
Abstract
Asthma and chronic obstructive pulmonary disease are inflammatory airway diseases for which alternative therapeutic strategies are urgently needed. Interestingly, HDAC inhibitors show anti-inflammatory effects in mouse models for these diseases. Here we explore underlying mechanisms that may explain these effects. In previous studies, effects of HDAC inhibitors on histone acetylation are often correlated with their effects on gene expression. However, effects of HDAC inhibitors on transcription factors and their acetylation status may be particularly important in explaining these effects. These effects are also cell type-specific. Recent developments (including chemoproteomics and acetylomics) allow for a more detailed understanding of the selectivity of HDAC inhibitors, which will drive their further development into applications in inflammatory airway diseases.
First draft submitted: 23 February 2017; Accepted for publication: 12 April 2017; Published online: 15 June 2017
Asthma and chronic obstructive pulmonary disease (COPD) are common inflammatory airway diseases. These diseases affect millions of people world-wide, and globally, the incidence is increasing. For asthma, the clinical symptoms include among others wheezing and shortness of breath. COPD is characterized by shortness of breath upon exercise and a largely irreversible and progressive airflow limitation [Citation1–4]. In asthma the larger airways are mainly affected, whereas in COPD, the lung parenchyma and peripheral airways are mainly affected [Citation3]. The immunopathology of asthma and COPD is characterized by different types of inflammatory cells that are at play. For example, asthma is driven by T helper 2 (Th2) cells, dendritic cells and is characterized by eosinophilic inflammation. In asthma there is mast-cell sensitization by immunoglobulin E (IgE), and multiple bronchoconstrictors are released [Citation1,Citation2]. On the other hand, COPD is characterized by T helper 1 (Th1) cells, cytotoxic T cells and neutrophilic inflammation [Citation5,Citation3]. The inflammation in COPD is also characterized by increased numbers of macrophages, neutrophils and innate lymphoid cells recruited from the circulation. A variety of pro-inflammatory mediators, including cytokines, chemokines, growth factors and lipid mediators are secreted by these cell types [Citation5]. Currently, glucocorticoids are the cornerstone in the treatment of asthma and COPD, however, this is not effective in all patients. Severe asthmatics display glucocorticoid resistance [Citation6], and the effectivity of glucocorticoids in COPD patients is subject to debate [Citation7,Citation8]. Alternative therapeutic strategies are needed for these patients, which currently form a major health burden for society due to high socioeconomic costs [Citation1–4,Citation9]. Studying the underlying molecular mechanisms may enable the identification of new therapeutic targets.
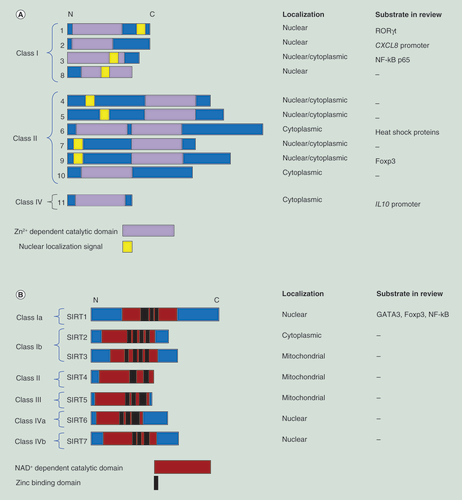
These underlying processes may include epigenetic regulation, such as lysine acetylation. Lysine acetylation is a post-translational modification of proteins, which is crucial in the regulation of cellular processes such as signal transduction and gene expression [Citation10,Citation11]. Lysine acetylation was initially discovered as a modification of histone proteins [Citation12], where it plays a role in facilitating an open chromatin structure and gene transcription [Citation13]. Lysine acetylation is regulated by histone acetyltransferases (HATs, also known as lysine acetyltransferases or KATs) as writers and histone deacetylases (HDACs, also known as lysine deacetylases or KDACs) as erasers. Dysregulation in the balance between the activities of HDACs and HATs can result in disease, such as inflammatory diseases and cancer [Citation14–17]. Mammalian HDACs are classified into four main groups based on their homology with yeast orthologs [Citation18]. Class I HDACs, including HDAC1, 2, 3 and 8 are predominantly found within the nucleus, due to the presence of a nuclear localization sequence and the absence of a nuclear export signal sequence within HDAC1, 2 and 8. However, HDAC3 has both a nuclear import and export signal, allowing for localization in both the cytoplasm and the nucleus [Citation18,Citation19]. Class I HDACs have a ubiquitous tissue distribution [Citation18]. Class II HDACs are subdivided into two groups, IIA (HDAC4, 5, 7, 9) and IIB (HDAC6 and 10), and are predominantly found in the cytoplasm [Citation20]. Class II HDACs are able to shuttle between the cytoplasm and the nucleus, and have a more tissue-specific distribution than class I HDACs [Citation18]. Class IV consists only of HDAC11, which shares similarities with both Class I and II HDACs. Class I, II and IV HDACs are zinc dependent. Class III HDACs are also called sirtuins (SIRT1–SIRT7) and are found in the nucleus, cytoplasm or mitochondria. Sirtuins act via different mechanisms and require the cofactor NAD+ for their activity [Citation21]. HDAC enzymes act on nonhistone proteins as well, and can exist in multiprotein complexes that influence their activity [Citation22,Citation23]. A summary of the HDAC classification is given in .
(A) Zn2+-dependent HDACs. (B) NAD+-dependent HDACs.
HDAC: Histone deacetylase.
HDAC inhibitors (HDACi) can be grouped into different classes based on their chemical structure such as hydroxamic acids, aminoanilides, cyclic peptides and short-chain fatty acids. From these, the first two classes have been studied most intensively [Citation17]. Other classes include ketones, thiols, mercaptoacetamides and other types of inhibitors [Citation24,Citation25]. The distinct features of these classes have been discussed in other reviews [Citation26,Citation27]. Three HDACi that are nonselective among the Zn2+-dependent HDACs of the hydroxamic acid type, vorinostat, belinostat and panobinostat, obtained US FDA approval for treatment of hematological cancers [Citation28]. This is also true for the cyclic peptide romidepsin [Citation28]. After their introduction in cancer therapy, HDACi are gaining attention for application in other diseases as well. Their potential applications range from neurodegenerative (Alzheimer and Huntington disease) to inflammatory diseases such as asthma, rheumatoid arthritis and also viral infections [Citation29–32]. Interestingly, anti-inflammatory effects of HDACi have been reported in a number of studies using mouse models of asthma, mostly using the nonselective HDACi trichostatin A (TSA), which inhibits class I and II HDACs [Citation33]. TSA reduced T-cell infiltration and expression of the Th2 cytokines IL-4 and IL-5, and IgE in an ovalbumin sensitization and challenge mouse asthma model [Citation34]. In another study, the effects of TSA were also evaluated in an ovalbumin model. TSA-treated mice had a reduced number of total inflammatory cells and eosinophils in the bronchoalveolar lavage (BAL) fluid compared with vehicle-treated mice. Furthermore, airway remodeling changes were significantly reduced with TSA compared with vehicle-treated mice, with fewer goblet cells, less subepithelial collagen deposition and attenuated airway hyperresponsiveness induced by methacholine [Citation35]. Another study also showed that TSA inhibited methacholine-induced airway hyperresponsiveness in mice sensitized and challenged with Aspergillus fumigatus antigen. However, this was not related to anti-inflammatory effects of TSA, since no effects were observed on leukocyte trafficking or concentrations of cytokines in BAL fluid in antigen-challenged mice. Instead, using human precision-cut lung slices and airway smooth muscle cells, an inhibiting effect on smooth muscle contraction was observed upon TSA treatment [Citation36]. In a recent study using an Alternaria mouse allergic model, TSA downregulated the number of innate lymphoid group 2 cells (ILC2; an important source of the cytokines IL-5 and IL-13 which are critical to allergic airway inflammation) expressing IL-5, IL-13 and IL-33 upon Alternaria extract challenge, and reduced lung eosinophilia and mucus hypersecretion. TSA treatment also decreased Alternaria extract-induced pro-inflammatory cytokines and chemokines including KC (murine IL-8), TNF-α and granulocyte-macrophage colonystimulating factor [Citation37]. Another recent study using an ovalbumin mouse model demonstrated that TSA decreased IL-17 level in the BAL fluid [Citation38]. On the other hand, it was shown that TSA increased pro-inflammatory cytokine levels in the culture supernatant of memory T cells which were isolated from ovalbumin asthmatic mice; and when these memory T cells (treated with TSA) were adoptively transferred into naive mice, increased pro-inflammatory cytokine levels were also found in the BAL fluid [Citation39]. Another study did not use TSA, but other HDACi including tubastatin A (an HDAC6i), PCI-34051 (an HDAC8i) and the non-selective HDACi givinostat. These were tested in a mouse ovalbumin asthma model and all reduced inflammatory cell counts in the BAL fluid of these mice [Citation40]. Taken together, HDACi generally reduce inflammation in mouse asthma models, and different mechanisms have been reported for this.
