
Abstract
Aims: To investigate if DNA methylation pattern assay could be a new approach to identifying the primary tumor cell origin of the recurrent hepatocellular carcinoma (HCC). Materials & Methods: Methylation of 24 genes and expression of 22 cancer stem cell (CSC) biomarkers were quantitatively measured in 10 paired primary and recurrent HCC specimens. The HBV viral–host junctions were determined in six pairs of them with HBV infection. Results: Similar DNA methylation patterns were observed among nine of ten pairs of primary and recurrent tumors. In five of six paired specimens with HBV infection, exactly same HBV DNA integrations were identified in each paired tumors. The expression of seven CSC biomarkers increased significantly in either primary or recurrent tumor tissues. Conclusion: Recurrent HCCs mostly originate from their primary tumors. Assay of DNA methylation patterns could provide a new approach to determining the origin of recurrent HCC.
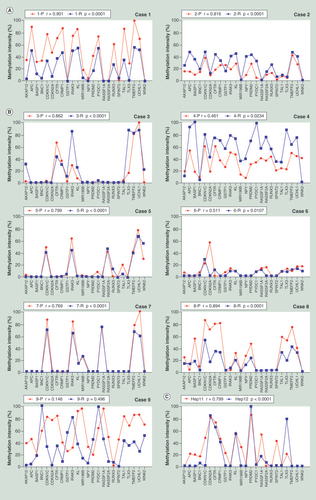
(A) The methylation patterns between the primary and recurrent tumors of Cases 1 and 2, who were HBV negative. (B) The methylation patterns between the primary and recurrent tumors of Cases 3 to 9, who were HBV infection related. (C) The methylation pattern between Hep-11 and Hep-12 cell lines. The Spearman’s rank correlation coefficient test was used to analyze the correlations, p < 0.05 was considered significant.
P: Primary; R: Recurrent.
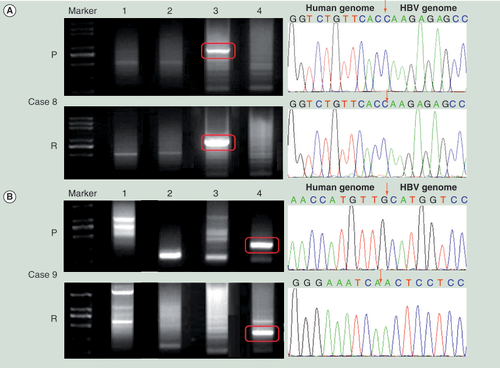
(A) The HBV DNA integration in Case 8. (B) The HBV DNA integration in Case 9. Left panel shows the Alu-PCR results detected by agarose gel electrophoresis separation. Lanes 1–4 represent different PCR results using different Alu-PCR primers. Lane 1: HBV X gene primer as forward and Alu (+) primer as reversed; Lane 2: HBV preC/C gene primer as forward and Alu (+) primer as reversed; Lane 3: HBV X gene primer as forward and Alu (−) primer as reversed; Lane 4: HBV preC/C gene primer as forward and Alu (−) primer as reversed. The right panel shows the sequencing results of the circled bands in the left panel. The arrows indicates the location of HBV–host genome junctions.
P: Primary hepatocellular carcinoma tumor; R: Recurrent hepatocellular carcinoma tumor.
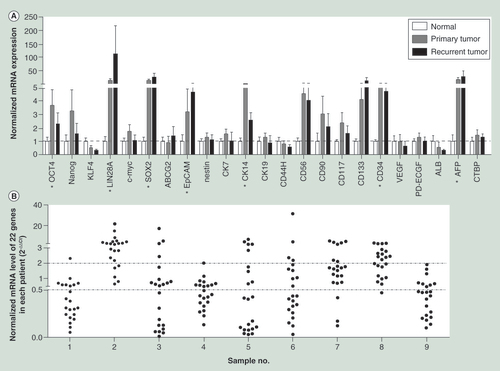
(A) The mRNA expression levels of the 22 cancer stem cell biomarkers in primary and recurrent hepatocellular carcinoma tissues normalized by that in normal liver tissues. The housekeeping gene, CTBP, was also detected as a quality control. (B) The normalized mRNA expression levels of 22 cancer stem cell biomarkers in recurrent tumors compared with that in the primary.
*indicates that the gene’s expression level of either the primary tumor or the recurrent was significantly higher than that of normal liver tissues. The two-tailed unpaired t-test was used to analyze the difference of expression levels in different groups.
p < 0.05 was considered significant.
