
Abstract
Aim: The detection of specific DNA methylation patterns bears great promise as biomarker for personalized management of cancer patients. Co-amplification at lower denaturation temperature-PCR (COLD-PCR) assays are sensitive methods, but have previously only been able to analyze loss of DNA methylation. Materials & methods: Enhanced (E)-ice-COLD-PCR reactions starting from 2 ng of bisulfite-converted DNA were developed to analyze methylation patterns in two promoters with locked nucleic acid (LNA) probes blocking amplification of unmethylated CpGs. The enrichment of methylated molecules was compared to quantitative (q)PCR and quantified using serial dilutions.Results: E-ice-COLD-PCR allowed the multiplexed enrichment and quantification of methylated DNA. Assays were validated in primary breast cancer specimens and circulating cell-free DNA from cancer patients. Conclusion: E-ice-COLD-PCR could prove a useful tool in the context of DNA methylation analysis for personalized medicine.
Graphical abstract
The analysis of mutations present at low proportions in biological material has recently attracted great interest to detect clinically relevant subpopulations or to follow the response and potential emergence of mutations conferring treatment resistance [Citation1–3]. Enrichment technologies such as co-amplification at lower denaturation temperature PCR (COLD-PCR) approaches combined with various read-out technologies facilitate the detection of molecules present at low to very low levels [Citation4]. However, the mutations to be followed need to be adapted to each individual patient and new subclones with genetic mutations may arise and escape detection. DNA methylation has been proposed as an alternative molecular alteration that might be used for the detection and follow-up of cancerous lesions [Citation5–9]. The frequent and localized presence of aberrant DNA hypermethylation patterns in the promoter regions of cancer-related genes bears therefore great promise for personalized medicine of cancer or other diseases. Changes in DNA methylation patterns can be analyzed using a number of sequencing based read-out technologies after bisulfite conversion, which ‘freezes’ the methylation state by deaminating unmethylated cytosines to uracils, while methylated cytosines remain unchanged [Citation10]. However, Sanger, next generation or pyrosequencing shows an analytical sensitivity of 20–5% for DNA methylation detection [Citation11]. Technologies such as methylation-specific PCR (MSP), quantitative (q)MSP or MethyLight allow the selective amplification of molecules with a specific DNA methylation profiles and detect methylated molecules at a sensitivity of 1:1000–10,000 [Citation12,Citation13]. However, these technologies will amplify a subpopulation of molecules, usually completely methylated molecules, which are complementary to the sequence of the amplification primers after bisulfite conversion. In the presence of frequently observed heterogeneous methylation patterns, they might underestimate or fail to detect the aberrantly methylated molecules [Citation13,Citation14]. COLD-PCR methods such as full-, fast-, ice- (improved and complete enrichment) and E-ice- (enhanced-ice) use a critical temperature (Tc) in the PCR to selectively denature wild-type mutant heteroduplexes allowing the enrichment of rare mutations [Citation4]. Recently, a fast-COLD-MS-PCR (co-amplification at lower denaturation temperature methylation-specific PCR) has been shown to enrich unmethylated genomic regions even in the presence of an excess of methylated DNA molecules [Citation15]. Due to the enrichment of the molecules melting at lower temperatures, COLD-PCR approaches can only be used for the selective enrichment of hypo- or unmethylated molecules restricting significantly the use of COLD-PCR for the detection of aberrant DNA methylation patterns. However, especially for cancer, research has largely focused on the analysis of hypermethylated loci.
In the present study, we demonstrate in proof-of-principle experiments that E-ice-COLD-PCR [Citation16] can be used to enrich methylated molecules, which can be subsequently read-out at single nucleotide resolution using pyrosequencing. LNA-containing blocker probes were designed to prevent amplification of unmethylated bisulfite-converted DNA enriching preferentially methylated DNA molecules during the E-ice-COLD-PCR reaction. The performance of E-ice-COLD-PCR assays was evaluated by comparison to conventional amplification assays and validated with clinical samples including circulating cell-free DNA (ccfDNA) samples. Accurate quantification was achieved using calibration standards, and assays could easily be multiplexed.
Methods
DNA samples
Unmethylated and methylated bisulfite-converted DNAs were purchased from Qiagen (Courtaboeuf, France) and unmethylated and methylated DNA standards from Zymo Research (CA, USA). Serial dilutions were used to obtain standards with a known degree of methylation (100, 75, 50, 20, 10, 5, 2.5, 1, 0.5, 0.25, 0.1 and 0% methylation).
17 anonymized primary breast cancer tumors were used to validate the enrichment of methylation by E-ice-COLD-PCR assays. Use of these samples was approved by the Norwegian Regional Committee for Medical Research Ethics (approval numbers 429-04148 and 2014/895), and patients have given written informed consent for the use of material for research purposes. DNA was isolated from snap frozen tumor tissue using phenol/chloroform extraction. DNA concentrations were determined using the Quant-iT™ dsDNA broad range assay kit (Thermo Fisher Scientific, CA, USA) and normalized to a concentration of 50 ng/μl.
Six ccfDNA samples, previously extracted from human plasma samples of two colon, two breast, one lung cancer patients and one healthy individual, were included in this study [Citation17]. Two plasma samples from kidney cancer patients were purchased from Seralab (Haywards Heath, West Sussex, UK) and extracted and quantified as previously described [Citation17].
Bisulfite conversion
The bisulfite conversion of DNA standards and the breast cancer samples was performed on 200 or 500 ng of genomic DNA using the EpiTect Fast DNA Bisulfite (50) kit on a QIAcube (Qiagen) according to the manufacturer’s instruction in an elution volume of 40 μl. The bisulfite conversion of cell-free circulating DNA was performed on 15–65 ng in 20 μl of ccfDNA using the EZ DNA Methylation-Gold Kit from Zymo Research according to the manufacturer’s instruction with an elution volume of 30 μl. A different bisulfite conversion kit was used for the bisulfite conversion of the cell-free DNA as this kit was previously found to yield higher recovery rates for low amounts of fragmented DNA [Citation18]. The quantity of bisulfite-converted DNA was estimated using a 75% recovery rate.
E-ice-COLD-PCR
qPCR and E-ice-COLD-PCR reactions were performed on a LightCycler 480 thermocycler (Roche Applied Science, Penzberg, Germany). Primers, probes and pyrosequencing primers () for the E-ice-COLD-PCR assays were purchased HPLC-purified from TIBMOLBIOL (Berlin, Germany).