Counterintuitive to these findings, asthma is characterized by increased HAT activity [Citation41]. For instance, it was demonstrated that there is increased HAT activity, and decreased HDAC activity and HDAC1 and HDAC2 expression, in bronchial biopsies and BAL macrophages obtained from asthmatics compared with healthy adults [Citation42,Citation43], thereby shifting the balance toward HAT activity. Another study showed that HAT activity is increased in peripheral blood mononuclear cells (PBMCs) obtained from mild and severe asthmatic children compared with healthy children, whereas HDAC activity is decreased [Citation44]. However, another study did not find decreased expression of HDAC1 and HDAC2 mRNA or protein in endobronchial biopsies in a large set of severe asthmatics compared with healthy controls [Citation45]. Another recent study found that in neutrophilic asthma, there was increased HAT and decreased HDAC activity in isolated PBMCs. However, there were no differences in the expression levels of the HATs p300, KAT2B, cAMP-response-element-binding-protein-binding-protein or the HDACs HDAC1, HDAC2 or HDAC3, which could indicate differences in activity due to post-translational effects [Citation46]. More recently, it has been identified that acetylation of histone H3 lysine 18 (H3K18) is elevated in asthma epithelium compared with healthy subjects [Citation47], which is in line with a shift toward increased HAT activity.
COPD is characterized by a loss of SIRT1 [Citation48], and HDAC2 expression and activity [Citation41,Citation49]. In more recent studies, decreased HDAC2 expression was also found in PBMCs and lymphocytes in COPD [Citation50,Citation51]. In COPD, the decreased HDAC2 expression and activity can be linked to glucocorticoid resistance [Citation52]. This is because glucocorticoids are dependent on HDAC2 for their immunosuppressive activity. Glucocorticoids diffuse through the cellular membrane and bind the glucocorticoid receptor (GR) which is present in the cytoplasm. The GR is normally bound by heat shock proteins, but upon binding of glucocorticoids, is released and translocated into the nucleus where it binds to target genes with glucocorticoid response elements. This includes multiple inflammatory genes. GR then recruits HDAC2, which leads to transcriptional repression [Citation52]. It is interesting to note that also in severe asthmatics, there are indications that decreased HDAC2 expression and activity can be linked to steroid resistance [Citation53], and that passive smoking reduced HDAC2 expression in severe asthmatic children [Citation54].
Altogether, even though HDACi have been reported to reduce inflammation in asthma and COPD models, it should be noted that not much is known about the expression and activity of individual HDAC isoenzymes in asthma or COPD. The roles of HDACs in asthma and COPD need to be more clearly defined. Here, we will explore HDACs, cell types, transcription factors and mechanisms that have been implicated, in order to shed more light on mechanisms by which HDACi could reduce inflammatory responses in asthma and COPD. Next to this, we also review important methodological developments that allow for a better characterization of HDACi targets and selectivity, which will aid in further development of HDACi as therapeutic agents for inflammatory lung diseases such as asthma and COPD. The topics addressed in the review are summarized in .
Cell type-specific effects
For several cell types, which have been implicated in the inflammatory component of asthma and COPD, it is known that these cell types are characterized by transcription factors that are crucial to their differentiation and functionality. These transcription factors are subject to regulation by acetylation, which determines for example the transcription factor stability and DNA binding capability. Specific HDACs have been implicated in these processes, which can thereby control inflammatory responses. Inhibitors for these enzymes can affect these biological processes and thereby modulate the outcome of inflammatory responses. This will be discussed in detail below and is schematically represented in A, and summarized in . While effects of HDACi on (promoter specific) histone acetylation have also been described (e.g., in T cells IL-1β induces histone acetylation at the TGF-β-1 promoter, which is further increased by TSA [Citation55]), effects on histone acetylation are not the focus of this review.
(A) Macrophages and various T lymphocytes have been implicated in inflammatory airway diseases such as asthma and chronic obstructive pulmonary disease. These cell types depend on specific transcription factors for their differentiation and functionality. Histone deacetylases (HDACs) can affect the acetylation of these transcription factors, which influences, for example, the transcription factor stability or its DNA binding capability. HDAC inhibitors (HDACi) can influence these biological processes and thereby influence inflammatory responses by these cell types. (B) Effects of HDACi on histone acetylation are often used as a read-out indicative of the effects of these compounds in cells. However, histone acetylation is not sufficient to (fully) explain the effects of HDACi on biological processes such as gene expression. Effects of HDACi on transcription factors and their acetylation status may also be particularly important in explaining these effects. (C) Interestingly, selectivity profiles of HDACi are commonly elucidated by testing for inhibition of recombinant HDACs. Importantly, however, in cells HDACs are known to be present in multiprotein complexes that influence their activity. Importantly, recently, HDACi have been reported to have different specificities for such complexes, which adds another layer of complexity to the selectivity profile. (D) Different classes of HDACi have been reported to have different binding kinetics with the HDACs. Benzamine-based inhibitors have slow binding kinetics, while hydroxamate-based inhibitors have fast binding kinetics. This has been demonstrated to translate into biological consequences.
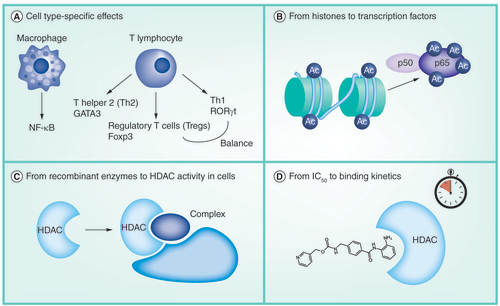
Table 1. The effects of histone deacetylase inhibitors on transcription factors and cell types implicated in lung inflammation models.
Macrophages: selective regulation of the NF-κB transcription factor acetylation status by HDAC3 inhibitors?
Macrophages play a central role in orchestrating inflammatory responses in asthma and COPD by expressing and secreting pro-inflammatory cytokines and chemokines, which then attract other inflammatory cell types [Citation56]. The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription factor plays an important role in this. Upregulated NF-κB activity has been reported in asthma [Citation57] and COPD [Citation51,Citation58]. NF-κB is subject to regulation by lysine acetylations [Citation59]. The p65 NF-κB subunit in particular is subject to dynamic lysine acetylations, which regulate its activity. For example, acetylation of p65 on lysine (K)310 is required for full transcriptional activity. Next to this, acetylations of p65 on K122 and K123 have been reported to diminish its activity due to increased binding to the IκB inhibitor [Citation60–62]. The main responsible enzyme for NF-κB acetylation is p300; and for deacetylation the enzymes HDAC3 and SIRT1 have been implicated [Citation60–62]. Interestingly, HDACi can regulate the NF-κB transcription factor. Nonselective HDACi have ambiguous effects on the NF-κB pathway [Citation15], and can either activate or diminish its activity, which results in either increased (as for example reported in [Citation63,Citation64]) or reduced (as for example reported in [Citation65]) pro-inflammatory gene expression in different model systems including mouse OP9 (bone marrow derived stromal) cells ([Citation63]), human leukemia cells [Citation64] or human non-small-cell lung cancer cells [Citation65].
