
Abstract
Cancer epigenetic mechanisms support the acquisition of hallmark characteristics during oncogenesis. EZH2 – an important histone methyltransferase that writes histone H3 lysine 27 trimethylation marks – is known to be dysregulated in cancer cells. However, the interactions between EZH2 and miRNAs that form a complex network of cross-talk and reciprocal regulation that enable cancer cells to acquire hallmark characteristics have been relatively poorly appreciated. The specific functions of EZH2 appear to be regulated by a vast array of miRNAs, which direct EZH2 toward regulation over the development of specific hallmark characteristics. This review discusses recent advances in the understanding of EZH2, focusing on its collaboration with miRNAs to orchestrate oncogenesis. These epigenetic processes promote the evasion of apoptosis/cell cycle arrest, cellular dedifferentiation and the establishment of a tumor microenvironment that facilitates local cancer cell invasion, anti-cancer drug resistance and evasion of the immune response.
Plain language summary
Cancer epigenetics involves cellular processes that ensure that gene expression changes that enable cancer cells to outcompete their neighboring normal tissues are passed on from one cancer cell to the next. One key epigenetic player is called EZH2, which is an enzyme that transfers methyl (CH3) groups from donor molecules to the histone proteins around which DNA is coiled. The transfer of methyl groups to histones – a process called histone methylation – silences the production of proteins from the genes coded in DNA. To achieve this goal, EZH2 is directed to affect specific genes through interactions with other molecules consisting of short sequences of RNA (a molecule similar in structure to DNA). The specific RNA molecules in question are called miRNAs or non-coding RNAs. These EZH2/miRNA interactions form a complex web of circuits that enables cancer cells to gain characteristics that give them a competitive advantage over their normal tissue neighbors. This review discusses recent advances in the understanding of the role of EZH2 in cancer formation, focusing on the interactions it has with miRNAs (and other non-coding RNAs) to organize many different processes linked with cancer formation. These processes include the avoidance of cell death or cell growth arrest, the development of stem cell properties and the formation of an environment around tumor cells that allows them to invade adjacent normal tissue (i.e., metastasize) and avoid being killed by anti-cancer drugs and the immune system.
Cancer is primarily a genetic disease arising through successive genetic mutations acquired over a person’s lifetime [Citation1]. However, the role of epigenetics in cancer development is now well established, with genetic mutations in epigenetic components and epigenetic aberrations in cell signaling pathways playing a major role in reshaping gene expression profiles and driving oncogenesis. The histone methyltransferase EZH2 is a major epigenetic player that reshapes the epigenetic landscape during oncogenesis. Recent research has highlighted that many interactions between EZH2 and RNAs, such as miRNAs and other non-coding RNAs, are important for this process. This review provides an overview of the recent evidence for such complex epigenetic regulatory mechanisms that enable cancer cells to avoid apoptosis and cell cycle checkpoint arrest, undergo dedifferentiation and form a successful cancer cell niche in the tumor microenvironment that promotes local invasion, anti-cancer drug resistance and evasion of the anti-cancer immune response.
DNA methylation & demethylation reactions
The earliest understood epigenetic modification involves the methylation of DNA. 5-methyl cytosine (5mC) was first isolated from nucleic acid samples in 1951 [Citation2]. It is formed from the transfer of a methyl group from the substrate S-adenosyl-L-methionine to the pyrimidine cytosine. The process of DNA methylation is catalyzed by a family of DNA methyltransferases (DNMT1/2/3) that ‘write’ methyl groups into the DNA backbone [Citation3]. In contrast, the family of epigenetic regulators known as ten-eleven translocation proteins (TET1/2/3) ’erase’ methyl groups from the DNA backbone by catalyzing DNA demethylation reactions that initially oxidize 5mC to 5-hydroxymethylcytosine (5hmC) [Citation4]. This step in TET-mediated demethylation requires the cofactors Fe(II) and 2-oxoglutarate (α-ketoglutarate) [Citation3], the latter of which is produced from the oxidative decarboxylation of isocitrate substrates from the citric acid cycle by the enzyme isocitrate dehydrogenase (IDH) [Citation5].
DNA hypermethylation at highly concentrated clusters of CpG dinucleotides, termed CpG islands (CGIs), restricts the binding of transcription factors to gene promoter regions. The consequence of hypermethylation of CGIs is 5mC-dependent silencing of gene expression. Conversely, CGI hypomethylation derepresses gene expression [Citation6], although recent evidence suggests that 5mC modifications at promoter and enhancer elements may also be permissive to transcription in certain contexts [Citation7]. After the oxidation product 5hmC is formed from 5mC, TET enzymes carry out a further conversion step, generating 5-formylcytosine (5fC) or 5-carboxylcytosine (5caC). Subsequently, 5fC and 5caC are excised from DNA in a thymine-DNA glycosylase-dependent manner [Citation8,Citation9].