+ denotes an LNA building block. PCR primers are indicated in yellow, LNA probes are underlined, pyrosequencing primers are noted in green and the sequence analyzed using pyrosequencing is in bold and italic for each assay.

Conditions for the E-ice-COLD-PCR were 1 × HotStar Taq buffer (Qiagen) supplemented with 1.6 mM MgCl2, 200 μM of each dNTP, 2.0 U of HotStar Taq polymerase, 200 nM of forward and reverse primers, 2 μM of SYTO9, 50 nM of blocker probe (AIM2-LNA1) for AIM2 or 40 nM of blocker probe (RARB2-LNA1) for RARB2 and 2 ng of bisulfite-converted DNA (corresponding to ∼700 DNA molecules) in a 25 μl volume (). The E-ice-COLD duplex PCR was performed under the same conditions except for the quantity of each primer, which was halved [Citation19]. The E-ice-COLD-PCR cycling conditions included an initial denaturation step for 10 min at 95°C, followed by six cycles of 30 s at 95°C, 20 s at 61°C and 10 s at 72°C, followed by 52 cycles of 20 s at 95°C, 30 s at 70°C, 20 s at 74°C, 20 s at 61°C and 10 s at 72°C, followed by a melting curve at 20 acquisition per degree from 65 to 95°C and a final cooling to 40°C. Melting curve analysis was performed as a quality control to detect amplification products not corresponding to the amplification product, for example, primer dimers. Optimal concentrations of blocker probes for each E-ice-COLD-PCR assay were determined by comparison to amplification reactions without the respective blocker probe using the same cycling conditions.
qPCRs for standard curves and samples analysis were performed under the same conditions without adding the blocker probes. The qPCR cycling included an initial denaturation step for 10 min at 95°C, followed by 52 cycles of 30 s at 95°C, 20 s at 61°C and 10 s at 72°C, followed by the above-described melting curve analysis. The methylation patterns of standards and samples were analyzed in sextuplicates and triplicates, respectively.
Pyrosequencing
Pyrosequencing was performed using PyroMark Gold SQA Q96 Reagents (Qiagen) as previously described [Citation20] using 4 pmol of the pyrosequencing primer (). DNA methylation sequencing was performed on a Pyromark® Q96 HS (Qiagen) and analyzed with the Pyromark CpG software (Qiagen). The enrichment factor was calculated using the percentage of methylation detected by pyrosequencing following E-ice-COLD-PCR divided by the methylation level of the sample without enrichment.
Results
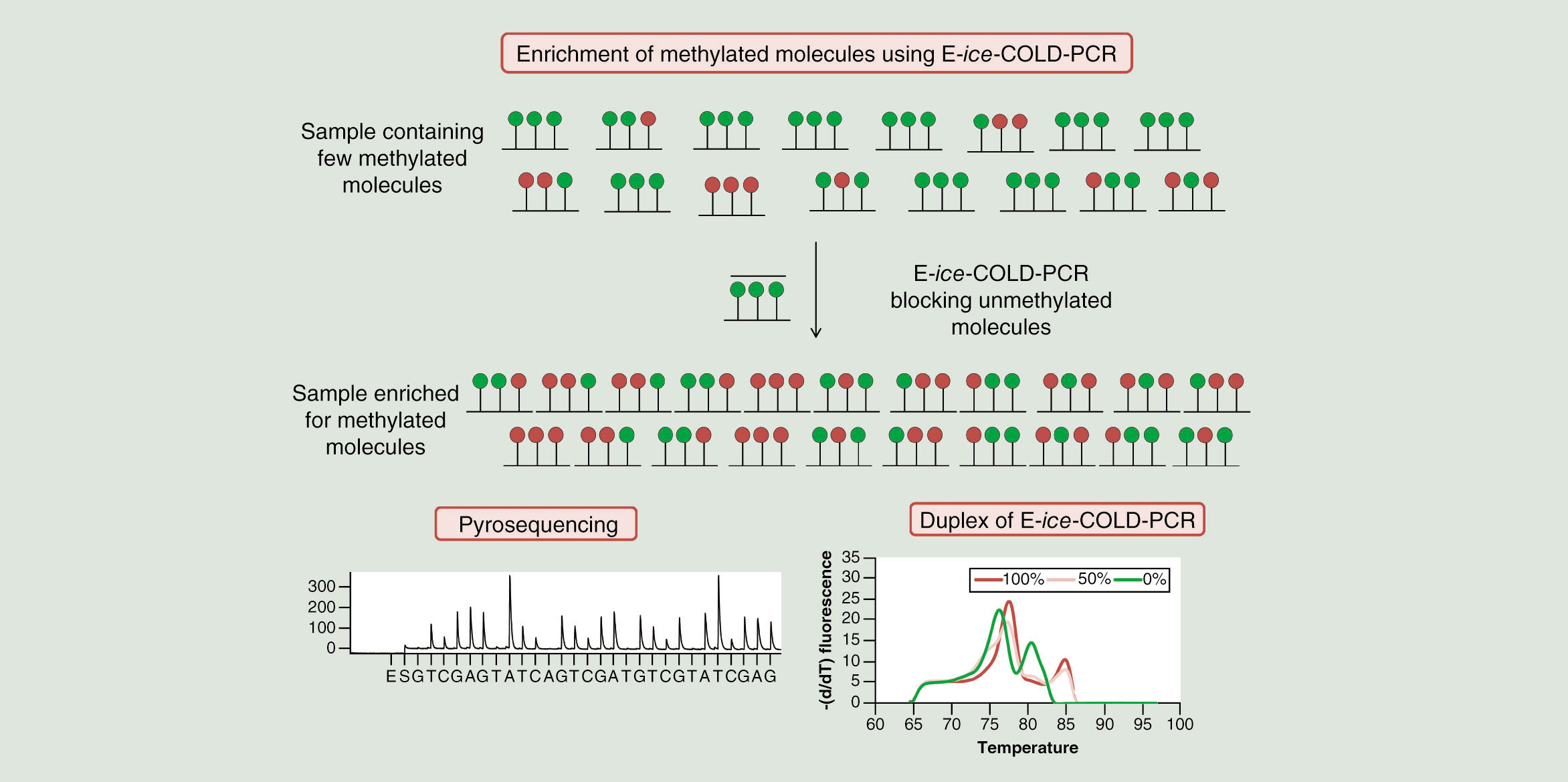
Due to the previously reported strong enrichment of mutations using E-ice-COLD-PCR [Citation16], we investigated in this proof-of-concept study if E-ice-COLD-PCR can – despite the unfavorable higher melting temperature of methylated molecules – be also used to enrich and facilitate the detection of rare methylated DNA molecules in the presence of unmethylated molecules in bisulfite-converted DNA. The method contains three steps: bisulfite conversion, the E-ice-COLD-PCR to enrich bisulfite-converted methylated DNA followed by pyrosequencing to analyze and quantify DNA methylation at single CpG resolution.