Also in macrophages, ambiguous effects have been described on NF-κB activity and pro-inflammatory gene expression for the more selective HDAC1–3 inhibitor MS-275. It was previously shown that this compound increases NF-κB p65 nuclear translocation, DNA binding, acetylation and activity in U937 human monocytes [Citation66]. However, MS-275 was shown to upregulate IL-10 protein levels, which is important in dampening inflammation, in RAW264.7 macrophages [Citation67], and to downregulate IL-1β. MS-275 also reduced nitric oxide concentration in the media and decreased mRNA levels of iNOS and TNF-α, showing anti-inflammatory effects [Citation67,Citation68]. MS-275 reduced LPS-induced IL-1β, IL-6, IL-18 and TNF-α secretion in THP-1 human monocytic cells; and nitric oxide secretion in RAW264.7 cells [Citation69]. On the other hand, in bone marrow derived macrophages MS-275 showed mainly pro-inflammatory effects on gene expression [Citation70]. Thus, MS-275 gives rise to either pro- or anti-inflammatory effects in these cells. In line with this, we demonstrated that MS-275 gives rise to mixed effects on pro- and anti-inflammatory gene expression in RAW264.7 macrophages with upregulation of both pro- and anti-inflammatory genes [Citation71]. Importantly, MS-275 increased anti-inflammatory IL10 expression, and we identified NF-κB as a regulator. MS-275 increased NF-κB promoter activity, acetylation, nuclear translocation and NF-κB p65 binding to the IL10 promoter in RAW264.7 macrophages [Citation71]. This mechanism is in line with literature that describes that LPS stimulation of macrophages activates NF-κB, which induces the expression of IL10 along with TNFα, IL1β and IL12b [Citation72], which is further enhanced by MS-275 in our study. Upon testing MS-275 in a mouse COPD (cigarette smoke exposure) model, anti-inflammatory effects were observed on several parameters including increased anti-inflammatory IL10 expression in lung macrophages, reduced KC (murine IL-8) expression and reduced neutrophilic influx upon MS-275 treatment, which is the first time that anti-inflammatory effects of an HDACi are being reported in a COPD mouse model [Citation71].
Compared with MS-275, more selective inhibitors may have more selective effects on NF-κB-mediated inflammatory responses in macrophages. To illustrate this, the isoform selectivity profiles on class I HDACs of the HDAC3 selective inhibitor RGFP966 and MS-275 are compared in . RGFP966 is an interesting candidate, based on the important role of HDAC3 in deacetylation of specific NF-κB p65 lysines (including K122 and K123) [Citation73], and our previous results [Citation74]. RGFP966 increased anti-inflammatory IL-10 expression, and reduced pro-inflammatory gene expression in murine precision-cut lung slices. In RAW264.7 murine macrophages, pro-inflammatory gene expression was decreased, which correlated with reduced NF-κB activity [Citation74]. An interesting hypothesis is that this could be attributed to the capability of more selectively regulating the acetylation status of individual NF-κB lysines such as K122 and K123 by selective HDAC3 inhibitors such as RGFP966.
Table 2. IC50 values of histone deacetylase 1–3-selective inhibitor MS-275 and histone deacetylase 3-selective inhibitor RGFP966 for recombinant class I human histone deacetylases.
Next to this, it is worthwhile to mention that the HDACs themselves have also been implicated in anti-inflammatory IL-10 expression by macrophages [Citation5,Citation56]. In RAW264.7 macrophages, human THP1 monocytic cells and primary human dendritic cells it was shown that HDAC11 siRNA upregulates IL10 expression; whereas overexpression of HDAC11 has the opposite effect. This translated to effects on the antigen-specific activation of T cells [Citation75]. In another study using RAW264.7 macrophages and THP1 monocytic cells, it was found that HDAC11 is a transcriptional repressor of IL10, whereas HDAC6 is required for IL10 gene transcription in these cells [Citation76]. Intriguingly, IL-10 itself can inhibit LPS-induced CXCL8 expression in monocytes, via a mechanism that involves HDAC2. IL-10 increased the association of HDAC2 with the CXCL8 promoter region, which resulted in reduced CXCL8 expression [Citation77]. This illustrates that individual HDACs have specific roles in the control of anti-inflammatory IL10 expression (with HDAC11 as an interesting possible target due to its inhibitory role in IL10 expression), and in the inhibition of CXCL8 expression by IL-10 in these cells.
Furthermore, there is impaired phagocytosis of bacteria and virus-infected cells by macrophages in asthma and COPD, which can cause adverse effects in patients [Citation78]. It was shown that depending on the treatment regime, HDACi can improve bacterial clearance of macrophages. The HDAC6-selective inhibitor tubastatin A promoted bacterial clearance from human macrophages, whereas the class I HDACi MS-275, which inhibits HDAC1–3, had no effect on intracellular bacterial loads. Although the mechanisms remain unclear, this suggests a role for HDAC6 in bacterial clearance by macrophages [Citation79].
In summary, macrophages are an important cell type in the inflammatory responses in asthma and COPD. The NF-κB transcription factor plays an important role in the pro-inflammatory gene expression in macrophages, and is subject to regulation by acetylation. Nonselective HDACi have ambiguous effects on the NF-κB transcription factor, which translates into either pro- or anti-inflammatory outcomes. This is also the case for the HDAC1–3 inhibitor MS-275 in macrophages. More selective inhibitors, such as the HDAC3 inhibitor RGFP966, however, may have more selective effects on gene expression due to more selective regulation of the acetylation status of the NF-κB transcription factor. This inhibitor causes downregulation of pro-inflammatory gene expression, but upregulates IL-10 expression. Next to this, HDACs also play an important role in anti-inflammatory IL-10 expression in macrophages. An example in this respect is the inhibitory role of HDAC11 in IL-10 expression. Furthermore, HDACi can also improve bacterial clearance in macrophages, which is relevant because bacterial clearance is impaired in asthma and COPD patients. Finally, the studies described above also demonstrate that the selectivity profile of an HDACi matters for the outcome on inflammatory responses in macrophages.
Th2 cells: regulation of the GATA3 transcription factor acetylation status by an SIRT1 inhibitor or activator
Th2 cells are considered to be important drivers of asthma due to production of cytokines such as IL-4 (which regulates the allergen-specific synthesis of IgE), and IL-5 (which recruits eosinophils) among others [Citation2]. Roles for HDACs have been described in Th2 cell mediated inflammatory responses in asthma. One study showed that conditional knockout of HDAC1 in T cells in a mouse ovalbumin asthma model resulted in increased eosinophil recruitment into the lung, mucus hypersecretion, parenchymal lung inflammation and enhanced airway resistance. This correlated with enhanced expression of Th2-related cytokines in T cells isolated from these mice [Citation80]. This indicates an inhibiting role for HDAC1 in Th2-related cytokine expression. Next to this, for the differentiation of T lymphocytes into Th2 cells, GATA3 is an essential transcription factor [Citation81], which is regulated by acetylation [Citation82]. GATA3 has been described to be acetylated by the HAT p300 on K305, which increases its activity [Citation82]. For deacetylation of GATA3, SIRT1 has been implicated in another study where the SIRT1 inhibitor sirtinol increased Th2-related expression in T cells from cord blood [Citation83]. This correlated with enhanced acetylation and activity of the GATA3 transcription factor in another T-cell line (HUT78 T lymphocytes) upon sirtinol treatment (although it was not studied on which lysine) [Citation83]. Importantly, this study also showed that SIRT1 is decreased in activity and expression at the protein level in whole cell extracts from PBMCs isolated from severe asthmatic patients. No decrease was observed for the other sirtuin members (SIRT2, SIRT3, SIRT6 and SIRT7). Sirtinol was tested in these PBMCs and increased Th2 cytokine expression. Furthermore, an SIRT1 activator (SRT2172) significantly reduced IL-4 mRNA expression in PBMCs from ten patients with severe asthma [Citation83]. Taken together SIRT1 plays a role in inhibiting Th2 differentiation in T cells, by regulating the acetylation status of GATA3. The decreased SIRT1 expression in severe asthmatics could contribute to sustained Th2 cytokine expression such as IL-4, raising interest for SIRT1 as a target for the treatment of severe asthma.
