerous alleles from a pooled population of cells such as white blood cells. Thus, measurement of DNAm levels warrants the careful use of various technical and statistical approaches.
Out of nine EWAS, three were diet and/or exercise interventions, and six GBP surgery interventions. One diet intervention [Citation34] and all six GBP studies [Citation35–40] measured pre- versus postintervention changes in DNAm to identify weight loss associated DMS and explore underlying epigenetic mechanisms. Overall, these mechanistic studies indicate that weight loss induces statistically significant but quantitatively small changes in DNAm levels (mostly of ≤5% magnitude) across the genome. The identified weight loss associated DMS were often enriched in genes implicated in obesity (PPARGC1A, SH2B1, FTO, BDNF, KCNQ1, WT1, PDK4, MC4R, SDCCAG8, TMEM18 [Citation27,Citation34–36,Citation38]), and T2DM (HHEX, IGF2BP2, JAZF1, TCF7L2 [Citation34]). Notably, some of these obesity-related genes displayed similar weight loss associated methylation changes in previous candidate-gene approach studies. This was the case for PDK4 [Citation18,Citation29,Citation35], KCNQ1 [Citation17,Citation34,Citation36], WT1 [Citation17,Citation27], PPARGC1A [Citation29,Citation35], SH2B1, FTO and BDNF [Citation21,Citation38]. Other DMS were found nearby or within genes with a wide diversity of biological functions, such as metabolism (CHST8 [Citation38]), immune response (CD44 [Citation27]) and regulation of transcription (CPEB4 [Citation34]).
Two dietary interventions [Citation32,Citation33] analyzed baseline and/or end point methylation differences between high versus low responders to the intervention in order to identify DNAm markers of weight loss response. These biomarker studies indicate that baseline methylation levels at several genomic locations can predict an individual’s propensity to lose weight in response to caloric restriction. Many of these genomic loci were proximal to genes with a broad range of functions such as membrane transport (AQP9: Aquaporin 9 [Citation33]), cell signaling (DUSP22: Dual Specificity Phosphatase 22, HIPK3: Homeodomain Interacting Protein Kinase 3 [Citation33]) and muscle contraction (TNNT1: Troponin T1, TNNI3: Troponin I3 [Citation33]).
We compared all the gene loci displaying statistically significant weight loss associated changes in DNAm within each of the nine EWAS across all of these studies to identify any overlap in loci (Supplementary Table 1). Out of 6091 genes cross-compared, 6 genes were identified across four different studies: GATA-Binding Protein 2 (GATA2) [Citation34–36,Citation38], Transcription Factor 7 Like 2 (TCF7L2) [Citation35–36,Citation38–39], Sidekick Homolog 1 (SDK1) [Citation35–38], Phosphatidylinositol-5-Phosphate 4-Kinase Type 2 Alpha (PIP4K2A) [Citation34–36,Citation38], Ras Responsive Element Binding Protein 1 (RREB1) [Citation35–38] and Histone Deacetylase 4 (HDAC4) [Citation34,Citation36–38]. Another 57 genes were identified as overlapping across three out of nine EWAS, and a further 720 genes across two out of nine EWAS. All of the studies across which overlapping results were found were mechanistic studies that analyzed pre- versus postintervention changes in DNAm in the same individuals. No overlap of results was identified between the two biomarker studies that measured differences in DNAm between high versus low responders to a dietary intervention [Citation32,Citation33].
One reason for this limited reproducibility of results may lie in the high heterogeneity of study design between the reviewed studies in terms of the type of intervention (i.e., diet and/or exercise or GBP surgery), characteristics of the study population and study duration. All these factors can indeed influence both weight loss predictor and DNAm outcomes. Moreover, studies on weight loss associated DNAm are based on the assumption that obesity-related DNAm marks are reversible within the same individuals – which may not be the case. This may lead to spurious associations due to random variation, thus affecting the reproducibility of results between different studies. Finally, differences in the tissue and methodology employed provide an additional source of variability between the different studies reviewed in this article.
Discussion
We have carried out a systematic review of candidate-gene and genome-wide studies of DNAm in overweight/obese patients before and after a weight loss intervention involving diet and/or exercise, or GBP surgery. We have focused on DNAm, which is by far the most investigated modification in epigenetic epidemiology. This is probably because the methods used to measure DNAm can quantitate epigenetic changes occurring in only a subset of alleles tested at single-nucleotide resolution, which is still not possible for methods used to measure other epigenetic modifications such as histone modifications. Our rationale behind the inclusion of only intervention studies in this review is that they overcome some possible sources of bias encountered in cross-sectional and, to a lesser extent, noninterventional longitudinal studies such as the potential for spurious and noncausal associations. Moreover, when measuring epigenetic changes in the same individuals, intervention studies can account for the effect of genotype on methylation status, which can explain 22–80% of the variability in DNAm between individuals [Citation44,Citation45]. Of the 25 studies included in this review, 16 used a candidate-gene approach, whereas 9 were EWAS. Each of the reviewed studies measured either pre- versus postintervention changes in DNAm, or differences in DNAm between high versus low responders to the intervention at baseline and/or end point.