E-ice-COLD-PCR assays
Two assays () were developed targeting the promoter regions of RARB2 (175 bp) and AIM2 (172 bp), which we and others have previously shown to present differential methylation in breast cancer samples [Citation21–23]. The size of the amplification product was kept relatively small to facilitate the analysis of fragmented DNA such as ccfDNA. E-ice-COLD-PCR reactions were developed to start from 2 ng of bisulfite converted DNA/reaction to make the method applicable to samples with limited DNA availability such as ccfDNA. Reactions were optimized using temperature gradients to define the optimal Tc. Gradients of the concentration of the blockers (0–100 nM) were performed to determine the best conditions for the enrichment and detection of methylated molecules using methylation standards with a known degree of DNA methylation (20, 10, 5, 1, 0.5–0.1, 0%).
LNA blocker probes for E-ice-COLD-PCR were designed to have six bases overlapping with one of the reverse PCR primers and had a minimum length of approximately 40 bases [Citation24]. Several LNA blocker probes blocking different numbers of CpGs were used for the evaluation of the enrichment by E-ice-COLD-PCR (). They were designed to block at least one unmethylated bisulfite-converted cytosine in the context of a CpG dinucleotide present in the sequence of AIM2 and RARB2 (). For the AIM2 assay, four blocker probes targeting the same sequence were compared using either four LNA bases to block the two unmethylated cytosine as well as the adjacent guanosine bases (AIM2-LNA1) of the two CpGs present in the analyzed sequence; two LNA bases to block each of the unmethylated cytosine of CpGs (AIM2-LNA2) or one LNA to block one of the two unmethylated cytosine of one of the CpGs (AIM2-LNA3 and AIM2-LNA4) (). For the RARB2 assay, five blocker probes targeting the same sequence were compared using five (RARB2-LNA1), four (RARB2-LNA2), three (RARB2-LNA3), two (RARB2-LNA4) and one (RARB2-LNA5) LNA bases to block unmethylated cytosines of CpGs (). The blocker probes AIM2-LNA3, AIM2-LNA4, RARB2-LNA4 and RARB2-LNA5 did not yield any enrichment, whereas all others enriched methylated DNA to different degrees. Assays using these blocker probes were subsequently compared with conventional qPCR. The strongest enrichment was obtained for AIM2 and RARB2 with LNA oligonucleotides that blocked all CpGs (AIM2-LNA1 and RARB2-LNA1) underlying the blocking probe sequence. For the RARB2 E-ice-COLD-PCR assay, there was also an enrichment using three or four of the five CpGs blocked (RARB2-LNA3 and LNA2), but the enrichment was lower compared with the LNA blocking five CpGs (RARB2-LNA1) yielding a 10.7-, 17.6- and 44.8-fold enrichment for the analysis of a 0.5% methylated DNA standard, respectively. For the AIM2 E-ice-COLD-PCR assay, there was a stronger enrichment using four LNAs (AIM2-LNA1) compared with two LNA bases (AIM2-LNA2) with a 32.6- and 12.8-fold enrichment for the analysis of a 0.5% methylated DNA standard, respectively. For higher methylation levels the efficiency of the enrichment decreased slightly, but showed the same trend with a 4.9-, 16.6- and 34.6-fold enrichment for RARB2 and 10.3- and 27.5-fold enrichment for AIM2, respectively. Increasing the amount of input material to 4 ng, led to a strong increase in the enrichment efficiency with a 61-fold enrichment for the 0.5% methylation standard for AIM2. For higher DNA quantities, the concentration of the blocker should be reduced to maintain the possibility for quantification. DNA methylation levels of 5% or below were indistinguishable after qPCR amplification without the blocking probe due to the limit of quantification of the pyrosequencer.
The Tc of E-ice-COLD-PCR using blocker probes of AIM2-LNA1 and RARB2-LNA1 () was 74°C for both assays and optimal concentrations of the blocker were 50 and 40 nM for AIM2 and RARB2, respectively, allowing the enrichment of methylated DNA present below 10% in the DNA methylation standards with minimal false positive detection of unmethylated DNA (Supplementary Figure 1).
Methylation enrichment using E-ice-COLD-PCR
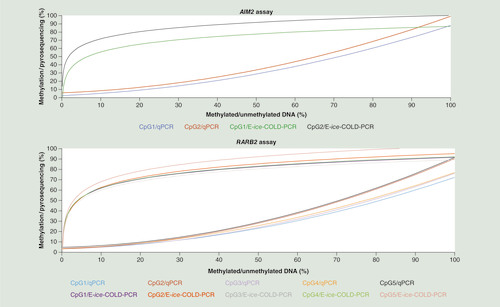
The enrichment of DNA methylation of both E-ice-COLD-PCR assays was then compared with the DNA methylation analysis using a methylation-independent qPCR assay without the blocking probe (). Standard curves of E-ice-COLD-PCR and qPCR assays for each CpG were established using serial dilutions (from 0 to 100%) of methylated DNA in sextuplicates. The standard curves of E-ice-COLD-PCR were logarithmic (R2 = 0.97 for AIM2 and R2 = 0.96 for RARB2) as previously shown for the enrichment of mutations [Citation16]. However, the standard curves of the qPCR assays fitted a second degree polynomial equation (R2 = 0.99 for AIM2 and R2 = 0.98 for RARB2) indicating the presence of the commonly observed preferential amplification of unmethylated molecules [Citation25].