Tregs: regulation of the Foxp3 transcription factor acetylation status by HDACi
Tregs can dampen inflammation through diminishing the function of other T cells, which is important in controlling immune homeostasis [Citation84]. A number of studies have suggested that Tregs are impaired in activity in asthma [Citation85,Citation86]. In COPD, there are indications that the number of Tregs is reduced [Citation1]. However, the role of Tregs in asthma and COPD has to be better defined. A study by Baru et al. demonstrated that the timing of the effects of Tregs is important to the outcome of inflammatory responses in an asthma model. The group demonstrated that Treg depletion during the allergen challenge phase did not change the airway inflammatory response, but there was an increased inflammatory response upon Treg depletion during the sensitization phase [Citation87]. Foxp3 is a transcription factor, which is crucial in the commitment of T cells in differentiating into Tregs. Reduced Foxp3 expression was observed in PBMCs from elderly asthmatics [Citation88]. There is an important role for acetylation and deacetylation of Foxp3 in the regulation of this transcription factor [Citation89,Citation90] including on K31, K262 and K267 [Citation91]. It has been identified that p300 and Tip60 can acetylate Foxp3 (intriguingly, p300 and Tip60 work together to acetylate one another and Foxp3) [Citation92]. Acetylation of Foxp3 increases its resistance to ubiquitination and proteasomal degradation, and increases DNA binding [Citation89]. Foxp3 is deacetylated by SIRT1 and HDAC9 which has the opposite effect, and therefore impairs Treg differentiation. Next to this, HDAC6 also plays an important indirect role, through the deacetylation of heat shock proteins, which can then decrease the stability of Foxp3 [Citation93]. More recently, it has been discovered that HDAC3 also plays a promoting role in the differentiation of Tregs by suppressing IFN-γ (ifng) and IL-12 production (which both need to be suppressed for proper differentiation) [Citation94]. Clearly HDACs play an important role in the differentiation and functionality of Tregs. Several reports have described that HDACi can affect the differentiation and regulatory capacity of Tregs, which has mostly been described for nonselective HDACi [Citation95]. More selective inhibitors have also been studied. One study used the HDAC6i ACY-738 and the SIRT1 inhibitor EX-527 in mice, and found these compounds to increase Treg-suppressive function (in combination and alone) [Citation96]. There has also been interest in the development of novel HDAC6i for augmentation of Treg-suppressive function [Citation97]. Furthermore, Sirt1 inhibitor EX-527 was used in a study where acetylation sites were identified on Foxp3 (K31, K262 and K267), and increased acetylation of these lysines as well as the stability and levels of the transcription factor [Citation91]. HDAC1–3 inhibitor MS-275 increased Foxp3 expression in T cells isolated from human blood [Citation98]. In other studies focusing on cancer models, however, downregulated Foxp3 expression by Tregs has also been reported upon MS-275 treatment [Citation99,Citation100]. These discrepancies can likely be explained by the different model systems, concentrations and methods of administration of MS-275 used. It could be that inhibition of HDAC6 or SIRT1 as opposed to inhibition of class I HDACs 1–3 gives rise to less ambiguous outcomes, and can augment Treg function and differentiation. This opens up an interesting novel line of research to define the effects of selective HDACi on Treg function in the diseases such as asthma and COPD.
Can HDACi alter the balance between Tregs & Th17 cells?
A more recently discovered subset of T cells are the Th17 cells, which are believed to play a role in asthma and COPD pathogenesis [Citation101,Citation102], although the role needs to be more clearly defined. Th17 cells are an important source of the pro-inflammatory cytokine IL-17, and IL-17 levels were found to be increased in sputum of asthmatic patients [Citation101–103]. The role of Th17 cells and IL-17 in COPD is much less clear [Citation101]. The RORγt transcription factor is essential for the differentiation of the Th17 cell type [Citation104], and has been reported to be regulated by acetylation in a recent study. It was demonstrated that p300 acetylates RORγt at K81 (increasing its activity), whereas HDAC1 reduces acetylation of RORγt (which decreases its activity, although it was not studied which lysine was involved) in HEK293T cells and isolated naive T cells from healthy human subjects [Citation105]. In line with this, MS-275 has been reported to enhance Th17 differentiation in mouse splenocytes [Citation106]. On the other hand, the HDACi givinostat downregulated the IL-6 receptor on T cells which reduced their ability to differentiate into Th17 cells, but instead skewed differentiation toward the Treg phenotype [Citation107]. Interestingly, in line with this, in a study using an ovalbumin challenge asthma mouse model, on the mRNA level TSA increased Foxp3 and decreased RoRγt levels. There were also decreased numbers of inflammatory cells in BAL fluid, and decreased IL-4, upon TSA treatment, as well as decreased IgE in the serum. In the same study, in Jurkat T cells, upon TSA treatment, the Foxp3 mRNA level was upregulated, while the RORγt mRNA level was downregulated. The Foxp3 protein level was also upregulated by TSA, on the whole suggesting that TSA can alter the balance of Th17/Treg cells in this study [Citation38]. Altogether, the hypothesis that HDACi could shift the balance from Th17 to Treg cells is interesting since this might have advantageous effects in the context of asthma and COPD, and calls for further research along this line. However, future investigations will need to further elucidate the specific HDACs involved, and demonstrate whether targeting the balance of Th17/Treg cells using HDACi could ultimately be a viable therapeutic strategy.
From histones to transcription factors
It is interesting to note that the effects of HDACi on histone acetylation are oftentimes used as a read-out indicating the effects of these inhibitors in cells. However, the studies discussed above indicate that effects of HDACi on transcription factors and their acetylation status are also important to the outcome in disease models, including those for asthma and COPD. Several studies also indicate that effects on histone acetylation do not always correlate with effects on biological processes such as gene expression. For example, upon HDACi treatment, some genes have been reported to be upregulated, while others go down (roughly as many go up as down), whereas global histone acetylation is generally pronouncedly increased [Citation108]. Interestingly, the balance between effects on histone acetylation and gene expression also changes depending on the employed incubation time [Citation109]. In HeLa cells (ATCC® CCL-2™) it was demonstrated that valproic acid and suberanilohydroxamic acid (SAHA) have an early onset effect on gene expression after 12 h of incubation. Upon 48 h of incubation, these changes in gene expression had returned to baseline levels, while increases in histone acetylation had not [Citation109]. Another study found that upon HDACi treatment there was a precisely timed increase in histone H3 lysine 27 trimethylation (H3K27me3) at transcription start sites, but little or no increase in histone acetylation, whose role seemed to be to provide a stable chromatin environment that allows transcription to be modified by other factors [Citation110]. In another study, the nonselective HDACi romidepsin and SAHA were tested in different cancer cell lines, and their effects on histone acetylation were investigated together with their effects on apoptosis [Citation111]. Treatment with romidepsin or SAHA for 6 h caused similar increases in histone acetylation in all cell lines, but not all of the cell lines underwent apoptosis upon romidepsin or SAHA treatment. Therefore, the effects on histone acetylation did not correlate with effects on apoptosis in all cell lines [Citation111]. In summary, recent research indicates that HDACi-induced changes in histone acetylation cannot (fully) explain their effects on gene expression and apoptosis. Effects of HDACi on the acetylation status of specific transcription factors may therefore be particularly important in explaining their effects in disease models, such as those for asthma and COPD (a schematic representation is given in B).
From recombinant enzymes to HDAC activity in cells
Isoenzyme selectivity of HDACi has important consequences for their effects. Hence, development of highly potent molecules selectively targeting specific HDACs or a select group of HDACs is important. This is likely important in obtaining selective effects on specific pathways such as the NF-κB pathway in the context of asthma and COPD models, which occurs through fine-regulation of its acetylation on specific lysines [Citation73]. A selectivity profile for HDACi is generally obtained by testing the inhibition on recombinant HDAC enzymes [Citation33]. However, it has become apparent that several other factors need to be taken into account. First, it has been demonstrated that binding characteristics of HDACi in cells differ from the profile on recombinant enzymes due to the fact that HDACs exist in multiprotein complexes, adding another level of complexity. For example, one study used a chemoproteomics strategy, where an affinity capture method was employed using Sepharose beads derivatized with HDACi hydroxamic acid analogs [Citation112]. This allowed for a competition binding assay, where extracts of cells treated with increasing HDACi concentration competed with the immobilized HDACi analog probe matrix for binding protein targets. The reduction in protein capture resulting from the competition with the increasing concentration of the ‘free inhibitor’ was quantified by mass spectrometry using isobaric tandem mass tags [Citation112]. Target protein complexes interacting with HDACi could be identified, and importantly, inhibitor selectivity for native drug target complexes deviated from literature values obtained using recombinant enzymes, revealing an unexpected degree of selectivity of certain HDACi. For example, aminoanilide-based inhibitors displayed a preference for the HDAC3–NCoR complex [Citation112]. As a side-note, interestingly, chemoproteomic approaches also enable the identification of possible off-targets of HDACi [Citation112]. Another option to investigate HDACi specificity is to analyze HDACi-induced changes at the level of all lysine acetylation sites (acetylome). A study from Scholz et al. elucidated the selectivity profile of HDACi at the level of the global acetylome [Citation113]. HeLa cells were treated with a panel of widely used HDACi and changes at the acetylome were monitored by quantitative bottom-up proteomics. Stable isotope labeling with amino acids in cell culture (SILAC) was combined with enrichment of acetylated peptides using anti-acetyllysine antibodies and analysis by mass spectrometry. The fraction of upregulated acetylation sites in HDACi-treated cells was greater than the fraction of downregulated sites [Citation113]. For several HDACi, the number of acetylation sites affected was not proportional to the number of HDACs they were found to inhibit when testing on recombinant HDACs, which is in line with chemoproteomics studies demonstrating unexpected selectivities of HDACi for multiprotein complexes (as for example described in [Citation112]). In principle, differential proteomics approaches as described by Scholz et al [Citation113] enable to elucidate HDACi selectivity profiles at the level of the nuclear, cytosolic or mitochondrial acetylome. We envision that a better characterization of HDACi targets and specificities is necessary for the further development of HDACi as potential therapeutics for diseases, such as asthma and COPD. Considering the described developments, this can be done by generating a selectivity profile on HDACs in cells, which can be complemented with acetylome analysis (a schematic representation is given in C).