Histone modifications
Genomes are tightly packaged structures that form a DNA–protein complex called chromatin. Chromatin contains approximately 30 million nucleosomes, basic units comprised of 147 base-pairs of DNA encircling eight histone proteins (two H2A-H2B dimers and one H3-H4 tetramer). Histone modifications are “written” onto the charged NH2 termini (tails) of histones by the GCN5, MYST and p300/CBP families of histone acetyltransferases (HATs) as well as the SET-domain-containing and non-SET-domain-containing lysine-specific and arginine-specific families of histone methyltransferases (HMTs) [Citation10]. In addition to acetylation and methylation, other histone modifications include phosphorylation, ubiquitylation, sumoylation and biotinylation, the effects of which are type- and site-specific. For example, ’activating’ modifications include the acetylation of lysine 27 in histone H3 (H3K27Ac), methylation of lysines 4 and 36 in histone H3 (H3K4me3 and H3K36me3) and demethylation of lysine 9 of histone H3 [Citation11], while ‘silencing’ modifications include the methylation of lysine 9 and 27 in histone H3 (M3K9me3 and H3K27me3) and sumoylation of lysine 59 in histone H4 (H4K59SUMO) [Citation11].
Histone modifications form docking sites in chromatin for the binding of a family of chromatin remodeling proteins called the polycomb group proteins (PcGs) [Citation12]. PcG-target genes often contain both silencing (H3K27me3) and activating (H3K4me3) histone modifications, which confers a poised, ready-to-transcribe state [Citation13]. Once docked, the E3 ligase of polycomb repressive complex 1 (PRC1) ubiquitylates lysine 119 of histone H2A (H2AK119Ub), causing the physical hinderance of RNA polymerase II, while polycomb repressive complex 2 (PRC2) possesses HMT activity that methylates lysine 27 of histone H3 (H3K27me3) [Citation12,Citation14]. The best characterized PRC2 subunit is EZH2, which plays a central role in oncogenesis.
EZH2, miRNAs & oncogenesis
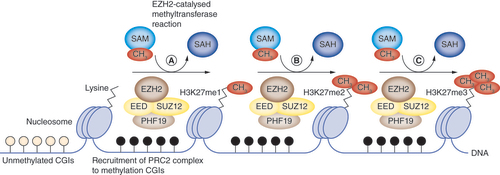
EZH2 is an evolutionarily conserved SET-domain-containing histone methyltransferase. The main epigenetic reactions catalyzed by EZH2 include the transfer of methyl groups from the substrate S-adenosyl-L-methionine to lysine 27 of histone H3, forming H3K27me3 and S-adenosyl-L-homocysteine (). These reactions are facilitated by binding with other core PRC2 complex proteins, EED and SUZ12. The H3K27me3 histone modifications catalyzed by EZH2 are critical in cancer development. Thus, a number of EZH2-targeted cancer therapeutics have been developed, including UNC1999, 3-deazaneplanocin A (DZnep), GSK126, tazemetostat (EPZ-6348), EI1, EPZ005687, GSK343 and DZnep, which inhibit the formation of H3K27me3 histone modifications. Although discussion of these pharmacological EZH2 inhibitors is outside the scope of this review, for a recent comprehensive review of epigenetic cancer therapeutics the reader is directed to Hillyar et al. [Citation15].
Following EZH2 recruitment via PHF19-binding to methylated CpG islands, EZH2-mediated H3K27me3 histone modifications proceed via the sequential transfer of methyl groups from S-adenosyl-L-methionine onto the N-termini of lysine residues of histones in the nucleosome complex, forming (A) H3K27me1, (B) H3K27me2 and finally (C) H3K27me3, as well as S-adenosyl-L-homocysteine.
CGI: CpG island; CH3: Methyl group; PRC2: Polycomb repressive complex 2; SAH: S-adenosyl-L-homocysteine; SAM: S-adenosyl-L-methionine.
EZH2, miRNAs & apoptosis/proliferation
The methyltransferase activity of EZH2 becomes dysregulated during oncogenesis (). In normal cells, EZH2 is regulated by the tumor suppressor BRCA1, mutations of which are associated with an increased risk for the development of breast and ovarian cancer of 54% and 30%, respectively, by age 60 years [Citation16]. In addition to its role in double-strand DNA break (DSB) repair, BRCA1 also controls the epigenetic regulation of gene expression by binding to EZH2 at its non-coding RNA-binding domain [Citation17]. Therefore, BRCA1 deficiency during oncogenesis results in the upregulation of EZH2 methyltransferase activity. This is associated with the downregulation of a tumor suppressive transcription factor, FOXO3, which binds conserved 5′-[AG]TAAA[TC]A-3′ sequences in the promoters of pro-apoptotic genes [Citation18]. In BRCA1-deficient cancer cells, EZH2 upregulation results in the decoration of the FOXO3 promoter with repressive H3K27me3 modifications, which causes FOXO3 downregulation and avoidance of apoptosis [Citation18].