Standard curves were obtained using serial dilution of 100, 75, 50, 20, 10, 5, 1, 0.25 and 0.1–0% of methylated and unmethylated bisulfite converted DNA standards in sextuplicates. For the AIM2 assay, the standard curve for CpG1 by E-ice-COLD-PCR was y = 13.602 ln (x) + 24.284 (R2 = 0.9701, green curve) and for CpG 2 was y = 12.512 ln (x) + 42.808 (R2 = 0.9714, black curve), for CpG1 by qPCR was y = 0.0066 x2 + 0.1957 x + 2.4394 (R2 = 0.9972, blue curve) and for CpG2 by qPCR was y = 0.0075 x2 + 0.1804 x + 5.8741 (R2 = 0.9965, red curve). For the RARB2 E-ice-COLD-PCR assay, the standard curve for CpG1 was y = 12.988 ln (x) + 31.723 (R2 = 0.973, purple curve), CpG2 y = 14.239 ln (x) + 29.63 (R2 = 0.9556, red curve), CpG3 y = 12.786 ln (x) + 30.307 (R2 = 0.9694, gray curve), CpG4 y = 13.133 ln (x) + 31.64 (R2 = 0.9606, green curve), CpG5 y = 15.02 ln (x) + 33.403 (R2 = 0.9505 faded red curve). For the qPCR assay, the standard curve for CpG1 by qPCR was y = 0.006 x2 + 0.0827 x + 3.8353 (R2 = 0.9851, blue curve), CpG2 y = 0.0076 x2 + 0.01033 x + 3.7873 (R2 = 0.9887, red curve), CpG3 y = 0.0063 x2 + 0.0105 x + 3.0971 (R2 = 0.9808, purple curve), CpG4 y = 0.0057 x2 + 0.1656 x + 3.113 (R2 = 0.9862, orange curve) and CpG5 y = 0.0077 x2 + 0.0931 x + 4.8262 (R2 = 0.9841, black curve).
qPCR: quantitative PCR.
The detection of DNA methylation was improved using E-ice-COLD-PCR, especially in samples/standards with a low to very low degree of DNA methylation (below 1%), compared with standard qPCR amplification. Furthermore, the limit of detection of E-ice-COLD-PCR coupled with pyrosequencing starting with 2 ng DNA/reaction was below 0.5%, tenfold less than the limit of detection of a standard PCR combined with pyrosequencing starting with 20 ng DNA per reaction, which was found to be around 5% concordant with previous publications [Citation26].
Quantification of clinical samples
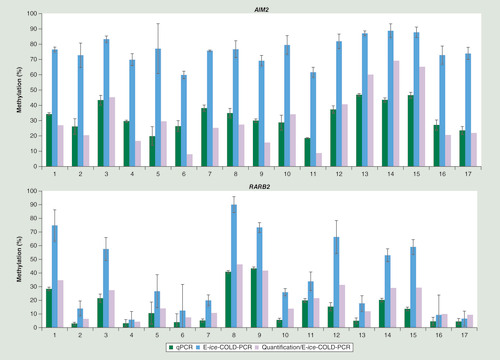
E-ice-COLD-PCR assays were validated using DNA from 17 breast cancer samples. All samples showed an enrichment for AIM2 and most samples for RARB2 (). The percentage of methylation was measured in triplicate and the average level of methylation and the standard deviation of the two CpGs of AIM2 and the five CpGs of RARB2 were calculated. The mean standard deviation was 4.6% for replicates of E-ice-COLD-PCR assays compared with 2.5% for the conventional qPCR for AIM2 and 7.8% compared with 2.3% for RARB2 showing a slight loss of accuracy probably due variation in the enrichment efficiency for the E-ice-COLD-PCR assays. Moreover, the DNA methylation levels were quantified using the E-ice-COLD-PCR standard curves for each CpG () taking the mean of all CpGs and triplicate experiments into account (). The quantification of methylation using E-ice-COLD-PCR yielded results in the same methylation range as the conventional qPCR assays or standard pyrosequencing assays starting with 20 ng of DNA.
Duplex of E-ice-COLD-PCR reaction
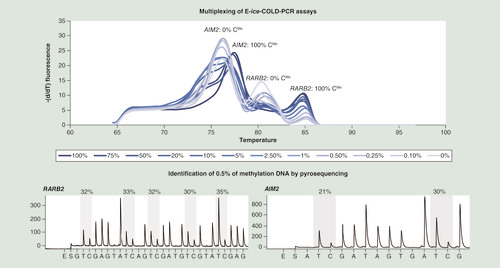
E-ice-COLD-PCR assays can be multiplexed if assays have a similar annealing (Ta) and Tc [Citation19]. Therefore, we evaluated the possibility to analyze the methylation of AIM2 and RARB2 in a duplex E-ice-COLD-PCR assay using a Ta of 61°C, a Tc of 74°C, and 50 and 40 nM of blocker probes for AIM2 and RARB2, respectively. The quantity of PCR primers in the duplex reaction was reduced to half compared with simplex reactions [Citation19].
High-resolution melting curves showed two peaks at approximately 76°C and approximately 80°C for unmethylated DNA and two peaks at approximately 78°C and approximately 85°C for methylated DNA for AIM2 and RARB2, respectively. The comparison of standard curves demonstrated the enrichment of both targets in the E-ice-COLD-PCR duplex. The E-ice-COLD-PCR duplex was followed by two separate pyrosequencing assays analyzing the AIM2 and RARB2 promoters to identify methylated DNA (). The percentage of the detected methylation levels of the seven CpGs in the two promoters after enrichment ranged between 20 and 35% starting from 2 ng of 0.5% methylated DNA demonstrating the efficient enrichment and straightforward detection of low to very low DNA methylation levels.
Upper panel: high-resolution melting curves of the duplex E-ice-COLD-PCR assay enriching methylation at the AIM2 and RARB2 promoters using 100, 75, 50, 20, 10, 5, 2.5, 1, 0.5, 0.25, 0.1 and 0% of methylated DNA. Lower panel: pyrograms after enrichment of a 0.5% methylated DNA standard using duplex E-ice-COLD-PCR combined with pyrosequencing assays analyzing AIM2 and RARB2.