From IC50 to binding kinetics
Next to these studies into selectivity profiles, there has been a study into the binding kinetics of HDACi with recombinant HDACs [Citation114]. A reporter displacement binding assay was used to quantify the association (kon) and dissociation (koff) kinetic rate constants as well as the binding constants (Kd) for a set of hydroxamic acid-based and aminoanilide-based inhibitors against HDAC1 and HDAC2. While Kd values were similar, the kon and koff were slow for the aminoanilides. Therefore, aminoanilides displayed slow, whereas hydroxamic acids displayed fast binding kinetics [Citation114]. The effects upon washout of the inhibitors were then tested in the neuroblastoma cell line SH-SY5Y; after a ‘pulse’ treatment with the inhibitors for 6 h and replacement of the medium with a drug-free medium. In line with slow binding kinetics for aminoanilides, a sustained state of histone hyperacetylation was observed for the aminoanilide-based inhibitor MS-275 after washout (which was still observed after 96 h), while upon treatment with the hydroxamic acid-based inhibitors, acetylation returned to baseline levels much faster (within 18 h) [Citation114]. Becher et al. reported similar findings using a chemoproteomic approach which allowed for a comparison of time-dependent binding of hydroxamic acid-based and aminoanilide-based HDACi [Citation115]. Also in line with this, another study using MS-275 found this compound to have a long lasting effect on histone acetylation of histone H4 lysine 12 (H4 K12) after washout [Citation116]. These studies indicate that the binding kinetics of HDACi have biological consequences, which is a point that needs to be taken into account next to the selectivity profile of HDACi, since it is important in evaluating their therapeutic utility (a schematic representation is given in D).
Conclusion & future perspective
Currently, HDACi are being used in the treatment of cancer. However, these compounds have anti-inflammatory properties at lower concentrations than those used for cancer treatment [Citation117]. Hence, HDACi have gained interest as therapeutic agents in other areas, including inflammatory airway diseases such as asthma and COPD. Studies indicate that HDACi can reduce inflammation in mouse asthma and COPD models. Counterintuitive to these observations, both diseases are characterized by increased HAT and decreased HDAC activity. However, little is known about the expression and activity of individual HDACs in asthma and COPD. Next to this, research should focus on further elucidating the role of protein acetylation and deacetylation in driving the initiation and progression of these diseases. It is therefore a field of study that urgently needs attention.
Interestingly, HDACi have been demonstrated to be able to affect the differentiation and functionality of immune cell types that have been implicated in asthma and COPD. These include macrophages and various T lymphocytes. A common phenomenon is that HDACs play a role in the regulation of transcription factors such as NF-κB and Foxp3 in these cell types. These transcription factors are crucial in determining cell functionality and the outcome in terms of inflammatory responses. Through deacetylation of critical lysine residues of these transcription factors, HDACs can affect, for example, transcription factor stability, nuclear translocation and DNA binding. HDACi can influence these biological processes and thereby affect inflammatory gene expression. It has become clear that these effects of HDACi are cell type specific, and that selective inhibition of HDACs is important. The effects of HDACi on transcription factors and their acetylation status may be particularly important. Previous studies oftentimes focused extensively on characterizing effects of HDACi on histone acetylation; however, this is not sufficient to fully explain the effects of HDACi on biological processes such as gene expression. Inhibitors that are highly potent as well as isoform selective can aid in elucidating the roles of individual HDACs, next to their potential as therapeutics for these diseases.
A number of important insights have been acquired in recent years. For example, it is important to study the selectivity profile of HDACi in cells, since the inhibition at the level of recombinant HDACs has been shown to vary from the inhibition on HDACs in cells which are present in multiprotein complexes. A selectivity profile can also be generated at the level of the acetylome. Mass spectrometry has been particularly useful in allowing these kind of analyses, which allows for a better characterization of HDACi targets and selectivity at different levels complementing each other. Next to studying IC50 values, it is important to take thermodynamic and kinetic parameters into account, such as the residence time of HDACi in recombinant HDACs, which has been shown to vary between hydroxamic acid and aminoanilide type HDACi.
Altogether, the studies discussed indicate that HDACi have great potential for the treatment of inflammatory airway diseases. However, a clinical application for HDACi in this field is still far from within reach. The recent insights as discussed above are crucial in the future development of more selective HDACi, and will drive the development of such HDACi. This is essential in broadening their application toward inflammatory lung diseases such as asthma and COPD. Finally, future steps must also include further elucidation of the effects of HDACi in more advanced disease models, particularly for COPD.
Background
Alternative treatments for the inflammatory airway diseases such as asthma and chronic obstructive pulmonary disease (COPD) can be found by studying underlying biological processes. This may include epigenetic regulation, such as lysine acetylation.
Lysine acetylation is a post-translational modification of proteins, which regulates protein activity. Lysine acetylation, in turn, is regulated by histone acetyltansferases (HATs) as writers and histone deacetylases (HDACs) as erasers.
The nonselective HDAC inhibitor (HDACi) trichostatin A (TSA) attenuates inflammation in mouse asthma models.
Counterintuitive to this, asthma and COPD are both characterized by increased HAT activity and decreased HDAC activity.
The expression and activity of individual HAT and HDAC isoforms in asthma and COPD need to be further elucidated.
The biological mechanisms by which HDACi can attenuate inflammation in models for airway inflammation need to be further elucidated.
Cell type-specific effects
HDACs play a role in the regulation of acetylation of transcription factors in specific cell types implicated in asthma and COPD, which determines cell functionality and the outcome in terms of inflammatory responses. HDACi can affect these processes.
Macrophages: selective regulation of the NF-κB transcription factor acetylation status by HDAC3 inhibitors?
In macrophages, nonselective HDACi have ambiguous effects on the NF-κB pathway, and can either up- or downregulate pro-inflammatory gene expression.
The HDAC1–3 inhibitor MS-275 upregulates both pro- and anti-inflammatory gene expression in macrophages.
The HDAC3 inhibitor RGFP966 gives rise to only anti-inflammatory effects. This could be due to more selective regulation of NF-κB acetylation by HDAC3 inhibitors such as RGFP966.
Th2 cells: regulation of the GATA3 transcription factor acetylation status by an SIRT1 inhibitor or activator
In Th2 cells, SIRT1 has been implicated in the deacetylation of GATA3.
SIRT1 is decreased in activity and expression in isolated cells from severe asthmatic patients.
A SIRT1 inhibitor (Sirtinol) enhances Th2-related cytokine expression, whereas an SIRT1 activator (SRT2172) reduces Th2-related cytokine expression, in these isolated cells, raising interest for SIRT1 as a target for the treatment of severe asthma.
Tregs: regulation of the Foxp3 transcription factor acetylation status by HDACi
In Tregs, SIRT1 and HDAC9 deacetylate Fop3, which reduces the stability and activity of this transcription factor.
In line with this, HDACi can augment the differentiation and regulatory capacity of Tregs, raising interest in further research along these lines.
Can HDACi alter the balance between Tregs & Th17 cells?
In Th17 cells, HDAC1 has recently been reported to deacetylate RORγt.
There are indications that HDACi can skew the differentiation of T lymphocytes from Th17 toward Tregs, which could be advantageous in the context of asthma and COPD, giving rise to interesting novel research directions along these lines.
From histones to transcription factors
Effects of HDACi on histone acetylation in cells are oftentimes used as a read-out indicative of the effects of HDACi in these cells, however, this does not always correlate with effects on biological processes such as gene expression.
Effects of HDACi on transcription factors and their acetylation status may also be particularly important in explaining the effects of these compounds on gene expression.
From recombinant enzymes to HDAC activity in cells
The selectivity profile of HDACi is oftentimes determined by measuring the inhibition of recombinant HDAC enzymes by these compounds, however, HDACs are part of multiprotein complexes in cells.