Hepatocellular carcinoma (HCC) is globally one of the most common and aggressive human malignancies. Despite some progress in treatment, hepatectomy remains the most effective and practical treatment for HCC patients [Citation1–5]. However, up to 70% of patients develop recurrences within 2 years of a hepatectomy [Citation6]. Following such recurrence, with current treatments, the 5-year survival rate is extremely poor [Citation7]. This means that there is a clear imperative to achieve a better understanding of the mechanisms relevant to the recurrent properties of HCC as this will be pivotal to the development of new therapeutic strategies.
The cancer stem cell (CSC) hypothesis indicates that a primary tumor is initiated and maintained by a small population of cancer cells with stem cell-like characteristics [Citation8]. Indeed, CSCs have been identified in a variety of human solid tumors, including HCC [Citation9,Citation10]. More importantly, CSCs shed from the primary tumor have been found to contribute to tumor recurrence [Citation11]. Consistent with this finding, circulating CSCs released from the primary tumor before surgery have been considered as a risk factor that might account for the high incidence of early recurrence in HCC [Citation12]. However, more experimental evidence is needed to support this hypothesis. The key to obtaining such evidence is the development of methodologies to confirm clonal linkages between recurrent and primary tumors.
The methods currently used to determine the clonal origin of a recurrent HCC include the detection of Hepatitis B virus (HBV) integration events, Southern blot analysis, loss of heterozygosity (LOH) assay, comparative genomic hybridization (CGH) and p53 mutant pattern assay, all of which reflect the genetic characteristics of HCC nodules [Citation13–15]. Among these, the pattern of HBV integration has been considered to be the gold standard for determining the clonal origin of tumors because the viral–host junctions created by HBV integration events are unique in different HBV-infected tissues. Consequently, the analysis of the detail of HBV integration events can be used to determinate the clonal origin of multiple HCC nodules, which is helpful particularly because of the levels of uncertainty associated with other molecular methods. It does of course have the disadvantage that it is not applicable for HCCs that are not associated with HBV infection. Similar to point mutations and insertion–deletion mutations, the methylation pattern of CpG islands is also a heritable epigenetic alteration. It has been shown that many genes can acquire DNA methylation in a cancer-specific manner and deregulated DNA methylation landscapes are ubiquitous in human cancers. Despite this, it is still not clear whether the methylation pattern in a recurrent HCC remains as the same as that seen in the primary HCC of the patient concerned. Therefore, the primary aim of this study was to define the relationship of methylation patterns found in paired primary and recurrent HCC tissues with the objective of providing important epigenetic evidence that is diagnostic of the clonal origin of recurrent HCC tumors.
Materials & methods
Primary HCC tissue samples & cell lines
Specimens of primary HCC and the paired recurrent tumor tissues (n = 18) were obtained from nine HCC patients who underwent routine curative surgery at the Henan Cancer Hospital in Zhengzhou, the capital of Henan Province in China, between 2008 and 2013. The tissues were immediately frozen in liquid nitrogen and stored at -80°C. All the patients were histologically diagnosed as HCC. Serum HBsAg was identified in seven of the nine patients before the first curative surgery. The clinico-pathological characteristics of the patients are summarized in . None of the patients received radiotherapy or chemotherapy before the resection surgery, but did receive chemotherapy post-surgery. The primary and recurrent cell lines Hep-11 and Hep-12, established previously from primary and recurrent cancers taken from the same HCC patient as previously described [Citation16], were kindly gifted by Z Zhang.
This study was approved by the institute ethics committee of Peking University Health Science Center, and informed consent was obtained from all patients and donors before the start of the study.