Circulating cell-free DNA samples
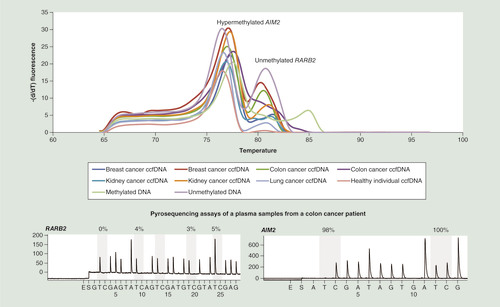
With the current interest in the analysis of ccfDNA for personalized patient management, we then applied the E-ice-COLD-PCR duplex to the detection of methylation in ccfDNA from eight plasma samples from different cancer patients (kidney, breast, colon and lung cancer) and one healthy individual (). ccfDNA was analyzed in triplicate and serial dilutions of methylated and unmethylated bisulfite-converted DNA were used as controls and compared with the duplex qPCR assay without LNA probes. High-resolution melting curves of the E-ice-COLD-PCR duplex showed that all ccfDNA were highly methylated for the AIM2 promoter and unmethylated for the RARB2 assay with the notable exception of one colon cancer sample, which showed low levels of DNA methylation (). Results were confirmed by two pyrosequencing assays of these genes (). The average percentage of the detected methylation levels in the plasma samples after enrichment was 93–98% for AIM2 and 25% for RARB2 in the ccfDNA from a colon cancer patient. The actual level of methylation in this sample using the standard curves was estimated DNA to be approximately 0.5% corresponding to a methylation degree that could not easily be detected by methods using methylation-independent amplification.
Discussion
In the current manuscript, we demonstrate that E-ice-COLD-PCR, which we initially developed to enrich low-abundant somatic mutations, can be also used for the enrichment of low-abundant methylation in bisulfite-converted DNA [Citation4,Citation16].
E-ice-COLD-PCR allowed the detection of methylated DNA using LNA-containing probes to block unmethylated cytosines and provides therefore a novel approach for the sensitive detection of, for example, promoter hypermethylation, which is currently of great interest for many clinical applications for the management of cancer patients using non-invasive liquid biopsies [Citation3,Citation7,Citation27,Citation28]. As unmethylated molecules have a lower melting temperature compared with methylated molecules as the bisulfite conversion leads to the replacement of unmethylated cytosines by thymines, they are more easily to enrich in COLD-PCR-based approaches and a fast-COLD-MS-PCR method has previously been reported [Citation15]. We demonstrate in this work that the LNAs contained in the blocking oligonucleotide will induce a stronger effect on the strength of the hybridisation compared to the melting temperature allowing for the enrichment under thermodynamic unfavorable conditions. Blocker probes must therefore be designed with several LNA bases and some more constraints are inevitable for the design of the assays. While in the initial ice-COLD-PCR publication, the blocking nucleotide was required to overlap with one of the amplification primer binding sites [Citation29], we have previously shown that this was not a prerequisite for the assay performance when using LNA containing blocking nucleotides [Citation16]. However, for the analysis of methylated DNA, blocking oligonucleotides that did not overlap primer binding sites did not efficiently block the amplification of unmethylated molecules. We have previously shown that successful E-ice-COLD-PCR assays for the enrichment of BRAF, KRAS and NRAS mutations contained at least three LNA bases per blocker probe covering the mutated codons [Citation16,Citation19,Citation30]. For the DNA methylation analysis shown in this study, it was not necessary that the LNA-containing blockers covered all CpGs in the region of interest. For the AIM2 assay only two of the four CpGs were covered by the blocking probe and therefore two LNAs complementary to invariable nucleotides were included to achieve optimal enrichment efficiency. For RARB2, five of the 11 CpGs were blocked and this allowed also the enrichment also at the five other CpGs as determined by pyrosequencing. It is probable that with fewer LNAs incorporated in the blocker probe, the decrease in the melting temperature due to the mismatch with partially or completely methylated molecules cannot outweigh the lower melting temperature of completely unmethylated molecules leading to a loss of the enrichment efficiency. As the enrichment will be determined by the DNA methylation status of the CpGs complementary to the blocker probe, a detailed knowledge of the CpG positions correlating best with the phenotype under investigation is required to determine the CpGs targeted by the blocker probe. Nonetheless, 90% closely neighbored CpGs with a distance of 50 base pairs or less show the same methylation status [Citation31] allowing some flexibility in the probe design.
While focusing on the technically more demanding detection of hypermethylation, blocker probes could also be used to block methylated DNA and enable the enrichment of unmethylated DNA in the presence of an excess of methylated DNA. This might be particularly useful for the analysis of complex diseases other than cancer such as the detection of hypomethylated molecules of the INS promoter in individuals with diseases inducing pancreatic β-cell death such as Type 1 diabetes [Citation11,Citation32].
E-ice-COLD-PCR combined with pyrosequencing has previously been used for the quantification of mutations in ccfDNA using standard curves of serial dilutions of known KRAS mutations and yielded data comparable to those obtained by digital PCR, but allowed a higher throughput, lower cost and shorter time-to-results [Citation33]. The possibility for quantification was here confirmed for DNA methylation analysis. The initial protocols devising the ice-COLD-PCR required a nested amplification approach totaling a first round of amplification of 35 cycles followed by the enrichment amplification using 25–40 cycles [Citation29]. E-ice-COLD PCR uses a single PCR, but requires a substantial number of amplification cycles to enrich for the methylated molecules, with detectable amplification levels only after 40 cycles as assessed by real-time monitoring of the enrichment and amplification. Nonetheless variation between technical replicates only slightly increased compared with conventional qPCR amplification. In the current study we have chosen to start from small amounts of DNA to be compatible even with ccfDNA samples from individuals with low levels of ccfDNA. Increasing the DNA input for patients with high levels could increase the accuracy and lower the detection limit of aberrantly methylated molecules.