Importantly, recent studies have shown that HDACi have an unexpected degree of selectivity toward HDACs in these different complexes in cell lysates.
Next to this, a selectivity profile of an HDACi can also be generated on the level of the acetylome.
From IC50 to binding kinetics
Different classes of HDACi have been reported to have different binding kinetics with the HDACs. Aminoanilide-based HDACi display slow binding kinetics, whereas hydroxamic acid-based HDACi display fast binding kinetics.
This translates into biological effects, with long-lasting effects of aminoanilide-based HDACi on histone acetylation.
Conclusion & future perspective
HDACi have great potential for the treatment of inflammatory airway diseases.
The recent insights as discussed above are crucial in the future development of more selective HDACi, and will drive the development of such HDACi.
This is essential in broadening their application toward inflammatory airway diseases such as asthma and COPD.
Future research must also elucidate the effects of HDACi in more advanced disease models, particularly for COPD.
Financial & competing interests disclosure
This research was financially supported by the European Research Council with an ERC starting grant (309782), and The Netherlands organization for scientific research (NWO) with a VIDI grant (723.012.005) to FJ Dekker. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Barnes PJ . Immunology of asthma and chronic obstructive pulmonary disease . Nat. Rev. Immunol.8 ( 3 ), 183 – 192 ( 2008 ).
- Kim HY , DeKruyffRH , UmetsuDT . The many paths to asthma: phenotype shaped by innate and adaptive immunity . Nat. Immunol.11 ( 7 ), 577 – 584 ( 2010 ).
- Barnes PJ . The cytokine network in asthma and chronic obstructive pulmonary disease . J. Clin. Invest.118 ( 11 ), 3546 – 3556 ( 2008 ).
- Caramori G , CasolariP , BarczykA , DurhamAL , Di StefanoA , AdcockI . COPD immunopathology . Semin. Immunopathol.38 ( 4 ), 497 – 515 ( 2016 ).
- Barnes PJ . Inflammatory mechanisms in patients with chronic obstructive pulmonary disease . J. Allergy Clin. Immunol.138 ( 1 ), 16 – 27 ( 2016 ).
- Keenan CR , SalemS , FietzER , GualanoRC , StewartAG . Glucocorticoid-resistant asthma and novel anti-inflammatory drugs . Drug Discov. Today17 ( 17–18 ), 1031 – 1038 ( 2012 ).
- Telenga ED , KerstjensHA , PostmaDS , Ten HackenNH , van den BergeM . Inhaled corticosteroids in chronic obstructive pulmonary disease: a review . Expert Opin. Pharmacother.11 ( 3 ), 405 – 421 ( 2010 ).
- Babu KS , KastelikJA , MorjariaJB . Inhaled corticosteroids in chronic obstructive pulmonary disease: a pro-con perspective . Br. J. Clin. Pharmacol.78 ( 2 ), 282 – 300 ( 2014 ).
- Rabe KF , HurdS , AnzuetoAet al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary . Am. J. Respir. Crit. Care Med.176 ( 6 ), 532 – 555 ( 2007 ).
- Choudhary C , KumarC , GnadFet al. Lysine acetylation targets protein complexes and co-regulates major cellular functions . Science325 ( 5942 ), 834 – 840 ( 2009 ).
- Choudhary C , WeinertBT , NishidaY , VerdinE , MannM . The growing landscape of lysine acetylation links metabolism and cell signalling . Nat. Rev. Mol. Cell Biol.15 ( 8 ), 536 – 550 ( 2014 ).
- Allfrey VG , FaulknerR , MirskyAE . Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis . Proc. Natl Acad. Sci. USA51 , 786 – 794 ( 1964 ).
- Strahl BD , AllisCD . The language of covalent histone modifications . Nature403 ( 6765 ), 41 – 45 ( 2000 ).
- Dekker FJ , HaismaHJ . Histone acetyl transferases as emerging drug targets . Drug Discov. Today14 ( 19–20 ), 942 – 948 ( 2009 ).
- Dekker FJ , van den BoschT , MartinNI . Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases . Drug Discov. Today19 ( 5 ), 654 – 660 ( 2014 ).
- Mottamal M , ZhengS , HuangTL , WangG . Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents . Molecules20 ( 3 ), 3898 – 3941 ( 2015 ).
- Bolden JE , PeartMJ , JohnstoneRW . Anticancer activities of histone deacetylase inhibitors . Nat. Rev. Drug Discov.5 ( 9 ), 769 – 784 ( 2006 ).
- Yang XJ , SetoE . The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men . Nat. Rev. Mol. Cell Biol.9 ( 3 ), 206 – 218 ( 2008 ).
- de Ruijter AJ , van GennipAH , CaronHN , KempS , van KuilenburgAB . Histone deacetylases (HDACs): characterization of the classical HDAC family . Biochem. J.370 ( Pt 3 ), 737 – 749 ( 2003 ).
- Parra M , VerdinE . Regulatory signal transduction pathways for class IIa histone deacetylases . Curr. Opin. Pharmacol.10 ( 4 ), 454 – 460 ( 2010 ).
- Dali-Youcef N , LagougeM , FroelichS , KoehlC , SchoonjansK , AuwerxJ . Sirtuins: the ‘magnificent seven’, function, metabolism and longevity . Ann. Med.39 ( 5 ), 335 – 345 ( 2007 ).
- Moser MA , HagelkruysA , SeiserC . Transcription and beyond: the role of mammalian class I lysine deacetylases . Chromosoma123 ( 1–2 ), 67 – 78 ( 2014 ).
- Joshi P , GrecoTM , GuiseAJet al. The functional interactome landscape of the human histone deacetylase family . Mol. Syst. Biol.9 , 672 ( 2013 ).
- Marks PA . The clinical development of histone deacetylase inhibitors as targeted anticancer drugs . Expert Opin. Investig. Drugs19 ( 9 ), 1049 – 1066 ( 2010 ).
- Wang H , DymockBW . New patented histone deacetylase inhibitors . Expert Opin. Ther. Pat.19 ( 12 ), 1727 – 1757 ( 2009 ).
- Zagni C , FlorestaG , MonciinoG , RescifinaA . The search for potent, small-molecule HDACIs in cancer treatment: a decade after vorinostat . Med. Res. Rev. ( 2017 ).
- Roche J , BertrandP . Inside HDACs with more selective HDAC inhibitors . Eur. J. Med. Chem.121 , 451 – 483 ( 2016 ).
- Chun P . Histone deacetylase inhibitors in hematological malignancies and solid tumors . Arch. Pharm. Res.38 ( 6 ), 933 – 949 ( 2015 ).
- Fischer A . Targeting histone-modifications in Alzheimer’s disease. What is the evidence that this is a promising therapeutic avenue?Neuropharmacology80 , 95 – 102 ( 2014 ).
- Royce SG , KaragiannisTC . Histone deacetylases and their inhibitors: new implications for asthma and chronic respiratory conditions . Curr. Opin. Allergy Clin. Immunol.14 ( 1 ), 44 – 48 ( 2014 ).
- Grabiec AM , TakPP , ReedquistKA . Function of histone deacetylase inhibitors in inflammation . Crit. Rev. Immunol.31 ( 3 ), 233 – 263 ( 2011 ).
- Wightman F , EllenbergP , ChurchillM , LewinSR . HDAC inhibitors in HIV . Immunol. Cell Biol.90 ( 1 ), 47 – 54 ( 2012 ).
- Khan N , JeffersM , KumarSet al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors . Biochem. J.409 ( 2 ), 581 – 589 ( 2008 ).
- Choi JH , OhSW , KangMS , KwonHJ , OhGT , KimDY . Trichostatin A attenuates airway inflammation in mouse asthma model . Clin. Exp. Allergy35 ( 1 ), 89 – 96 ( 2005 ).
- Royce SG , DangW , YuanGet al. Effects of the histone deacetylase inhibitor, trichostatin A, in a chronic allergic airways disease model in mice . Arch. Immunol. Ther. Exp. (Warsz)60 ( 4 ), 295 – 306 ( 2012 ).
- Banerjee A , TrivediCM , DameraGet al. Trichostatin A abrogates airway constriction, but not inflammation, in murine and human asthma models . Am. J. Respir. Cell Mol. Biol.46 ( 2 ), 132 – 138 ( 2012 ).
- Toki S , GoleniewskaK , ReissSet al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation . Thorax71 ( 7 ), 633 – 645 ( 2016 ).