EZH2 is negatively regulated by factors that are often downregulated during oncogenesis, including the tumor suppressor BRCA1 and two miRNAs, miR-101 and miR-124. This results in the upregulation of EZH2 methyltransferase activity and silencing of EZH2-target genes, including FOXO3 (a pro-apoptotic transcription factor) and miR-29b and miR-30d, both of which enhance proliferation when suppressed through the upregulation of EZH2.
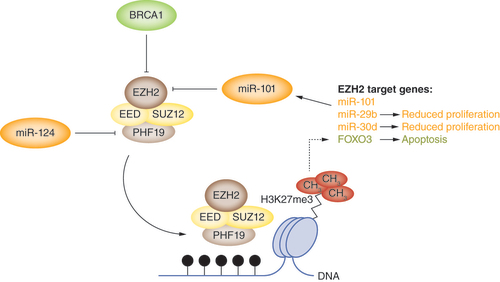
In addition to BRCA1, miRNAs and other non-coding RNAs are an important class of endogenous RNA molecules that interact with EZH2 and alter its control over cellular processes in order to promote oncogenesis. For example, EZH2 has been shown to bind to miR-101 and miR-124 via its RNA-binding domain. In retinoblastoma cells, miR-101 downregulation reduces the binding of miR-101 to the 3′-untranslated region (3′-UTR) of EZH2 mRNA [Citation19,Citation20]. This reduces miRNA-mediated repression of EZH2 expression and allows EZH2 to write H3K27me3 modifications. Indeed, through a negative feedback loop, EZH2 is then able to write H3K27me3 modifications in the miR-101 promoter [Citation20], further suppressing the expression of miR-101. In laryngeal squamous cell carcinoma cells, the upregulation of EZH2 expression following miR-101 downregulation promotes cell proliferation and evasion of apoptosis [Citation21]. In contrast, Lu et al. demonstrated that the upregulation of miR-124 expression in glioma cells results in the downregulation of PHF19, a PcG subunit that is involved in the trimethylation of lysine 36 of histone H3 (H3K36me3) [Citation22]. PHF19 forms a complex with EZH2 in order to facilitate the recruitment of the PRC2 complex to CGI regions.
Yin et al. has also reported that the EZH2 inhibitors DZnep and GSK343 significantly increased the expression of the miRNAs miR-29b and miR-30d in breast cancer cells [Citation23]. In contrast, knockdown of the PRC2 subunits SUZ12 and EED, which form a complex with EZH2, resulted in the overexpression of miR-29b and miR-30d. Results from chromatin immunoprecipitation experiments have indicated that EZH2 directly binds to, and induces H3L27me3 modification in, the promoter regions of these miRNAs. The consequence of miR-29b and miR-30d overexpression was reduced LOX4L expression, which correlated with inhibition of proliferation and metastasis of breast cancer cells in vivo.
EZH2, miRNAs & dysregulation of the cell cycle
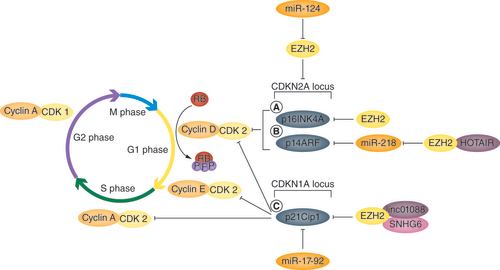
During oncogenesis, EZH2 plays a central role in the dysregulation of multiple cyclin-dependent kinases (CDKs), including p16INK4A (CDKN2A), p14ARF (CDKN2A) and p21Cip1 (CDKN1A). Recently, Takanlu et al. demonstrated that lentiviral expression of miR-124 in multiple myeloma cells caused CDKN2A-dependent G1 phase arrest through transcriptional repression of EZH2 [Citation23]. An overview of the role that EZH2 plays in dysregulation of the cell cycle is shown in .
During oncogenesis, EZH2 upregulation enables cancer cells to avoid cell cycle arrest through the inhibition of (A) p16INK4A, (B) p14ARF and (C) p21Cip1. This is achieved through EZH2-mediated H3K27me3 modifications in the CDKN2A and CDK1A loci. Interactions between EZH2 and the non-coding RNAs HOTAIR, linc01088 and SNHG6 ensure that modifications are directed in such a way that regulation of CDKN1A/2A-specific splice variants is achieved.