E-ice-COLD-PCR will be extremely useful for the detection of low levels of DNA methylation while for very high methylation levels, the quantification was improved using standard qPCR because of the logarithmic curves for E-ice-COLD-PCR leading to a plateau with no further enrichment. We have previously shown that the concentration of blocker probe could be adapted to the range of mutation levels and a similar approach could be taken for methylation. Furthermore, E-ice-COLD-PCR assays can be easily optimized using gradients of temperature and blocker concentrations. Various concentrations of blocker probes could be used to improve the resolution of the standard curves for different ranges of methylation levels and to enhance the quantification as we have previously shown for the detection of mutations [Citation33]. We have previously shown, that the COLD-PCR cycling protocol is essential for obtaining the enrichment observed for E-ice-COLD-PCR assays, as using a wild-type blocking PCR assay [Citation34] targeting the same sequence and using the same blocker, did achieve much lower rates of enrichment [Citation16]. In the future, it will be important to better understand the molecular nature of the enriched molecules. While our approach should enrich all molecules that contain at least some methylated positions at the CpG positions covered by the blocker, our read-out technology does not allow an analysis at the level of individual molecules. Further development is necessary to combine E-ice-COLD-PCR with next-generation sequencing (or cloning and conventional sequencing) as read-out technology. This would allow counting of the individual molecular patterns before and after enrichment and – using templates with heterogeneous levels of DNA methylation on the CpGs covered by the blocker – assessing the impact of DNA methylation at the different positions of the blocker as well as neighboring CpGs for the enrichment.
MSP- and qMSP-based methods, such as SMART-MSP, MethyLight, HeavyMethyl or Quantitative Analysis of Methylated Alleles, have proven well suited for the detection of cancer-specific methylation patterns in primary tumor tissues, but also for the detection of very low levels of methylation in body fluids in contact with the tumor site and in circulating DNA [Citation4,Citation10,Citation35]. These technologies are most commonly applied to validate and analyze DNA methylation biomarkers. While MSP and MethyLight will specifically amplify methylated molecules in the presence of a 1000–10,000-fold excess of unmethylated molecules, the efficient amplification relies on a predefined consistent methylation profile underlying the bindings sites for the amplification primers and the hydrolysis probe (for MethyLight) usually completely methylated molecules [Citation12,Citation36]. We and others have previously shown, that in the presence of heterogeneous DNA methylation patterns, the assays fail to amplify partially methylated molecules and significantly underestimate the proportion of methylated samples [Citation13,Citation14,Citation37]. Further, a recent large-scale comparison of locus-specific DNA methylation technologies for potential DNA methylation-based biomarker assessment, MethyLight was the most quantitative method among the relative methylation assays, but still failed to detect a considerable number of differences in the analyzed samples raising concerns on its use for the analysis of samples in a clinical setting [Citation38]. While HeavyMethyl uses small blocking oligonucleotides complementary to unmethylated molecules to compete with methylation-independent amplification primers to favor the amplification of methylated molecules, the hydrolysis probe will be complementary to completely methylated molecules [Citation39]. This technology is currently used in the US FDA-approved commercial Epi proColon® assay for the detection of colorectal cancer through the analysis of DNA hypermethylation of the v2 region in the SEPT9 gene [Citation40]. While highly specific and sensitive in proof-of-principle experiments [Citation39,Citation41], the assay displays in clinical practice only moderate sensitivity (positive detection of 48% of patients with a colorectal cancer) while maintaining high specificity [Citation42]. Furthermore, the design requires five oligonucleotides with some constraints on the availability and position of CpG complicates assay design. The design of E-ice-COLD-PCR assays is fundamentally different from these assays as it impedes the amplification of the normal, unmethylated state, but does not make any requirements on the degree or the patterns of DNA methylation in the amplified target region. All molecules different from the blocked pattern will be amplified and the subsequent sequencing-based analysis give detailed information on the molecules that have been enriched. While head-to-head comparisons with the HeavyMethyl technology will be required, it could be speculated that E-ice-COLD-PCR might have an increased sensitivity, as it does not require any specific co-methylation patterns in the analyzed region. In the current study we have been using pyrosequencing for its quantitative accuracy and short time to results, the enrichment could also be combined with next generation sequencing (NGS)-based read-out, which would allow the detailed analysis of DNA methylation patterns on individual molecules.
Methylation analysis of tumor-derived ccfDNA from different liquid biopsies has shown to bear information of great clinical value for diagnostics, early detection, prognostics and treatment decisions including early detection of neoplastic or preneoplastic lesions as well as the time detection of metastasis, recurrence or development of treatment resistance [Citation3,Citation7,Citation27,Citation28,Citation43]. The assays described within this manuscript have not the purpose to be used for clinical purposes, but solely as proof-of-principle of the technical feasibility of the developed approach. Of note, AIM2 shows significant methylation levels in blood cells, making it unsuitable for diagnostic assays due to the potential contamination of the samples through lysed blood cells. While assays like the above described Epi proColon V2 test analyzes the DNA methylation levels at a single locus, increased sensitivity and specificity can be obtained using signatures combining the DNA methylation levels at several CpGs in both the primary tumor and liquid biopsies [Citation44–47]. DNA methylation signatures will be difficult to analyze using real-time MSP or digital PCR assays requiring a single target per reaction. E-ice-COLD-PCR methylation assays can be designed to be multiplexed using the same Ta and Tc allowing the methylation analysis as shown with a duplex assay for in this study and previously for a triplex assay for mutation detection which demonstrated that E-ice-COLD-PCR can be used for the analysis of a gene panel [Citation19]. Therefore, for smaller signatures the approach described here will be sufficient, which is further supported by the fact that the commercial MX-ICP kits for mutation detection (Precipio, CT, USA), which rely on the same approach, allow the multiplexed analysis of a larger number of amplicons. For larger signatures, protocols for multiplexing COLD-PCR methods have been devised [Citation48,Citation49] which should allow the analysis of even larger number of target regions or assays might be complemented with alternative approaches such as Name-Pro [Citation50].
Conclusion
In summary, in this proof-of-concept study, we demonstrated that E-ice-COLD-PCR greatly facilitates the detection of low-abundant mutations or methylation patterns and could be a well-suited method for individualized patient management. E-ice-COLD-PCR can detect, identify and quantify low-abundant methylated molecules, requires little starting material, can be easily multiplexed and performed at high-throughput with a short time to results making it compatible with the time frame of clinical decisions. Additional work using next-generation sequencing is required to fully understand the nature of the enriched molecules and the impact of heterogeneous DNA methylation on DNA methylation.
DNA methylation patterns contain valuable information for diagnosis, prognosis and treatment decisions for the clinical management of cancer patients.
COLD-PCR assays have shown to enrich mutated alleles as well as hypomethylated molecules, but could due to unfavorable thermodynamic conditions not analyze hypermethylation.