- Hou X , WanH , AiXet al. Histone deacetylase inhibitor regulates the balance of Th17/Treg in allergic asthma . Clin. Respir. J.10 ( 3 ), 371 – 379 ( 2016 ).
- Zhang HP , WangL , FuJJ , FanT , WangZL , WangG . Association between histone hyperacetylation status in memory T lymphocytes and allergen-induced eosinophilic airway inflammation . Respirology21 ( 5 ), 850 – 857 ( 2016 ).
- Ren Y , SuX , KongLet al. Therapeutic effects of histone deacetylase inhibitors in a murine asthma model . Inflamm. Res.65 ( 12 ), 995 – 1008 ( 2016 ).
- Barnes PJ , AdcockIM , ItoK . Histone acetylation and deacetylation: importance in inflammatory lung diseases . Eur. Respir. J.25 ( 3 ), 552 – 563 ( 2005 ).
- Ito K , CaramoriG , LimSet al. Expression and activity of histone deacetylases in human asthmatic airways . Am. J. Respir. Crit. Care Med.166 ( 3 ), 392 – 396 ( 2002 ).
- Cosio BG , MannB , ItoKet al. Histone acetylase and deacetylase activity in alveolar macrophages and blood mononocytes in asthma . Am. J. Respir. Crit. Care Med.170 ( 2 ), 141 – 147 ( 2004 ).
- Su RC , BeckerAB , KozyrskyjAL , HayglassKT . Altered epigenetic regulation and increasing severity of bronchial hyperresponsiveness in atopic asthmatic children . J. Allergy Clin. Immunol.124 ( 5 ), 1116 – 1118 ( 2009 ).
- Butler CA , McQuaidS , TaggartCCet al. Glucocorticoid receptor beta and histone deacetylase 1 and 2 expression in the airways of severe asthma . Thorax67 ( 5 ), 392 – 398 ( 2012 ).
- Gunawardhana LP , GibsonPG , SimpsonJL , PowellH , BainesKJ . Activity and expression of histone acetylases and deacetylases in inflammatory phenotypes of asthma . Clin. Exp. Allergy44 ( 1 ), 47 – 57 ( 2014 ).
- Stefanowicz D , LeeJY , LeeKet al. Elevated H3K18 acetylation in airway epithelial cells of asthmatic subjects . Respir. Res.16 , 95–015 – 0254-y ( 2015 ).
- Rajendrasozhan S , YangSR , KinnulaVL , RahmanI . SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease . Am. J. Respir. Crit. Care Med.177 ( 8 ), 861 – 870 ( 2008 ).
- Ito K , ItoM , ElliottWMet al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease . N. Engl. J. Med.352 ( 19 ), 1967 – 1976 ( 2005 ).
- Hodge G , JersmannH , TranHBet al. Lymphocyte senescence in COPD is associated with decreased histone deacetylase 2 expression by pro-inflammatory lymphocytes . Respir. Res.16 , 130–015 – 0287–2 ( 2015 ).
- Tan C , XuanL , CaoS , YuG , HouQ , WangH . Decreased histone deacetylase 2 (HDAC2) in peripheral blood monocytes (PBMCs) of COPD patients . PLoS ONE11 ( 1 ), e0147380 ( 2016 ).
- Barnes PJ . Glucocorticosteroids: current and future directions . Br. J. Pharmacol.163 ( 1 ), 29 – 43 ( 2011 ).
- Hew M , BhavsarP , TorregoAet al. Relative corticosteroid insensitivity of peripheral blood mononuclear cells in severe asthma . Am. J. Respir. Crit. Care Med.174 ( 2 ), 134 – 141 ( 2006 ).
- Kobayashi Y , BossleyC , GuptaAet al. Passive smoking impairs histone deacetylase-2 in children with severe asthma . Chest145 ( 2 ), 305 – 312 ( 2014 ).
- Lee KY , ItoK , HayashiR , JazrawiEP , BarnesPJ , AdcockIM . NF-kappaB and activator protein 1 response elements and the role of histone modifications in IL-1beta-induced TGF-beta1 gene transcription . J. Immunol.176 ( 1 ), 603 – 615 ( 2006 ).
- Barnes PJ . Alveolar macrophages as orchestrators of COPD . COPD1 ( 1 ), 59 – 70 ( 2004 ).
- Caramori G , OatesT , NicholsonAGet al. Activation of NF-kappaB transcription factor in asthma death . Histopathology54 ( 4 ), 507 – 509 ( 2009 ).
- Edwards MR , BartlettNW , ClarkeD , BirrellM , BelvisiM , JohnstonSL . Targeting the NF-kappaB pathway in asthma and chronic obstructive pulmonary disease . Pharmacol. Ther.121 ( 1 ), 1 – 13 ( 2009 ).
- Chen L , FischleW , VerdinE , GreeneWC . Duration of nuclear NF-kappaB action regulated by reversible acetylation . Science293 ( 5535 ), 1653 – 1657 ( 2001 ).
- Chen L- , MuY , GreeneWC . Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB . EMBO J.21 ( 23 ), 6539 – 6548 ( 2002 ).
- Kiernan R , BrèsV , NgRWMet al. Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65 . J. Biol. Chem.278 ( 4 ), 2758 – 2766 ( 2003 ).
- Yeung F , HobergJE , RamseyCSet al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase . EMBO J.23 ( 12 ), 2369 – 2380 ( 2004 ).
- Sato T , KotakeD , HiratsukaM , HirasawaN . Enhancement of inflammatory protein expression and nuclear factor Kappab (NF-Kappab) activity by trichostatin A (TSA) in OP9 preadipocytes . PLoS ONE8 ( 3 ), e59702 ( 2013 ).
- Rosato RR , KollaSS , HockSKet al. Histone deacetylase inhibitors activate NF-kappaB in human leukemia cells through an ATM/NEMO-related pathway . J. Biol. Chem.285 ( 13 ), 10064 – 10077 ( 2010 ).
- Imre G , GekelerV , LejaA , BeckersT , BoehmM . Histone deacetylase inhibitors suppress the inducibility of nuclear factor-kappaB by tumor necrosis factor-alpha receptor-1 down-regulation . Cancer Res.66 ( 10 ), 5409 – 5418 ( 2006 ).
- Dai Y , RahmaniM , DentP , GrantS . Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation . Mol. Cell. Biol.25 ( 13 ), 5429 – 5444 ( 2005 ).
- Zhang ZY , SchluesenerHJ . HDAC inhibitor MS-275 attenuates the inflammatory reaction in rat experimental autoimmune prostatitis . Prostate72 ( 1 ), 90 – 99 ( 2012 ).
- Zhang ZY , SchluesenerHJ . Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model . J. Neuropathol. Exp. Neurol.72 ( 3 ), 178 – 185 ( 2013 ).
- Choo QY , HoPC , TanakaY , LinHS . Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells . Rheumatology (Oxford)49 ( 8 ), 1447 – 1460 ( 2010 ).
- Halili MA , AndrewsMR , LabzinLIet al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS . J. Leukoc. Biol.87 ( 6 ), 1103 – 1114 ( 2010 ).
- Leus NG , van den BoschT , van der WoudenPEet al. HDAC1–3 inhibitor MS-275 enhances IL10 expression in in RAW264.7 macrophages and reduces cigarette smoke-induced airway inflammation in mice . Sci. Rep.7 , 45047 ( 2017 ).
- Saraiva M , O’GarraA . The regulation of IL-10 production by immune cells . Nat. Rev. Immunol.10 ( 3 ), 170 – 181 ( 2010 ).
- Leus NG , ZwindermanMR , DekkerFJ . Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-kappaB-mediated inflammation . Curr. Opin. Chem. Biol.33 , 160 – 168 ( 2016 ).
- Leus NG , van der WoudenPE , van den BoschTet al. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-kappaB p65 transcriptional activity . Biochem. Pharmacol.108 , 58 – 74 ( 2016 ).
- Villagra A , ChengF , WangHWet al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance . Nat. Immunol.10 ( 1 ), 92 – 100 ( 2009 ).
- Cheng F , LienlafM , Perez-VillarroelPet al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells . Mol. Immunol.60 ( 1 ), 44 – 53 ( 2014 ).
- Castellucci M , RossatoM , CalzettiFet al. IL-10 disrupts the Brd4-docking sites to inhibit LPS-induced CXCL8 and TNF-alpha expression in monocytes: implications for chronic obstructive pulmonary disease . J. Allergy Clin. Immunol.136 ( 3 ), 781.e9 – 791.e9 ( 2015 ).