Ten candidate-gene approach studies and seven EWAS analyzed weight loss associated changes in DNAm pre- and postintervention to explore mechanisms of epigenetic remodeling in response to weight loss [Citation17–18,Citation21–25,Citation29–31,Citation34–40]. Overall, these studies indicate that weight loss is associated with small but extensive changes in DNAm across the genome in proximity to obesity- and diabetes-related genes, as well as genes with a wide variety of functions ranging from immune defense to gene transcription. Seven obesity-related genes displayed similar weight loss associated methylation changes across candidate-gene approach and EWAS studies: PDK4 [Citation18,Citation29,Citation35], KCNQ1 [Citation17,Citation34,Citation36], WT1 [Citation17,Citation27], PPARGC1A [Citation29,Citation35], SH2B1, FTO and BDNF [Citation21,Citation38]. Moreover, six genes showed weight loss associated changes in DNAm across four different mechanistic EWAS: GATA2, TCF7L2, SDK1, PIP4K2A, RREB1, HDAC4. These findings support the hypothesis that several epigenetic marks might be modifiable, not only by changing exposures in utero such as the maternal diet [Citation46–49], but also by lifestyle changes in adult life. This implies that there is the potential to design targeted interventions in adult life to revert disease-associated epigenetic patterns toward those observed in healthy subjects.
Six candidate-gene approach studies and two EWAS analyzed baseline and/or end point DNAm in high versus low responders to the dietary or GBP intervention to identify DNAm biomarkers of weight loss response [Citation16,Citation20,Citation26–28,Citation31–33]. Studies of DNAm biomarkers measure epigenetic markers that vary between different individuals, but which are stable and potentially permanent, within the same individual. These studies indicate that baseline methylation levels at several genomic locations can predict an individual’s propensity to lose weight on a calorie-restricted diet. These genomic loci are located in proximity to obesity-associated genes such as LEP, TNF-α, BDNF and NPY, and nearby other genes within a variety of cellular pathways from cell signaling to membrane transport. These epigenetic signatures may potentially be used in precision medicine as baseline biomarkers to predict the effectiveness of a weight loss intervention.
There was limited overlap of results between the studies included in this review. Among the candidate-gene approach studies, reproducible results were reported only for the TNF-α gene, for which lower methylation in the promoter region was consistently associated with greater weight loss response in two different diet intervention studies [Citation16,Citation19]. Out of 6091 genes analyzed in the 9 EWAS, only 6 genes were identified as overlapping across four different studies (GATA2, TCF7L2, SDK1, PIP4K2A, RREB1, HDAC4) and 57 genes across three studies (Supplementary Table 1).
One reason for this limited reproducibility may lie in the nature of epigenetic modifications, and the difficulty in eliminating sources of bias in epigenetic studies. Any epigenetic change associated with weight loss may, in fact, reflect weight loss associated physiological events that can never be fully accounted for in the study design. Weight loss induces a wide range of physiological events that can affect epigenetic modifications, and display a high degree of variability between individuals depending on the type of intervention (i.e., diet and/or exercise or GBP surgery), characteristics of the study population and study duration.
Diet and GBP surgery induce drastically different weight loss responses. These are stronger for GBP interventions, which likely mimic starvation effects reported to affect epigenetic changes in studies of populations exposed to famine [Citation49,Citation50]. In addition, the populations of diet and GBP studies often differ in terms of grade of adiposity, presence of associated metabolic disturbances and the use of pharmacological drugs at baseline, since bariatric surgery is indicated for subjects with higher BMI who are often under prescription for drugs to improve the severity of comorbidities associated with higher adiposity. Furthermore, other factors inherent to the surgical procedure might affect DNAm outcomes, such as alterations of the gastrointestinal tract (changes in hormone secretion and the microbiome), and the prescription of vitamin/mineral supplements to avoid nutritional deficiencies after the intervention. Finally, in some of the reviewed studies the subjects followed a calorie-restricted diet prior to the GBP surgery, and hence the observed changes in DNAm reflect a consequence of both interventions [Citation29,Citation31]. All these factors may contribute to the lack of overlap between weight loss associated DNAm markers identified by diet and GBP intervention studies. In some cases diet and GBP studies show even contrasting results. For instance, IL-6 methylation increased after a diet intervention, and decreased after GBP surgery [Citation31]. Not only weight loss associated DNAm but also DNAm markers of weight loss response show discrepancy between diet and GBP intervention studies. For instance, although SERPINE-1 displayed different baseline methylation levels between high versus low responders to both a diet [Citation26] and a GBP intervention [Citation31], the reported methylation differences show opposite directions in the two studies: higher baseline SERPINE-1 methylation was associated with greater weight loss in response to a calorie restricted diet, and lower weight loss after GBP surgery.