DNA extraction & gene CGI methylation assay
Genomic DNA was isolated from frozen liver tissues using the DNeasy Blood & Tissue Kit (Qiagen Cat No. 69504, USA) and was quantified using a NanoDrop 2000 (NanoDrop, Wilmington, USA). Methylation sensitive restriction enzyme based quantitative PCR assay (MSRE-qPCR) was carried out as described previously [Citation17]. In brief, 1.3 μg DNA from each sample was divided into two equal aliquots after being diluted in NEB CutSmart Buffer. The first aliquot served as a control and only water was added while the second aliquot was digested with 100U HhaI (NEB, Inc. USA) in a final reaction volume of 60 μl. Digestion was at 37°C for 6 h and was terminated by heating the samples to 95°C for 10 min. The sequences targeted in the PCR were rich in CpG dinucleotides and contained at least two HhaI digestion sites between the primers used. Details of the primers used in MSRE-qPCR are listed in Supplementary Table 1 (see online: www.tandfonline.com/doi/full/10.2217/EPI.15.23). The amplification protocol consisted of a first denatured step at 95°C for 10 min, followed by 45 cycles of denaturation at 95°C for 10 s, with annealing and extension both being at 74°C for 60 s in total. Comparison of the Ct values obtained for the control and the methylation-sensitive enzyme digested sample indicated the amount of template DNA that remained after HhaI digestion. The methylation intensity was calculated by using the following equation:Equation 1
Identification of viral–host junctions
Alu-PCR was performed to amplify the viral–host junctions as previously described [Citation18]. In brief, HBV specific primers located either in the HBX gene for the forward and the preC/C gene for the reverse primer were used in combination with human Alu sequences specific primers, respectively, to amplify viral–host junctions. The PCR products were gel purified, subcloned into the TA cloning vector (Genstar, Beijing, China) and sequenced. The viral–host sequences were analyzed by using the NCBI Blast tool and UCSC database hg38 to identify viral genome sequences and to map the integration sites in the human genome.
RNA isolation & the gene expression analysis
Total RNA was prepared following extraction with TRIZOL reagent (Life Technologies, Gaithersburg, MD, USA) in accordance with the manufacturer’s instructions. Real time quantitative PCR was performed to measure the expression levels of 22 CSC biomarkers [OCT4, Nanog, Kruppel-likefactor 4 (KLF4), lin-28 homolog A (LIN28A), c-myc, sex determining region Y-box 2 (SOX2), albumin (ALB), AFP, ATP-binding cassette sub-family G member 2 (ABCG2), epithelial cell adhesion molecule (EpCAM), Nestin, cytokeratin (CK) 7, CK14, CK19, CD44H, CD56, CD90, CD117, CD133, CD34, vascular endothelial growth factor (VEGF), endothelial cell growth factors (PD-ECGF)] using Roche lightcycle 480 sequence detection system (Roche, Mannheim, Germany) according to the manufacturer’s instructions. Primers of OCT4, Nanog, KLF4, LIN28A, c-myc, SOX2, ALB and AFP are shown in Supplementary Table 2 and the others were as described in previous reports [Citation17,Citation19]. The amplification protocol consisted of a first denaturation step at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 10 s, annealing at 60°C for 30 s and extension at 72°C for 30 s. The expression level of each gene was determined using the comparative Ct method (2-△Ct) after normalization to beta-actin.
Statistical analyses
The differences of expression levels between groups were analyzed by the two-tailed paired or unpaired t-test. The correlation between the primary and recurrent HCC methylation status was tested using the Spearman’s rank correlation coefficient test. All of the statistical analyses were performed using SAS 9.1. In all cases, a p value of less than 0.05 was considered significant.
Results
Pathological features of paired primary & recurrent HCC tissues
Among the nine HCC patients whose paired samples were used, the primary tumors of Cases 1–7 were all monocentric, that is there was only one nodule in the primary HCC, while for Cases 8 and 9 the primary tumors were double-centric. The recurrent nodules in all Cases were found in the same or a nearby liver segment to that in which the original primary tumor occurred. The patients’ serum AFP concentrations immediately before the second hepatectomy were almost the same as that at the diagnosis of primary HCC diagnosis ().