Enhanced-ice-COLD-PCR allows for the enrichment up to 40-fold of methylated molecules and enables the sensitive detection and accurate quantification of DNA methylation levels.
DNA methylation can be analyzed by E-ice-COLD-PCR in both primary tumor specimens as well as liquid biopsies.
Multiplexing enables the analysis of DNA methylation signatures, which are promising tools for personalized medicine.
Authors’ contributions
F Mauger and J Tost designed the study, supervised experiments, analyzed the data and wrote the manuscript. M Kernaleguen and C Lallemand performed experiments and analyzed the data. VN Kristensen and J-F Deleuze provided clinical samples and intellectual input. All authors contributed to the finalization of the manuscript and approved the final version.
Ethical conduct of research
Use of breast cancer samples was approved by the Norwegian Regional Committee for Medical Research Ethics (approval numbers 429-04148 and 2014/895), and patients have given written informed consent for the use of material for research purposes.
Supplemental Figure 1
Download TIFF Image (1.6 MB)Supplementary
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/full/10.2217/epi-2017-0166
Financial & competing interests disclosure
The study was funded from the institutional budget of the Centre National de Recherche en Génomique Humaine (CNRGH). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Schmitt MW , LoebLA , SalkJJ . The influence of subclonal resistance mutations on targeted cancer therapy . Nat. Rev. Clin. Oncol.13 ( 6 ), 335 – 347 ( 2016 ).
- Wan JCM , MassieC , Garcia-CorbachoJet al. Liquid biopsies come of age: towards implementation of circulating tumour DNA . Nat. Rev. Cancer17 ( 4 ), 223 – 238 ( 2017 ).
- Heitzer E , UlzP , GeiglJB . Circulating tumor DNA as a liquid biopsy for cancer . Clin. Chem.61 ( 1 ), 112 – 123 ( 2015 ).
- Mauger F , How-KitA , TostJ . COLD-PCR technologies in the area of personalized medicine: methodology and applications . Mol. Diagn. Ther.21 ( 3 ), 269 – 283 ( 2017 ).
- How Kit A , NielsenHM , TostJ . DNA methylation based biomarkers: practical considerations and applications . Biochimie94 ( 11 ), 2314 – 2337 ( 2012 ).
- Costa-Pinheiro P , MontezumaD , HenriqueR , JeronimoC . Diagnostic and prognostic epigenetic biomarkers in cancer . Epigenomics7 ( 6 ), 1003 – 1015 ( 2015 ).
- Warton K , MahonKL , SamimiG . Methylated circulating tumor DNA in blood: power in cancer prognosis and response . Endocr. Relat. Cancer23 ( 3 ), R157 – R171 ( 2016 ).
- Kang S , LiQ , ChenQet al. CancerLocator: non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA . Genome Biol.18 ( 1 ), 53 ( 2017 ).
- Guo S , DiepD , PlongthongkumN , FungHL , ZhangK , ZhangK . Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA . Nat. Genet.49 ( 4 ), 635 – 642 ( 2017 ).
- Tost J . Current and emerging technologies for the analysis of the genome-wide and locus-specific DNA methylation patterns . Adv. Exp. Med. Biol.945 , 343 – 430 ( 2016 ).
- Lehmann-Werman R , NeimanD , ZemmourHet al. Identification of tissue-specific cell death using methylation patterns of circulating DNA . Proc. Natl Acad. Sci. USA113 ( 13 ), e1826 – e1834 ( 2016 ).
- Herman JG , GraffJR , MyohanenS , NelkinBD , BaylinSB . Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands . Proc. Natl Acad. Sci. USA93 ( 18 ), 9821 – 9826 ( 1996 ).
- Alnaes GI , RonnebergJA , KristensenVN , TostJ . Heterogeneous DNA methylation patterns in the GSTP1 promoter lead to discordant results between assay technologies and impede its implementation as epigenetic biomarkers in breast cancer . Genes (Basel)6 ( 3 ), 878 – 900 ( 2015 ).
- Mikeska T , CandiloroIL , DobrovicA . The implications of heterogeneous DNA methylation for the accurate quantification of methylation . Epigenomics2 ( 4 ), 561 – 573 ( 2010 ).
- Castellanos-Rizaldos E , MilburyCA , KaratzaE , ChenCC , MakrigiorgosGM , MerewoodA . COLD-PCR amplification of bisulfite-converted DNA allows the enrichment and sequencing of rare un-methylated genomic regions . PLoS ONE9 ( 4 ), e94103 ( 2014 ).
- How Kit A , MazaleyratN , DaunayA , NielsenHM , TerrisB , TostJ . Sensitive detection of KRAS mutations using enhanced-ice-COLD-PCR mutation enrichment and direct sequence identification . Hum. Mutat.34 ( 11 ), 1568 – 1580 ( 2013 ).
- Mauger F , DularyC , DaviaudC , DeleuzeJF , TostJ . Comprehensive evaluation of methods to isolate, quantify, and characterize circulating cell-free DNA from small volumes of plasma . Anal. Bioanal. Chem.407 ( 22 ), 6873 – 6878 ( 2015 ).
- Moison C , AssematF , DaunayA , ArimondoPB , TostJ . DNA methylation analysis of ChIP products at single nucleotide resolution by pyrosequencing(R) . Methods Mol. Biol.1315 , 315 – 333 ( 2015 ).
- Mauger F , DaunayA , DeleuzeJF , TostJ , How-KitA . Multiplexing of E-ice-COLD-PCR assays for mutation detection and identification . Clin. Chem.62 ( 8 ), 1155 – 1158 ( 2016 ).
- Tost J , GutIG . DNA methylation analysis by pyrosequencing . Nat. Protoc.2 ( 9 ), 2265 – 2275 ( 2007 ).
- Tao MH , ShieldsPG , NieJet al. DNA hypermethylation and clinicopathological features in breast cancer: the Western New York Exposures and Breast Cancer (WEB) Study . Breast Cancer Res. Treat.114 ( 3 ), 559 – 568 ( 2009 ).
- Klajic J , BusatoF , EdvardsenHet al. DNA methylation status of key cell-cycle regulators such as CDKNA2/p16 and CCNA1 correlates with treatment response to doxorubicin and 5-fluorouracil in locally advanced breast tumors . Clin. Cancer Res.20 ( 24 ), 6357 – 6366 ( 2014 ).