The lack of reproducible results even within diet or GBP surgery studies may be explained by differences in study population and duration between different studies. The populations of the reviewed studies varied in ethnicity, age, gender-ratio, degree of obesity and presence of co-morbidities. All of these factors can influence both weight loss response and DNAm outcome, thereby affecting any associations between the two events. Also the timeline of the designed interventions can influence both weight loss response and DNAm outcomes. Weight loss is an exquisitely dynamic process, characterized by different stages and degrees of cellular catabolism. Therefore, the underlying physiological processes of weight loss, and the epigenetic changes that reflect them, can differ significantly at different time points during and after the intervention. The diet interventions included in this review have different durations ranging from 14 days to 1 year, while studies on GBP surgery collected samples from subjects at different time-points after the intervention, ranging from 1 week to 2 years. These different time snapshots capture different physiological events, which may result in different epigenetic outcomes. Finally, there is a limitation inherent to all intervention-based studies of weight loss associated changes in DNAm, which may contribute to the lack of reproducibility in their results. These studies are based on the assumption that obesity-related DNAm marks are indeed reversible within the same individuals. But what if weight loss reverts the obesity phenotype but not the associated epigenotype to the normal-weight state? For example, the obesity-associated leptin gene (LEP), whose promoter methylation displays lower baseline levels in high versus low responders to a calorie restricted diet [Citation16] did not change upon weight loss within the same individuals [Citation30]. If obesity-associated epigenetic marks, once established, remain stable within the individual, could this contribute to the susceptibility to subsequent weight regain among formerly obese/overweight subjects?
Another reason for the limited reproducibility of results may lie in different methodological approaches applied between the different studies. Candidate gene studies measured weight loss associated DNAm at specific putative obesity susceptibility genes using diverse quantitative methylation assays including pyrosequencing, methylation-sensitive single-nucleotide primer extension and MassARRAY EpiTYPER. The nine EWAS analyzed weight loss associated DMS or DMR using a variety of techniques to measure the relative levels of DNAm on a ‘genome-wide’ scale, each of which incur some bias in CpG sites sampled. The majority of the EWAS used the Infinium 27 k or 450 k arrays (Illumina), which focus primarily on a few individual CpG sites within promoter regions, under the assumption these are representative of the methylation status of the region encompassing them (the methylation state of neighboring CpG sites tend to correlate) [Citation51]. Another study used methylated DNA immunoprecipitation [Citation35], which is biased toward the capture of CpG rich sequences such as CpG island promoters. The study by Multhaup et al. used McrBC digestion to cleave methylated DNA, followed by comprehensive high-throughput arrays-based relative methylation analysis to interrogate over 25% of the CpGs in the human genome [Citation39]. This methodology was applied to both a mouse model of obesity and to humans in a creative cross-species approach to identify loci that displayed weight loss associated changes in DNAm that were conserved between species. Another study employed reduced representation bisulfite sequencing [Citation38], which aims to minimize bisulfite sequencing to the most informative fraction of the genome – the regions containing CpG sites – but in doing so favors CpG rich regions such as CpG island promoters. The ‘gold standard’ EWAS to identify DNAm changes associated with obesity or weight loss would entail an unbiased interrogation of the entire DNA methylome in appropriate samples; that is, measurement of the absolute levels of methylation at all 28 million CpG sites contained within the human genome. Whole genome bisulfite sequencing at a depth of coverage that would enable the deconvolution of heterogeneous cell types within any given sample, and the detection of 5% changes in DNAm between pre- and postintervention samples from each study subject after correction for multiple testing, would be the optimal method. However, this method is currently cost-prohibitive and, therefore, impracticable for application to a large sample size.