P16INK4A
A illustrates that the tumor suppressor p16INK4A – one of three splice variants of the CDKN2A gene – inhibits cyclin-dependent kinase 4, a serine/threonine protein kinase involved in the regulation of G1 to S phase transition through the phosphorylation of retinoblastoma protein [Citation24]. Recently, Lu et al. demonstrated that EZH2 knockdown in ovarian cancer cells upregulates p16INK4A mRNA and protein levels [Citation25]. This leads to reduced cellular proliferation and migration in vitro and suppression of tumor formation in vivo [Citation25]. In cells expressing EZH2, downregulation of p16INK4A is associated with transcriptionally repressive H3K27me3 modifications in the p16INK4A promoter [Citation26]. These histone modifications are catalyzed by EZH2 [Citation27]. Interestingly, mutation of lysine 27 in histone H3 to methionine renders the p16INK4A promoter refractory to EZH2-mediated modifications [Citation26]. Thus, EZH2 overexpression during oncogenesis has been found to enhance gliomagenesis and the proliferation of neurospheres through the repression of p16INK4A.
P14ARF
B illustrates that the tumor suppressor p14ARF is another splice variant of the CDKN2A gene, which inhibits E3 ubiquitin ligase (MDM2)-dependent p53 degradation, leading to p53 stabilization and cell cycle inhibition at the G1 checkpoint [Citation28]. Fu et al. demonstrated that the lncRNA HOTAIR (HOX antisense intergenic RNA) plays a role in the dysregulation of p14ARF by directing EZH2 to repress the tumor suppressive miRNA miR-218 [Citation29]. By binding to PRC2, HOTAIR promotes EZH2-dependent miR-218 promoter methylation. Knockdown of HOTAIR induces G1 phase arrest in vitro and suppresses tumor growth in vivo through derepression of miR-218 expression [Citation29]. Upregulation of the miRNA miR-218 was associated with the activation of p14ARF (and p16INK4A) as well as the post-transcriptional repression of the PRC1 subunit BMI1 in human hepatocellular carcinoma cells [Citation29]. In gastric cancer cells, EZH2 knockdown has been demonstrated to reduce H3K27me3 modifications at the CDKN2A locus and increase the expression of p14ARF (and p16INK4A), resulting in cellular senescence [Citation30]. Thus, changes in EZH2 activity through HOTAIR-dependent and -independent mechanisms enable cancer cells to evade cell cycle arrest through p14ARF-mediated MDM2-dependent degradation of p53.
P21Cip1
C illustrates that the tumor suppressor p21Cip1 (CDKN1A) inhibits cyclin–CDK2/4 complexes, promoting p53-dependent G1 phase arrest [Citation31]. Transcriptionally repressive H3K27me3 modifications in the p21Cip1 promoter are induced by ankylphenol 4-nonylphenol, which increases the expression of EZH2 [Citation32]. Liu et al. demonstrated that EZH2 binds the lncRNA linc01088, which directs transcriptional repression of p21Cip1 [Citation33]. Upregulation of p21Cip1 by EZH2 knockdown or linc01088 knockdown was found to inhibit tumor growth of non-small cell lung cancer in vivo [Citation33]. Upregulation of the snoRNA SNHG6 in gastric cancer cells also inhibits p21Cip1 expression through interaction with EZH2 [Citation34]. In non-small cell lung cancer cells, miR-17-92 causes downregulation of p21Cip1 [Citation35]; however, whether this involves an interaction with EZH2 has yet to be explored. Thus, interactions among EZH2, linc01088 and SNHG6 lead to the repression of p21Cip1 transcription, driving tumor growth.
EZH2, miRNAs & cellular dedifferentiation
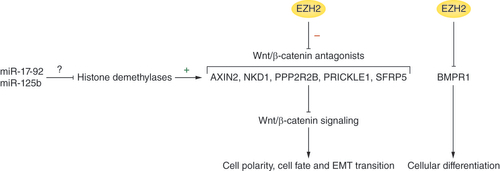
Recent evidence suggests that EZH2 disrupts cellular differentiation pathways to facilitate malignant transformation. The Wnt/β-catenin signaling pathway controls cell polarity and cell fate [Citation36]. Cheng et al. demonstrated that EZH2 represses the Wnt/β-catenin antagonists AXIN2, NKD1, PPP2R2B, PRICKLE1 and SFRP5 through H3K27me3 modifications [Citation37]. Knockdown of EZH2 has been found to reduce the proliferation of hepatocellular carcinoma cells by restoring inhibition of Wnt/β-catenin signaling [Citation37]. In non-small cell lung cancer cells, the miRNA miR-17-92 has been reported to cause an increase in Wnt/β-catenin signaling [Citation38]. Similarly, a study by Nie et al. claimed that inhibition of miR-125b suppressed the proliferation, migration and epithelial–mesenchymal transition (EMT) of triple negative breast cancer cells, through a reduction in Wnt/β-catenin signaling [Citation39]. It has yet to be explored whether these mechanisms involve interactions that allow miR-17-92 and miR-125b to direct EZH2-mediated histone modifications to suppress specific antagonists of the Wnt/β-catenin pathway. However, a study by Jing et al. hints that the mechanism that enables miRNAs to antagonize the effects of EZH2 might involve the suppression of lysine demethylase enzymes that could enhance the activity of antagonists of the Wnt/β-catenin pathway. For example, the negative effects of miR-125b on Wnt/β-catenin signaling are mediated, at least in part, through the suppression of the lysine demethylase KDM4B [Citation40].