- Bediaga NG , Acha-SagredoA , GuerraIet al. DNA methylation epigenotypes in breast cancer molecular subtypes . Breast Cancer Res.12 ( 5 ), R77 ( 2010 ).
- How-Kit A , TostJ . Pyrosequencing(R)-based identification of low-frequency mutations enriched through enhanced-ice-COLD-PCR . Methods Mol. Biol.1315 , 83 – 101 ( 2015 ).
- Warnecke PM , StirzakerC , MelkiJR , MillarDS , PaulCL , ClarkSJ . Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA . Nucleic Acids Res.25 ( 21 ), 4422 – 4426 ( 1997 ).
- Tost J , DunkerJ , GutIG . Analysis and quantification of multiple methylation variable positions in CpG islands by pyrosequencing . Biotechniques35 ( 1 ), 152 – 156 ( 2003 ).
- Warton K , SamimiG . Methylation of cell-free circulating DNA in the diagnosis of cancer . Front. Mol. Biosci.2 , 13 ( 2015 ).
- Schrock A , LeisseA , de VosLet al. Free-circulating methylated DNA in blood for diagnosis, staging, prognosis, and monitoring of head and neck squamous cell carcinoma patients: an observational prospective cohort study . Clin. Chem.63 ( 7 ), 1288 – 1296 ( 2017 ).
- Milbury CA , LiJ , MakrigiorgosGM . Ice-COLD-PCR enables rapid amplification and robust enrichment for low-abundance unknown DNA mutations . Nucleic Acids Res.39 ( 1 ), e2 ( 2011 ).
- How-Kit A , LebbeC , BousardAet al. Ultrasensitive detection and identification of BRAF V600 mutations in fresh frozen, FFPE, and plasma samples of melanoma patients by E-ice-COLD-PCR . Anal. Bioanal. Chem.406 ( 22 ), 5513 – 5520 ( 2014 ).
- Eckhardt F , LewinJ , CorteseRet al. DNA methylation profiling of human chromosomes 6, 20 and 22 . Nat. Genet.38 ( 12 ), 1378 – 1385 ( 2006 ).
- Zhang K , LinG , HanY , XieJ , LiJ . Circulating unmethylated insulin DNA as a potential non-invasive biomarker of β cell death in Type 1 diabetes: a review and future prospect . Clin. Epigenetics9 , 44 ( 2017 ).
- Sefrioui D , MaugerF , LeClereLet al. Comparison of the quantification of KRAS mutations by digital PCR and E-ice-COLD-PCR in circulating-cell-free DNA from metastatic colorectal cancer patients . Clin. Chim. Acta465 , 1 – 4 ( 2017 ).
- Dominguez PL , KolodneyMS . Wild-type blocking polymerase chain reaction for detection of single nucleotide minority mutations from clinical specimens . Oncogene24 ( 45 ), 6830 – 6834 ( 2005 ).
- Kristensen LS , MikeskaT , KrypuyM , DobrovicA . Sensitive melting analysis after real time-methylation specific PCR (SMART-MSP): high-throughput and probe-free quantitative DNA methylation detection . Nucleic Acids Res.36 ( 7 ), e42 ( 2008 ).
- Eads CA , DanenbergKD , KawakamiKet al. MethyLight: a high-throughput assay to measure DNA methylation . Nucleic Acids Res.28 ( 8 ), e32 ( 2000 ).
- Vinarskaja A , GoeringW , IngenwerthM , SchulzWA . ID4 is frequently downregulated and partially hypermethylated in prostate cancer . World J. Urol.30 ( 3 ), 319 – 325 ( 2012 ).
- BLUEPRINT consortium . Quantitative comparison of DNA methylation assays for biomarker development and clinical applications . Nat. Biotechnol.34 ( 7 ), 726 – 737 ( 2016 ).
- Cottrell SE , DistlerJ , GoodmanNSet al. A real-time PCR assay for DNA-methylation using methylation-specific blockers . Nucleic Acids Res.32 ( 1 ), e10 ( 2004 ).
- Molnar B , TothK , BartakBK , TulassayZ . Plasma methylated septin 9: a colorectal cancer screening marker . Expert Rev. Mol. Diagn.15 ( 2 ), 171 – 184 ( 2015 ).
- de Vos T , TetznerR , ModelFet al. Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer . Clin. Chem.55 ( 7 ), 1337 – 1346 ( 2009 ).
- Church TR , WandellM , Lofton-DayCet al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer . Gut63 ( 2 ), 317 – 325 ( 2014 ).
- Tost J . The clinical potential of enhanced-ice-COLD-PCR . Expert Rev. Mol. Diagn.16 ( 3 ), 265 – 268 ( 2016 ).
- Qiu J , PengB , TangYet al. CpG methylation signature predicts recurrence in early-stage hepatocellular carcinoma: results from a multicenter study . J. Clin. Oncol.35 ( 7 ), 734 – 742 ( 2017 ).
- Sandoval J , Mendez-GonzalezJ , NadalEet al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer . J. Clin. Oncol.31 ( 32 ), 4140 – 4147 ( 2013 ).
- Geybels MS , WrightJL , BibikovaMet al. Epigenetic signature of Gleason score and prostate cancer recurrence after radical prostatectomy . Clin. Epigenetics8 , 97 ( 2016 ).
- Rasmussen SL , KrarupHB , SunesenKGet al. Hypermethylated DNA, a circulating biomarker for colorectal cancer detection . PLoS ONE12 ( 7 ), e0180809 ( 2017 ).
- Castellanos-Rizaldos E , MilburyCA , MakrigiorgosGM . Enrichment of mutations in multiple DNA sequences using COLD-PCR in emulsion . PLoS ONE7 ( 12 ), e51362 ( 2012 ).
- Castellanos-Rizaldos E , LiuP , MilburyCAet al. Temperature-tolerant COLD-PCR reduces temperature stringency and enables robust mutation enrichment . Clin. Chem.58 ( 7 ), 1130 – 1138 ( 2012 ).
- Liu Y , SongC , LadasI , Fitarelli-KiehlM , MakrigiorgosGM . Methylation-sensitive enrichment of minor DNA alleles using a double-strand DNA-specific nuclease . Nucleic Acids Res.45 ( 6 ), e39 ( 2017 ).