In parallel to these epidemiological and methodological considerations, the choice of the tissue to analyze and the heterogeneity of its cell composition cast an additional layer of complexity in the interpretation of these studies. Easily accessible and routinely sampled in large-scale studies, blood is the most frequently used source of DNA in the studies included in this review, and in epigenetic studies in general. A potential problem with using blood in studies of DNAm is that it consists of a mixture of different cell types with distinct methylation profiles. Differences in the proportion of these cell types could obscure or confound the association between DNAm and the phenotype being studied. However, this variability in cell composition has been shown to affect methylation at a minority of CpGs [Citation52], while leaving unaffected methylation at most CpGs [Citation53]. Moreover, it is possible to statistically correct for the effects of this cellular heterogeneity on methylation patterns by quantifying cell subtype composition using cell biology and molecular approaches [Citation54–56]. Another possible concern is that DNAm measured in blood may not reflect the epigenetic state of disease-relevant tissues. However, a recent study shows that DNAm markers of BMI detected in blood can be replicated in adipose tissue, which is a highly disease-relevant tissue in obesity [Citation57]. Adipose tissue not only is the primary tissue for energy storage, but it also exerts important endocrine activities, which are often deregulated in obese subjects. A minority of the reviewed studies used adipose tissue to measure weight loss associated changes in DNAm. Some of these studies indicate that DNAm profiles in adipose tissue change significantly after a diet, GBP or exercise intervention [Citation34,Citation58–59], and display differences between low- and high-responders to the weight loss interventions [Citation16]. This corroborates the notion that studies of DNAm in adipose tissues are of high relevance, and warrant further investigation. However, adipose tissue also displays a high degree of cell-type heterogeneity, being composed of adipocytes that make up about 20–40% of the total cellular content, fibroblasts, preadipocytes, stem cells and immune cells. During weight loss, this cell composition will change, and these changes will affect observed levels of cell type-specific DNAm. Therefore, in order to avoid spurious associations and understand causal relationships between weight loss and DNAm, it is important to use algorithms to deconvolute cell mixtures or analyze isolated adipocytes. On the other hand, if the goal of the study is not to propose causal mechanisms but to develop biomarkers for predictive, prognostic or diagnostic use, correcting for cell-subtype composition is not necessary, and may be in fact detrimental. Robust biomarkers, either in blood or adipose tissue, can result from reliable but noncausal associations, independently of whether these are due to epigenetic modifications in the cells tested or reflective of cell subtype effects on DNAm [Citation60].
All complicating and confounding factors mentioned above emphasize the importance of large populations in studies aiming to identify DNAm markers affected by, or able to predict, weight loss outcome. The sample sizes of the studies included in this review range from 8 to 155 subjects for candidate-gene approach studies, and from 6 to 47 subjects for EWAS. These numbers are likely not large enough to minimize potential confounding factors and random variation, which affect reproducibility. In addition, it is important to note that most of the changes in DNAm identified by the reviewed studies were <5% in magnitude, which is the minimum change detectable with good reproducibility for most platforms. In EWAS, only a few of these changes remained significant after multiple testing corrections, as the number of subjects was low compared with the number of sites tested.
For all the above reasons it is imperative to analyze and interpret the results of these studies with extreme caution and avoid any overinterpretation. Most of the reviewed studies lack appropriate replication testing in different cohorts and tissues, functional assays (such as gene expression correlates) and controls (i.e., longitudinal data of healthy individuals over the same time frame). Because of all these potential sources of bias, the findings of these studies, especially when reporting quantitatively small changes in DNAm, might not be related to any clear biological mechanism. Despite representing the gold standard of research designs, intervention studies should fulfill several quality standards in order to provide the strongest evidence of association between exposure and outcome. These standards include adequate population size and study duration, random assignment to intervention groups and making sure all those originally randomized to the intervention and control groups are retained in their group when analyzing study results. When intervention studies fail to meet these standards, as in the case of many of the studies reviewed herein, their findings can be less robust and reliable than those produced by a well-designed large-scale longitudinal study. An example of such a study is a recently published EWAS of BMI and DNAm by Wahl et al. In this study, BMI-associated DNAm was measured in blood samples from European (n = 2707) and Asian-Indian (n = 2680) populations and replicated in separate cohorts of European and Asian-Indian populations (n = 4874). Most of the 187 BMI-associated DNAm markers that were identified and successfully replicated were found to be a consequence of increased BMI, rather than a cause, based on genetic-association analyses. Most importantly, in further longitudinal analyses (n = 2664) baseline methylation at 62 of these markers predicted the onset of T2DM after 7 years. The T2DM risk associated with these DNAm markers was independent from, and for some markers greater than, those estimated by traditional risk factors such as overweight, increased fasting glucose and levels of glycated hemoglobin (HbA1c). The most significant association was found for a CpG (cg06500161) within a CpG island shore in the ABCG1 locus, a gene involved in insulin secretion and pancreatic β-cell function [Citation61,Citation62]. The association between methylation of this specific CpG and BMI was replicated across all the cohorts, validated in different tissues (blood, adipocytes, liver) and functionally reflected by an inverse correlation with ABCG1 gene expression. Strikingly, one of the EWAS reviewed herein found that this particular CpG of ABCG1 was differentially methylated in sperm cells from obese versus lean, and pre- versus post-GBP men [Citation38]. Furthermore, another of the reviewed studies found that a different CpG within the ABCG1 locus (cg10192877) was differentially methylated in postobese versus never-obese women in isolated adipocytes, and that these differences also corresponded with changes in gene expression [Citation37]. Three other T2DM markers identified by Wahl et al. displayed weight loss associated DNAm in five of the studies reviewed herein: ATP10A, TNFA and CTP1A. The ATP10A gene showed higher baseline methylation in high-responders versus low responders to a diet intervention [Citation27], whereas lower promoter methylation of TNFA was consistently associated with greater weight loss response in two different diet intervention studies [Citation16,Citation19]. Finally, methylation at the CTP1A locus was found to be significantly different between lean versus pre-GBP subjects [Citation35], and, like in the case of cg06500161 of ABCG1, between obese versus lean, and pre- versus post-GBP men [Citation38]. Interestingly, in a recent cross-sectional EWAS the same CpGs identified by Wahl et al. in CPT1A and ABCG1 displayed highly significant associations with BMI across populations of African American adults (n = 2,097) and Europeans (n = 3,368), both in blood and adipose tissue [Citation57]. These exciting findings make ABCG1, CPT1A, TNFA and ATP10A extremely promising candidates for future intervention studies of weight loss associated DNAm. The longitudinal study by Wahl et al. sets a new gold standard for these future studies, which should include larger populations, longer study duration, appropriate replication testing in different cohorts/tissues, correlative functional assays and genetic analyses that address causality relationships.