In another study, by Lee et al., it was demonstrated that EZH2 represses BMPR1B, a transmembrane serine/threonine kinase involved in cellular differentiation and endochondral bone formation [Citation41]. Therefore, EZH2 overexpression has been found to inhibit astroglial differentiation and promote glioblastoma tumorigenesis [Citation41]. Astroglial differentiation was restored by BMPR1B promoter demethylation [Citation41]. Thus, EZH2 represses the activity of Wnt/β-catenin and BMPR1B signaling, leading to tumorigenesis by promoting cellular dedifferentiation ().
EZH2 inhibits antagonists of Wnt/β-catenin signaling and BMPR1, which promotes cellular dedifferentiation. The pro-oncogenic effects of EZH2 may be enhanced by miRNAs (e.g., miR-17-92 and miR-125b) that inhibit the histone demethylases that erase histone modifications.
EMT: Epithelial–mesenchymal transition.
EZH2, miRNAs & the tumor microenvironment
EZH2 interacts, along with other epigenetic factors such as miRNAs, with the tumor microenvironment. This interplay between intracellular and extracellular signaling impacts extracellular matrix remodeling that facilitates metastatic spread, releases cancer cell exosomes that confer anti-cancer drug resistance and promotes evasion of the anti-cancer immune response ().
Table 1. Effect on the tumor microenvironment of upregulation and downregulation of EZH2 during oncogenesis.
Extracellular matrix remodeling
miRNAs play an important role in regulating EZH2-dependent extracellular matrix (ECM) remodeling. Recently, Zhao et al. reported that the miRNA miR-26a promoted the invasive behavior of hepatocellular carcinoma cells by negatively regulating EZH2 expression. The suppression of EZH2 activity resulted in the upregulation of the activity of the matrix metalloproteinases (MMPs) MMP1, MMP2, MMP9 and MMP10 [Citation42]. These MMPs play a crucial role in ECM remodeling and tumor invasiveness by mediating the digestion of type I, II and III collagens in the ECM, which facilitates the metastatic dissemination of cancer cells [Citation43]. Thus, Zhao et al. have discovered a novel mechanism of ECM remodeling, which involves miR-26a-mediated inhibition of EZH2-dependent suppression of MMP activity. In contrast to other studies where oncogenesis is associated with the upregulation of EZH2 activity, these new insights suggest that EZH2 activity may also be downregulated in order to promote metastatic spread through the invasion of cancer cells into adjacent tissue.
Cancer cell-released exosomes
Recently, Yang et al. demonstrated that the knockdown of EZH2 inhibited chemotherapy-induced upregulation of the miRNAs miR378a-3p and miR-378d, both in breast cancer cells and in cancer cell-released exosomes [Citation44]. The consequences of reducing the release of cancer cell exosomes containing these miRNAs were reduced miRNA-dependent stemness and drug resistance to both doxorubicin and paclitaxel, through interference with the EZH2/STAT pathway, which regulates the expression of these miRNAs. Conversely, the overexpression of EZH2 in mice resulted in an increase in the levels of miR-378a-3p and miR-378d in exosomes extracted from these mice. Treatment of an EZH2-overexpressing tumor xenograft mouse model with the EZH2 inhibitor tazemetostat (Tazverik®), which specifically prevents the methylation of H3K27, resulted in the reversal of chemotherapy-elicited miRNA-containing exosome-induced drug resistance.
Anti-cancer immune response
Epigenetic modifications mediated by EZH2 play an important role in the regulation of the anti-cancer function of immune cells, such as T and B lymphocytes. Recently, Zhao et al. demonstrated that knockdown of EZH2 reduces the expression of PD-L1. PD-L1 is an immune checkpoint immunoglobulin that induces tolerance in anti-tumor T and B cells. PD-L1 downregulation via reduced EZH2 activity resulted in an enhanced anti-tumor immune response [Citation45]. EZH2 downregulation has also been associated, in a review by Shao et al., with increased activity of CD4+ and CD8+ cells in the tumor microenvironment. This translated into a significant reduction in tumor growth due to enhanced production of the cytokines IFNγ, TNF-α and IL-2 [Citation46]. Therefore, the upregulation of EZH2 activity during oncogenesis is likely to enable cancer cells to evade the anti-cancer immune response via mechanisms that induce immune tolerance and reduce the production of anti-cancer cytokines.