- Liang Z , ZhangQ , ThomasCMet al. Impaired macrophage phagocytosis of bacteria in severe asthma . Respir. Res.15 , 72–9921 – 15–72 ( 2014 ).
- Ariffin JK , das GuptaK , KapetanovicRet al. Histone deacetylase inhibitors promote mitochondrial reactive oxygen species production and bacterial clearance by human macrophages . Antimicrob. Agents Chemother.60 ( 3 ), 1521 – 1529 ( 2015 ).
- Grausenburger R , BilicI , BoucheronNet al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production . J. Immunol.185 ( 6 ), 3489 – 3497 ( 2010 ).
- Wang C , CollinsM , KuchrooVK . Effector T cell differentiation: are master regulators of effector T cells still the masters?Curr. Opin. Immunol.37 , 6 – 10 ( 2015 ).
- Yamagata T , MitaniK , OdaHet al. Acetylation of GATA-3 affects T-cell survival and homing to secondary lymphoid organs . EMBO J.19 ( 17 ), 4676 – 4687 ( 2000 ).
- Colley T , MercadoN , KunoriYet al. Defective sirtuin-1 increases IL-4 expression through acetylation of GATA-3 in patients with severe asthma . J. Allergy Clin. Immunol.137 ( 5 ), 1595.e7 – 1597.e7 ( 2016 ).
- Wing K , SakaguchiS . Regulatory T cells exert checks and balances on self tolerance and autoimmunity . Nat. Immunol.11 ( 1 ), 7 – 13 ( 2010 ).
- Matsumoto K , InoueH , FukuyamaSet al. Frequency of Foxp3+CD4CD25+ T cells is associated with the phenotypes of allergic asthma . Respirology14 ( 2 ), 187 – 194 ( 2009 ).
- Hamzaoui A , AmmarJ , HamzaouiK . Regulatory T cells in induced sputum of asthmatic children: association with inflammatory cytokines . Multidiscip Respir. Med.5 ( 1 ), 22 – 30 ( 2010 ).
- Baru AM , GaneshV , KrishnaswamyJKet al. Absence of Foxp3+ regulatory T cells during allergen provocation does not exacerbate murine allergic airway inflammation . PLoS ONE7 ( 10 ), e47102 ( 2012 ).
- Vale-Pereira S , Todo-BomA , GeraldesL , Schmidt-WeberC , AkdisCA , Mota-PintoA . FoxP3, GATA-3 and T-bet expression in elderly asthma . Clin. Exp. Allergy41 ( 4 ), 490 – 496 ( 2011 ).
- Beier UH , AkimovaT , LiuY , WangL , HancockWW . Histone/protein deacetylases control Foxp3 expression and the heat shock response of T-regulatory cells . Curr. Opin. Immunol.23 ( 5 ), 670 – 678 ( 2011 ).
- Akimova T , BeierUH , LiuY , WangL , HancockWW . Histone/protein deacetylases and T-cell immune responses . Blood119 ( 11 ), 2443 – 2451 ( 2012 ).
- Kwon HS , LimHW , WuJ , SchnolzerM , VerdinE , OttM . Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells . J. Immunol.188 ( 6 ), 2712 – 2721 ( 2012 ).
- Xiao Y , NagaiY , DengGet al. Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60 . Cell. Rep.7 ( 5 ), 1471 – 1480 ( 2014 ).
- de Zoeten EF , WangL , ButlerKet al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells . Mol. Cell. Biol.31 ( 10 ), 2066 – 2078 ( 2011 ).
- Wang L , LiuY , HanRet al. FOXP3+ regulatory T cell development and function require histone/protein deacetylase 3 . J. Clin. Invest.125 ( 3 ), 1111 – 1123 ( 2015 ).
- Wang L , de ZoetenEF , GreeneMI , HancockWW . Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells . Nat. Rev. Drug Discov.8 ( 12 ), 969 – 981 ( 2009 ).
- Beier UH , WangL , HanR , AkimovaT , LiuY , HancockWW . Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms . Sci. Signal.5 ( 229 ), ra45 ( 2012 ).
- Segretti MC , ValleriniGP , BrochierCet al. Thiol-based potent and selective HDAC6 inhibitors promote tubulin acetylation and T-regulatory cell suppressive function . ACS Med. Chem. Lett.6 ( 11 ), 1156 – 1161 ( 2015 ).
- Lucas JL , MirshahpanahP , Haas-StapletonE , AsadullahK , ZollnerTM , NumerofRP . Induction of Foxp3+ regulatory T cells with histone deacetylase inhibitors . Cell. Immunol.257 ( 1–2 ), 97 – 104 ( 2009 ).
- Shen L , CiesielskiM , RamakrishnanSet al. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models . PLoS ONE7 ( 1 ), e30815 ( 2012 ).
- Bridle BW , ChenL , LemayCGet al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy . Mol. Ther.21 ( 4 ), 887 – 894 ( 2013 ).
- Alcorn JF , CroweCR , KollsJK . TH17 cells in asthma and COPD . Annu. Rev. Physiol.72 , 495 – 516 ( 2010 ).
- Aujla SJ , AlcornJF . T(H)17 cells in asthma and inflammation . Biochim. Biophys. Acta1810 ( 11 ), 1066 – 1079 ( 2011 ).
- Bullens DM , TruyenE , CoteurLet al. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir. Res. 7 , 135 ( 2006 ).
- Ivanov II , McKenzieBS , ZhouLet al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells . Cell126 ( 6 ), 1121 – 1133 ( 2006 ).
- Wu Q , NieJ , GaoYet al. Reciprocal regulation of RORgammat acetylation and function by p300 and HDAC1 . Sci. Rep.5 , 16355 ( 2015 ).
- Chen RY , FanYM , ZhangQet al. Estradiol inhibits Th17 cell differentiation through inhibition of RORgammaT transcription by recruiting the ERalpha/REA complex to estrogen response elements of the RORgammaT promoter . J. Immunol.194 ( 8 ), 4019 – 4028 ( 2015 ).
- Glauben R , SonnenbergE , WetzelM , MascagniP , SiegmundB . Histone deacetylase inhibitors modulate interleukin 6-dependent CD4+ T cell polarization in vitro and in vivo . J. Biol. Chem.289 ( 9 ), 6142 – 6151 ( 2014 ).
- Glaser KB , StaverMJ , WaringJF , StenderJ , UlrichRG , DavidsenSK . Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines . Mol. Cancer Ther.2 ( 2 ), 151 – 163 ( 2003 ).
- Mackmull MT , IskarM , ParcaLet al. Histone deacetylase inhibitors (HDACi) cause the selective depletion of bromodomain containing proteins (BCPs) . Mol. Cell. Proteomics14 ( 5 ), 1350 – 1360 ( 2015 ).
- Halsall JA , TuranN , WiersmaM , TurnerBM . Cells adapt to the epigenomic disruption caused by histone deacetylase inhibitors through a coordinated, chromatin-mediated transcriptional response . Epigenetics Chromatin8 , 29–015 – 0021–9 ; eCollection; 8, 29 ( 2015 ).
- Luchenko VL , LitmanT , ChakrabortyARet al. Histone deacetylase inhibitor-mediated cell death is distinct from its global effect on chromatin . Mol. Oncol.8 ( 8 ), 1379 – 1392 ( 2014 ).
- Bantscheff M , HopfC , SavitskiMMet al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes . Nat. Biotechnol.29 ( 3 ), 255 – 265 ( 2011 ).
- Scholz C , WeinertBT , WagnerSAet al. Acetylation site specificities of lysine deacetylase inhibitors in human cells . Nat. Biotechnol.33 ( 4 ), 415 – 423 ( 2015 ).
- Lauffer BE , MintzerR , FongRet al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability . J. Biol. Chem.288 ( 37 ), 26926 – 26943 ( 2013 ).
- Becher I , DittmannA , SavitskiMM , HopfC , DrewesG , BantscheffM . Chemoproteomics reveals time-dependent binding of histone deacetylase inhibitors to endogenous repressor complexes . ACS Chem. Biol.9 ( 8 ), 1736 – 1746 ( 2014 ).
- Ito T , UmeharaT , SasakiKet al. Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors . Chem. Biol.18 ( 4 ), 495 – 507 ( 2011 ).
- Dinarello CA , FossatiG , MascagniP . Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer . Mol. Med.17 ( 5–6 ), 333 – 352 ( 2011 ).