Future perspective
Future studies should take into account several factors that are crucial for the identification of causal associations between weight loss and DNAm, and for the development of DNA biomarkers of weight loss response. A first key factor is a robust study design, which should include a clear hypothesis, and a solid methodological approach to test it. While biomarker studies may use surrogate tissues such as blood, mechanistic studies may require cross-tissue replication in a disease relevant cell population such as adipose tissue with high cell-type purity or appropriate cell-type deconvolution. Functional genomics, gene expression and genetic analyses are also important to assess biological relevance of epigenetic changes and their causality relationships with adiposity. An additional crucial factor in the study design is the duration of a weight loss intervention. Ideally, diet and/or exercise studies should last at least 1 year to cover both the weight loss phases, which typically hits a plateau at 6 months, and the subsequent maintenance phase. GBP studies should monitor patients for 1–2 years at different time points after surgery to ensure representation of different stages in the wide spectrum of catabolic processes associated with GBP-induced weight loss. Finally, adequate power is key to overcome the potential of spurious associations and produce robust and reliable evidence. For future EWAS, the next challenge is to find next-generation sequencing methods at a reasonable cost, in order to enable large enough population size and adequate power.
Table 1. Candidate-gene approach studies: diet interventions.
Table 2. Candidate-gene approach studies: gastric bypass surgery interventions.
Table 3. Epigenome-wide association studies: diet and/or exercise interventions.
Table 4. Epigenome-wide association studies: gastric bypass surgery interventions.
Out of 2694 studies on obesity & DNA methylation only 25 are intervention studies
Twenty-five articles, out of 2694 found through PubMed, SCOPUS and the Cochrane Library, were included in this study. These studies analyzed DNAm at baseline and end point of a weight loss intervention (either diet and/or exercise interventions, or a gastric bypass surgery) in overweight/obese subjects free of hereditary disease and cancer.
Sixteen of these studies used a candidate-gene approach, wherein the relationship between weight loss and DNAm was tested at specific putative obesity susceptibility genes.
The other nine studies were epigenome-wide association studies (EWAS) of obesity-associated DNAm, in which a genome-wide approach was used to identify weight loss associated CpGs, termed differentially methylated sites.
Weight loss is associated with small but extensive DNAm changes across the genome
Ten candidate-gene studies and seven EWAS-analyzed weight loss associated changes in DNAm to explore mechanisms of epigenetic remodeling in response to weight loss.
Weight loss was associated with extensive changes in DNAm at several genomic loci, including obesity- and diabetes-related genes. However, these were small in magnitude (mostly <5% difference in methylation levels).
Baseline DNAm at several genomic loci were associated with weight loss outcome
Six candidate-gene approach studies and two EWAS-analyzed baseline DNAm in high versus low diet responders to identify DNAm biomarkers of weight loss response.
Baseline DNAm levels at several genomic loci were associated with weight loss outcome at end point. Some of these loci are located in proximity of obesity-associated genes such as LEP, TNF-α, BDNF and NPY.
Limitations
The studies included in this review show limited overlap of results. The only reproducible results were: an association between weight loss and changes in DNAm at the genes GATA2, TCF7L2, SDK1, PIP4K2A, RREB1, HDAC4 (which were identified in four out of nine EWAS) and at the obesity-related genes PDK4, KCNQ1, WT1, SH2B1, FTO, BDNF and PPARGC1A (which were identified across EWAS and candidate gene studies); an association between greater weight loss response to a diet intervention and lower methylation levels of the TNF-α gene promoter at baseline.
This limited overlap of results may be due to the high heterogeneity of study design between the reviewed studies in terms of the type of intervention (i.e., diet and/or exercise or gastric bypass surgery), characteristics of the study population, study duration, tissue type and methodology employed.