Finally, Zhao et al. demonstrated that EZH2 was upregulated in hepatocellular carcinoma compared with surrounding normal tissue. EZH2 overexpression reduced the expression level of the miRNAs miR-144 and miR-451a in hepatocellular carcinoma cells [Citation47]. In these cells, overexpression of EZH2 induced H3K27me3 modifications, which inhibited the expression of miR-144 and miR-451a. However, overexpression of miR-144 resulted in the downregulation of EZH2, which is a target for miR-144 post-translational regulation. The impact of the upregulation of these miRNAs was found to include reduced secretion of macrophage migratory inhibitory factor (MIF) by hepatoma cells. The levels of miR-144 and miR-451a were negatively correlated with the M2-polarization marker CD163 but positively correlated with M1-polarizing marker HLA-DR on infiltrating tumor-associated macrophages (TAMs) in a hepatoma mouse model. This suggested that EZH2 upregulation in cancer cells promotes an M2 phenotype in TAMs that consequently facilitates tumor growth, metastasis, tissue remodeling and immunosuppression.
Conclusion
EZH2, in collaboration with miRNAs and other non-coding RNAs, plays a critical role in cancer epigenetics. The interactions between EZH2 and miRNAs are numerous and complex, creating an elaborate network of cross-talk and reciprocal regulation that, during oncogenesis, provides multiple redundant pathways for cells to acquire the hallmark characteristics of cancer. The specific functions of EZH2 appear to be regulated by a vast array of miRNAs (and other non-coding RNAs), which direct EZH2 toward regulation over the development of specific cancer cell hallmark characteristics during the process of oncogenesis. The evidence suggests that EZH2/miRNA interactions are involved in the evasion of apoptosis and cell cycle checkpoint arrest, cellular dedifferentiation and the establishment of a successful tumor microenvironment that promotes local cancer cell invasion, anti-cancer drug resistance and evasion of the anti-cancer immune response.
Future perspective
This review highlights that a number of unsolved questions exist for further research. These include understanding how specific miRNAs antagonize the effects of EZH2 on Wnt/β-catenin signaling, which regulates cell polarity, cell fate and EMT. Another area that deserves attention involves understanding why in certain contexts EZH2 expression levels are increased (to avoid apoptosis and cell cycle checkpoint arrest), while in other contexts EZH2 expression is decreased (to promote MMP-dependent local invasion of cancer cells). Focusing on these questions will help to improve the understanding of how EZH2 and miRNAs determine the evolution of specific cancer cell clones that occupy different niches within the tumor microenvironment.
The detection of EZH2 and specific miRNAs with which EZH2 interacts may provide a clinical application for the evidence summarized in this review, through identifying the prognostic value of the detection of various epigenetic components in malignant tissue. Indeed, further investigation of cancer cell exosomes released into circulation may also enable the non-invasive detection of prognostic biomarkers. Advances in the understanding of the mechanisms involving EZH2 and miRNA interactions might also lead to clinical trials that investigate a role for novel compounds that either reduce or increase the levels of specific EZH2-interacting miRNAs, or enhance or inhibit their interactions with EZH2, in order to improve the clinical efficacy of EZH2 inhibitor anti-cancer drugs for treating a wide range of malignancies. Thus, targeting EZH2/miRNA interactions may not only serve as a means for understanding the mechanisms underpinning the acquisition of hallmark characteristics during oncogenesis but also provide an opportunity to improve on the early detection of cancer and to effect better cancer cures.
EZH2 is an evolutionarily conserved SET-domain-containing histone methyltransferase that catalyzed the transfer of methyl groups from the substrate S-adenosyl-L-methionine to lysine 27 of histone H3, forming H3K27me3 and S-adenosyl-L-homocysteine.
A number of EZH2-targeted cancer therapeutics have been developed, including UNC1999, 3-deazaneplanocin A (DZnep), GSK126, tazemetostat (EPZ-6348), EI1, EPZ005687, GSK343, and DZnep, which inhibit the formation of H3K27me3 histone modifications.
EZH2, miRNAs & apoptosis/proliferation
EZH2 is negatively regulated by BRCA1 and the miRNAs miR-101 and miR-124, which are frequently dysregulated during oncogenesis.
Dysregulation of EZH2 due to BRCA1 mutation results in the suppression of the pro-apoptotic transcription factor FOXO3, while miR-101 and miR-124 downregulation results in reduced cancer cell proliferation.
EZH2, miRNAs & dysregulation of the cell cycle
During oncogenesis, EZH2 upregulation enables cancer cells to avoid cell cycle arrest through the inhibition of the cyclin-dependent kinase inhibitors p16INK4A, p14ARF and p21Cip1. This is achieved through EZH2-mediated H3K27me3 modifications in the CDKN2A and CDK1A loci.
Interactions between EZH2 and the non-coding RNAs HOTAIR, linc01088 and SNHG6 ensure that modifications are directed in such a way that regulation of CDKN1A/2A-specific splice variants is achieved.
EZH2, miRNAs & cellular dedifferentiation
EZH2 inhibits antagonists of Wnt/β-catenin signaling and BMPR1, which promotes cellular dedifferentiation. The pro-oncogenic effects of EZH2 are enhanced by the miRNAs miR-17-92 and miR-125b, which inhibit the histone demethylases that erase histone modifications.