Conclusion
Only 25 studies of DNAm have analyzed mechanisms of epigenetic remodeling or epigenetic biomarkers of weight loss outcome in response to a weight loss intervention. Weight loss induces small (mostly <5%) changes in DNAm across the genome, and weight loss outcome is associated with interindividual differences in DNAm at several genomic loci.
The identified DNAm markers show poor reproducibility between studies, likely due to the high heterogeneity of study design, population and duration between different studies.
The identified weight loss associated DNAm markers, especially those showing reproducibility across different studies, warrant validation by further studies with robust design and adequate power.
Acknowledgements
Thanks to Christopher Stave at the Lane Medical Library at the Stanford School of Medicine for helping us develop our search strategy, guiding us through the screening process and providing valuable input to the Methods section.
Financial & competing interests disclosure
This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no 701944. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- World Health Organisation . www.who.int/mediacentre/factsheets/fs311/en/
- Pi-Sunyer X . The medical risks of obesity . Postgrad. Med.121 ( 6 ), 21 – 33 ( 2009 ).
- Wang YC , McphersonK , MarshT , GortmakerSL , BrownM . Health and economic burden of the projected obesity trends in the USA and the UK . Lancet378 ( 9793 ), 815 – 825 ( 2011 ).
- Misra A , SinghalN , KhuranaL . Obesity, the metabolic syndrome, and Type 2 diabetes in developing countries: role of dietary fats and oils . J. Am. Coll. Nutr.29 ( 3 Suppl. ), s289 – s301 ( 2010 ).
- Bouchard C . Gene-environment interactions in the etiology of obesity: defining the fundamentals . Obesity16 ( Suppl. 3 ), S5 – S10 ( 2008 ).
- Yang J , BakshiA , ZhuZet al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index . Nat. Genet.47 ( 10 ), 1114 – 1120 ( 2015 ).
- Llewellyn CH , TrzaskowskiM , PlominR , WardleJ . Finding the missing heritability in pediatric obesity: the contribution of genome-wide complex trait analysis . Int. J. Obes. (Lond.)37 ( 11 ), 1506 – 1509 ( 2013 ).
- Elks CE , Den HoedM , ZhaoJHet al. Variability in the heritability of body mass index: a systematic review and meta-regression . Front. Endocrinol.3 , 29 ( 2012 ).
- Fleisch AF , WrightRO , BaccarelliAA . Environmental epigenetics: a role in endocrine disease?J. Mol. Endocrinol.49 ( 2 ), R61 – R67 ( 2012 ).
- De Carvalho DD , YouJS , JonesPA . DNA methylation and cellular reprogramming . Trends Cell Biol.20 ( 10 ), 609 – 617 ( 2010 ).
- Jones PA . Functions of DNA methylation: islands, start sites, gene bodies and beyond . Nat. Rev. Genet.13 ( 7 ), 484 – 492 ( 2012 ).
- Losko M , KotlinowskiJ , JuraJ . Long noncoding RNAs in metabolic syndrome related disorders . Mediators Inflamm.2016 , 5365209 ( 2016 ).
- Feil R , FragaMF . Epigenetics and the environment: emerging patterns and implications . Nat. Rev. Genet.13 ( 2 ), 97 – 109 ( 2012 ).
- Drummond EM , GibneyER . Epigenetic regulation in obesity . Curr. Opin. Clin. Nutr. Metab. Care16 ( 4 ), 392 – 397 ( 2013 ).
- Wahl S , DrongA , LehneBet al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity . Nature541 ( 7635 ), 81 – 86 ( 2017 ).
- Cordero P , CampionJ , MilagroFIet al. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet . J. Physiol. Biochem.67 ( 3 ), 463 – 470 ( 2011 ).
- Abete I , Gomez-UrizAM , MansegoMLet al. Epigenetic changes in the methylation patterns of KCNQ1 and WT1 after a weight loss intervention program in obese stroke patients . Curr. Neurovasc. Res.12 ( 4 ), 321 – 333 ( 2015 ).
- Amaral CLD , MilagroFI , CuriR , MartínezJA . DNA methylation pattern in overweight women under an energy-restricted diet supplemented with fish oil . BioMed Res. Int.2014 , 675021 ( 2014 ).
- Campión J , MilagroFI , GoyenecheaE , MartínezJA . TNF-α promoter methylation as a predictive biomarker for weight-loss response . Obesity17 ( 6 ), 1293 – 1297 ( 2009 ).
- Crujeiras AB , CampionJ , Diaz-LagaresAet al. Association of weight regain with specific methylation levels in the NPY and POMC promoters in leukocytes of obese men: a translational study . Regul. Pept.186 , 1 – 6 ( 2013 ).
- Mansego ML , MilagroFI , ZuletMA , MartinezJA . SH2B1 CpG-SNP is associated with body weight reduction in obese subjects following a dietary restriction program . Ann. Nutr. Metab.66 ( 1 ), 1 – 9 ( 2014 ).
- Martin-Nunez GM , Cabrera-MuleroR , Rubio-MartinEet al. Methylation levels of the SCD1 gene promoter and LINE-1 repeat region are associated with weight change: an intervention study . Mol. Nutr. Food Res.58 ( 7 ), 1528 – 1536 ( 2014 ).
- Milagro FI , Gómez-AbellánP , CampiónJ , MartínezJA , OrdovásJM , GarauletM . CLOCK, PER2 and BMAL1 DNA methylation: association with obesity and metabolic syndrome characteristics and monounsaturated fat intake . Chronobiol. Int.29 ( 9 ), 1180 – 1194 ( 2012 ).
- Samblas M , MilagroFI , Gómez-AbellánP , MartínezJA , GarauletM . Methylation on the Circadian Gene BMAL1 Is Associated with the Effects of a Weight Loss Intervention on Serum Lipid Levels . J. Biol. Rhythms31 ( 3 ), 308 – 317 ( 2016 ).
- De La Iglesia R , MansegoML , Sanchez-MunizFJ , ZuletMA , MartinezJA . Arylesterase activity is associated with antioxidant intake and paraoxonase-1 (PON1) gene methylation in metabolic syndrome patients following an energy restricted diet . EXCLI J.13 , 416 – 426 ( 2014 ).
- Lopez-Legarrea P , MansegoML , ZuletMA , MartinezJA . SERPINE1, PAI-1 protein coding gene, methylation levels and epigenetic relationships with adiposity changes in obese subjects with metabolic syndrome features under dietary restriction . J. Clin. Biochem. Nutr.53 ( 3 ), 139 – 144 ( 2013 ).
- Milagro FI , CampiónJ , CorderoPet al. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss . FASEB J.25 ( 4 ), 1378 – 1389 ( 2011 ).
- Perez-Cornago A , MansegoML , ZuletMA , MartinezJA . DNA hypermethylation of the serotonin receptor type-2A gene is associated with a worse response to a weight loss intervention in subjects with metabolic syndrome . Nutrients6 ( 6 ), 2387 – 2403 ( 2014 ).
- Kirchner H , NylenC , LaberSet al. Altered promoter methylation of PDK4, IL1 B, IL6, and TNF after Roux-en y gastric bypass . Surg. Obes. Relat. Dis.10 ( 4 ), 671 – 678 ( 2014 ).
- Marchi M , LisiS , CurcioMet al. Human leptin tissue distribution, but not weight loss-dependent change in expression, is associated with methylation of its promoter . Epigenetics6 ( 10 ), 1198 – 1206 ( 2011 ).
- Nicoletti CF , NoninoCB , De OliveiraBaPet al. DNA methylation and hydroxymethylation levels in relation to two weight loss strategies: energy-restricted diet or bariatric surgery . Obes. Surg.26 ( 3 ), 603 – 611 ( 2016 ).
- Bouchard L , Rabasa-LhoretR , FarajMet al. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction . Am. J. Clin. Nutr.91 ( 2 ), 309 – 320 ( 2010 ).
- Moleres A , CampionJ , MilagroFIet al. Differential DNA methylation patterns between high and low responders to a weight loss intervention in overweight or obese adolescents: the EVASYON study . FASEB J.27 ( 6 ), 2504 – 2512 ( 2013 ).
- Ronn T , VolkovP , DavegardhCet al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue . PLoS Genet.9 ( 6 ), e1003572 ( 2013 ).
- Barres R , KirchnerH , RasmussenMet al. Weight Loss after Gastric Bypass Surgery in Human Obesity Remodels Promoter Methylation . Cell Rep.3 ( 4 ), 1020 – 1027 ( 2013 ).
- Benton MC , JohnstoneA , EcclesDet al. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss . Genome Biol.16 , 8 ( 2015 ).
- Dahlman I , SinhaI , GaoHet al. The fat cell epigenetic signature in post-obese women is characterized by global hypomethylation and differential DNA methylation of adipogenesis genes . Int. J. Obes. (Lond.)39 ( 6 ), 910 – 919 ( 2015 ).
- Donkin I , VersteyheS , IngerslevLRet al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans . Cell Metab.23 ( 2 ), 369 – 378 ( 2016 ).
- Multhaup ML , SeldinMM , JaffeAEet al. Mouse-human experimental epigenetic analysis unmasks dietary targets and genetic liability for diabetic phenotypes . Cell Metab.21 ( 1 ), 138 – 149 ( 2015 ).