EZH2, miRNAs & the tumor microenvironment
EZH2 plays an important role in extracellular matrix remodeling by regulating the activity of the matrix metalloproteinases MMP2, MMP9 and MMP10. The miRNA miR-26a negatively regulates EZH2-dependent matrix metalloproteinase activity.
EZH2 upregulation promotes the release of miRNA-containing exosomes, which confer anti-cancer drug resistance.
EZH2 regulates the anti-cancer immune response by inducing T and B cell tolerance and by causing the cancer-supportive M2-polarization of tumor-associated macrophages through the downregulation of the miRNAs miR-144 and miR-451a.
Author contributions
CRT Hillyar drafted, edited and revised the manuscript; SS Kanabar edited and revised the manuscript; KS Rallis edited and revised the manuscript. JS Varghese supervised the work. All authors read and approved the final version of the manuscript.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Pomerantz MM , FreedmanML. The genetics of cancer risk. Cancer J.17(6), 416–422 (2011).
- Wyatt GR . Recognition and estimation of 5-methylcytosine in nucleic acids. Biochem. J.48(5), 581–584 (1951).
- Ludwig AK , ZhangP , CardosoMC. Modifiers and readers of DNA modifications and their impact on genome structure, expression, and stability in disease. Front. Genet.7, 115 (2016).
- Bayraktar G , KreutzMR. The role of activity-dependent DNA demethylation in the adult brain and in neurological disorders. Front. Mol. Neurosci.11, 169 (2018).
- Scourzic L , MoulyE , BernardOA. TET proteins and the control of cytosine demethylation in cancer. Genome Med.7(1), 9 (2015).
- Bird A . DNA methylation patterns and epigenetic memory. Genes Dev.16(1), 6–21 (2002).
- Angeloni A , BogdanovicO. Enhancer DNA methylation: implications for gene regulation. Essays Biochem.63(6), 707–715 (2019).
- Ito S , ShenL , DaiQet al. TET proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science333(6047), 1300–1303 (2011).
- Hahn MA , SzaboPE , PfeiferGP. 5-Hydroxymethylcytosine: a stable or transient DNA modification?Genomics104(5), 314–323 (2014).
- Bannister AJ , KouzaridesT. Regulation of chromatin by histone modifications. Cell Res.21(3), 381–395 (2011).
- Zhao Y , GarciaBA. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol.7(9), a025064 (2015).
- Aranda S , MasG , DiCroce L. Regulation of gene transcription by polycomb proteins. Sci. Adv.1(11), e1500737 (2015).
- Messier TL , BoydJR , GordonJA , SteinJL , LianJB , SteinGS. Oncofetal epigenetic bivalency in breast cancer cells: H3K4 and H3K27 tri-methylation as a biomarker for phenotypic plasticity. J. Cell. Physiol.231(11), 2474–2481 (2016).
- Gan L , YangY , LiQ , FengY , LiuT , GuoW. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark. Res.6, 10 (2018).
- Hillyar C , RallisKS , VargheseJ. Advances in epigenetic cancer therapeutics. Cureus12(11), e11725 (2020).
- Easton DF , FordD , BishopDT. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am. J. Hum. Genet.56(1), 265–271 (1995).
- Wang L , ZengX , ChenSet al. BRCA1 is a negative modulator of the PRC2 complex. EMBO J.32(11), 1584–1597 (2013).
- Gong C , YaoS , GomesARet al. BRCA1 positively regulates FOXO3 expression by restricting FOXO3 gene methylation and epigenetic silencing through targeting EZH2 in breast cancer. Oncogenesis5, e214 (2016).
- Jin Q , HeW , ChenL , YangY , ShiK , YouZ. MicroRNA-101-3p inhibits proliferation in retinoblastoma cells by targeting EZH2 and HDAC9. Exp. Ther. Med.16(3), 1663–1670 (2018).
- Jiang M , XuB , LiXet al. O-GlcNAcylation promotes colorectal cancer metastasis via the miR-101-O-GlcNAc/EZH2 regulatory feedback circuit. Oncogene38(3), 301–316 (2019).
- Chen L , JiaJ , ZangY , LiJ , WanB. MicroRNA-101 regulates autophagy, proliferation and apoptosis via targeting EZH2 in laryngeal squamous cell carcinoma. Neoplasma66(4), 507–515 (2019).
- Lu J , JiH , TangH , XuZ. MicroRNA-124a suppresses PHF19 over-expression, EZH2 hyper-activation, and aberrant cell proliferation in human glioma. Biochem. Biophys. Res. Commun.503(3), 1610–1617 (2018).
- Sabour Takanlu J , AghaieFard A , MohammdiS , HosseiniRad SMA , AbrounS , NikbakhtM. Indirect tumor inhibitory effects of microRNA-124 through targeting EZH2 in the multiple myeloma cell line. Cell J.22(1), 23–29 (2020).