- Nilsson EK , ErnstB , VoisinSet al. Roux-en Y gastric bypass surgery induces genome-wide promoter-specific changes in DNA methylation in whole blood of obese patients . PLoS ONE10 ( 2 ), e0115186 ( 2015 ).
- Campion J , MilagroFI , GoyenecheaE , MartinezJA . TNF-alpha promoter methylation as a predictive biomarker for weight-loss response . Obesity17 ( 6 ), 1293 – 1297 ( 2009 ).
- Irizarry RA , Ladd-AcostaC , CarvalhoBet al. Comprehensive high-throughput arrays for relative methylation (CHARM) . Genome Res.18 ( 5 ), 780 – 790 ( 2008 ).
- Boyle P , ClementK , GuHet al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling . Genome Biol.13 ( 10 ), R92 ( 2012 ).
- Bell JT , PaiAA , PickrellJKet al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines . Genome Biol.12 ( 1 ), R10 ( 2011 ).
- Gertz J , VarleyKE , ReddyTEet al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation . PLoS Genet.7 ( 8 ), e1002228 ( 2011 ).
- Cooper WN , KhulanB , OwensSet al. DNA methylation profiling at imprinted loci after periconceptional micronutrient supplementation in humans: results of a pilot randomized controlled trial . FASEB J.26 ( 5 ), 1782 – 1790 ( 2012 ).
- Drake AJ , McphersonRC , GodfreyKMet al. An unbalanced maternal diet in pregnancy associates with offspring epigenetic changes in genes controlling glucocorticoid action and foetal growth . Clin. Endocrinol. (Oxf.)77 ( 6 ), 808 – 815 ( 2012 ).
- Heijmans BT , TobiEW , SteinADet al. Persistent epigenetic differences associated with prenatal exposure to famine in humans . Proc. Natl Acad. Sci. USA105 ( 44 ), 17046 – 17049 ( 2008 ).
- Tobi EW , GoemanJJ , MonajemiRet al. DNA methylation signatures link prenatal famine exposure to growth and metabolism . Nat. Commun.5 , 5592 ( 2014 ).
- Heijmans BT , TobiEW , SteinADet al. Persistent epigenetic differences associated with prenatal exposure to famine in humans . Proc. Natl Acad. Sci. USA105 ( 44 ), 17046 – 17049 ( 2008 ).
- Eckhardt F , LewinJ , CorteseRet al. DNA methylation profiling of human chromosomes 6, 20 and 22 . Nat. Genet.38 ( 12 ), 1378 – 1385 ( 2006 ).
- Moverare-Skrtic S , MellstromD , VandenputL , EhrichM , OhlssonC . Peripheral blood leukocyte distribution and body mass index are associated with the methylation pattern of the androgen receptor promoter . Endocrine35 ( 2 ), 204 – 210 ( 2009 ).
- Bjornsson HT , SigurdssonMI , FallinMDet al. Intra-individual change over time in DNA methylation with familial clustering . JAMA299 ( 24 ), 2877 – 2883 ( 2008 ).
- Heijmans BT , MillJ . Commentary: the seven plagues of epigenetic epidemiology . Int. J. Epidemiol.41 ( 1 ), 74 – 78 ( 2012 ).
- Liu Y , AryeeMJ , PadyukovLet al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis . Nat. Biotechnol.31 ( 2 ), 142 – 147 ( 2013 ).
- Koestler DC , ChristensenB , KaragasMRet al. Blood-based profiles of DNA methylation predict the underlying distribution of cell types: a validation analysis . Epigenetics8 ( 8 ), 816 – 826 ( 2013 ).
- Demerath EW , GuanW , GroveMLet al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci . Hum. Mol. Genet.24 ( 15 ), 4464 – 4479 ( 2015 ).
- Bouchard L , Rabasa-LhoretR , FarajMet al. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction . Am. J. Clin. Nutr.91 ( 2 ), 309 – 320 ( 2010 ).
- Feinberg AP , IrizarryRA , FradinDet al. Personalized epigenomic signatures that are stable over time and covary with body mass index . Sci. Transl. Med.2 ( 49 ), 49ra67 ( 2010 ).
- Birney E , SmithGD , GreallyJM . Epigenome-wide association studies and the interpretation of disease -omics . PLoS Genet.12 ( 6 ), e1006105 ( 2016 ).
- Aron-Wisnewsky J , JuliaZ , PoitouCet al. Effect of bariatric surgery-induced weight loss on SR-BI-, ABCG1-, and ABCA1-mediated cellular cholesterol efflux in obese women . J. Clin. Endocrinol. Metab.96 ( 4 ), 1151 – 1159 ( 2011 ).
- Johansson LE , DanielssonAP , ParikhHet al. Differential gene expression in adipose tissue from obese human subjects during weight loss and weight maintenance . Am. J. Clin. Nutr.96 ( 1 ), 196 – 207 ( 2012 ).