- Peurala E , KoivunenP , HaapasaariKM , BloiguR , Jukkola-VuorinenA. The prognostic significance and value of cyclin D1, CDK4 and p16 in human breast cancer. Breast Cancer Res.15(1), R5 (2013).
- Lu F , XuH , WangQ , LiM , MengJ , KuangY. Inhibition of enhancer of zeste homolog 2 increases the expression of p16 and suppresses the proliferation and migration of ovarian carcinoma cells in vitro and in vivo. Oncol. Lett.15(3), 3233–3239 (2018).
- Cordero FJ , HuangZ , GrenierCet al. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol. Cancer Res.15(9), 1243–1254 (2017).
- Vire E , BrennerC , DeplusRet al. The polycomb group protein EZH2 directly controls DNA methylation. Nature439(7078), 871–874 (2006).
- Ichimura K , BolinMB , GoikeHM , SchmidtEE , MoshrefA , CollinsVP. Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res.60(2), 417–424 (2000).
- Fu WM , ZhuX , WangWMet al. HOTAIR mediates hepatocarcinogenesis through suppressing miRNA-218 expression and activating P14 and P16 signaling. J. Hepatol.63(4), 886–895 (2015).
- Jie B , WeilongC , MingCet al. Enhancer of zeste homolog 2 depletion induces cellular senescence via histone demethylation along the INK4/ARF locus. Int. J. Biochem. Cell Biol.65, 104–112 (2015).
- Martin A , OdajimaJ , HuntSLet al. CDK2 is dispensable for cell cycle inhibition and tumor suppression mediated by p27(Kip1) and p21(Cip1). Cancer Cell7(6), 591–598 (2005).
- Ghosh K , ChatterjeeB , MaheswariU , AthifaM , KanadeSR. 4-Nonylphenol-enhanced EZH2 and RNF2 expression, H3K27me3 and H2AK119ub1 marks resulting in silencing of p21(CDKN1A) in vitro. Epigenomics11(8), 899–916 (2019).
- Liu JQ , FengYH , ZengS , ZhongMZ. linc01088 promotes cell proliferation by scaffolding EZH2 and repressing p21 in human non-small cell lung cancer. Life Sci.241, 117134 (2020).
- Li Y , LiD , ZhaoMet al. Long noncoding RNA SNHG6 regulates p21 expression via activation of the JNK pathway and regulation of EZH2 in gastric cancer cells. Life Sci.208, 295–304 (2018).
- Wong P , IwasakiM , SomervailleTCet al. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res.70(9), 3833–3842 (2010).
- MacDonald BT , TamaiK , HeX. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell17(1), 9–26 (2009).
- Cheng AS , LauSS , ChenYet al. EZH2-mediated concordant repression of Wnt antagonists promotes beta-catenin-dependent hepatocarcinogenesis. Cancer Res.71(11), 4028–4039 (2011).
- Guinot A , Oeztuerk-WinderF , VenturaJJ. miR-17-92/p38alpha dysregulation enhances Wnt signaling and selects Lgr6+ cancer stem-like cells during lung adenocarcinoma progression. Cancer Res.76(13), 4012–4022 (2016).
- Nie J , JiangHC , ZhouYCet al. miR-125b regulates the proliferation and metastasis of triple negative breast cancer cells via the Wnt/beta-catenin pathway and EMT. Biosci. Biotechnol. Biochem.83(6), 1062–1071 (2019).
- Jing JC , FengZ , ChenZHet al. KDM4B promotes gastric cancer metastasis by regulating miR-125b-mediated activation of Wnt signaling. J. Cell. Biochem. doi:10.1002/jcb.28065 (2018).
- Lee J , SonMJ , WoolardKet al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell13(1), 69–80 (2008).
- Zhao WT , LinXL , LiuYet al. miR-26a promotes hepatocellular carcinoma invasion and metastasis by inhibiting PTEN and inhibits cell growth by repressing EZH2. Lab. Invest.99(10), 1484–1500 (2019).
- Deryugina EI , QuigleyJP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev.25(1), 9–34 (2006).
- Yang Q , ZhaoS , ShiZet al. Chemotherapy-elicited exosomal miR-378a-3p and miR-378d promote breast cancer stemness and chemoresistance via the activation of EZH2/STAT3 signaling. J. Exp. Clin. Cancer Res.40(1), 120 (2021).
- Zhao Y , WangXX , WuWet al. EZH2 regulates PD-L1 expression via HIF-1alpha in non-small cell lung cancer cells. Biochem. Biophys. Res. Commun.517(2), 201–209 (2019).
- Shao FF , ChenBJ , WuGQ. The functions of EZH2 in immune cells: principles for novel immunotherapies. J. Leukoc. Biol.110(1), 77–87 (2021).
- Zhao J , LiH , ZhaoSet al. Epigenetic silencing of miR-144/451a cluster contributes to HCC progression via paracrine HGF/MIF-mediated TAM remodeling. Mol. Cancer20(1), 46 (2021).