Determination of the clonal origin by methylation pattern in paired primary & recurrent HCC cell lines & tumor tissues
To detect the DNA CpG island (CGI) methylation patterns of primary and recurrent HCC, a total of 24 genes were used, including A kinase anchor protein 12 (AKAP12), adenomatosis polyposis coli (APC), brain abundant membrane attached signal protein 1 (BASP1), basonuclin 1 (BNC1), cyclin-dependent kinase inhibitor (CDKN) 1C, CDKN2A, cystic fibrosis transmembrane conductance regulator (CFTR), collapsin response mediator protein 1 (CRMP1), glutathione S-transferase pi 1 (GSTP1), interleukin-1 receptor-associated kinase 3 (IRAK3), klotho (KL), microRNA 196b (MIR196B), neuropeptide Y (NPY), PR domain containing 2 (PRDM2), pyrin domain containing 1 (PYDC1), Ras association domain family member (RASSF) 1A, RASSF2A, runt-related transcription factor 3 (RUNX3), serine peptidase inhibitor Kunitz type 2 (SPINT2), T-cell acute lymphocytic leukemia 1 (TAL1), T-cell leukemia homeobox 3 (TLX3), transmembrane protein with EGF-like and two follistatin-like domains 2 (TMEFF2), ubiquitin carboxyl-terminal esterase L1 (UCHL1), WNK lysine deficient protein kinase 2 (WNK2). The choice of those genes was based on their high methylation frequencies and specificities in HCC according to previous reports both from others and our own laboratory findings [Citation17,Citation20–29]. The methylation status of these 24 genes in paired primary HCC and corresponding recurrent HCC tumor specimens was quantified by MSRE-qPCR. The methylation patterns were found to be similar between each paired primary and recurrent tissues for patients with monocentric primary HCC, and of these, Cases 1 and 2 were HBV negative () and the others were HBV infection related (). In the two paired cases with multicentral primary HCC, one showed a similar methylation pattern (Case 8) between the primary and the recurrent tumors, while the other (Case 9) showed a totally different methylation pattern. The methylation patterns in primary HCC cell line Hep-11 and its corresponding recurrent cell line Hep-12 were examined next. Consistent with the results obtained for the bulk of the tumor tissue samples, two similar and statistically related (p < 0.0001) methylation patterns were identified in these two cell lines ().
The practicability of using the CGI methylation assay to identify the clonal origin of tumor samples was evaluated next. First, the tumor methylation specificities of these genes were verified in a panel of 20 malignancy-free liver tissues which confirmed either no or very low methylation frequencies in all cases (Supplementary Figure 1A and B). Next, several tissue specimens were randomly picked up from the study cohort and repeat tests in replicate tumor tissues were carried out at different times. These repeat assays all showed the same methylation patterns as those obtained on initial screening, proving the reproducibility and precision of the CGI assay used (Supplementary Figure 1C). Finally, to further validate the specificity of this methylation-based assay, a comparison of the methylation patterns between randomly chosen pairs of tumor samples was undertaken. As expected, the methylation patterns from different patients were totally different (Supplementary Figure 1D).
Determination of the clonal origin by HBV integration pattern in paired primary & recurrent HCC cell lines & tumor tissues
The same clonal origin for the HCC cell lines Hep-11 and Hep-12 has been confirmed previously by the pattern of HBV DNA integration [Citation30]. To confirm that the similarity of methylation pattern between the primary and the recurrent HCC tumors indicated that they had the same clonal origin, the pattern of HBV DNA integration into the host cellular genome in all seven paired tumor tissue specimens derived from HCC patients with HBV infection was examined using an Alu-PCR assay. As shown in , the viral–host genome junction sequences were successfully determined in paired tissues from six cases. In Cases 3, 4, 6, 7 and 8 all of which had similar methylation patterns in paired primary and recurrent tumors, the agarose gel electrophoresis showed the same pattern of amplified DNA band(s) in the Alu-PCR assay for recurrent and primary tumors (Case 8 shown as an example in ). By contrast, in Case 9 which showed different methylation patterns in paired primary and recurrent tumors, the HBV integration pattern in the recurrent tumor was totally different from that found in primary tumor (). Taken together, these analyses using the accepted gold standard of HBV integration pattern served to validate the proposal that an CGI methylation assay can be used as an alternative approach to determining whether the primary and recurrent HCC share the same clonal origin.
The CGI methylation intensity of some genes varied between the primary & the recurrent tumors
Although the methylation patterns of the 24 genes subjected to CGI methylation analysis were similar in paired primary and recurrent HCC tumors, the methylation levels of some single genes were noticeably different between these tissues. Therefore, to optimize the DNA methylation assay, each gene’s methylation intensities between the primary and the recurrent tumor specimens amongst the eight cases with the same origin (Cases 1–8) was examined. As shown in , methylation levels of 15 of the genes being analyzed showed a statistical association between methylation intensities seen for the primary and its recurrent tumor. Of these, BASP1, TLX3 and SPINT2 showed the highest statistical association. By contrast, no correlations of AKAP12, CDKN2A and GSTP1 were found between the primary and recurrent tumors. As for BNC1, CRMP1, KL, MIR196B, PYDC1 and WNK2, an approximate correlation was apparent, but it did not reach statistical significance perhaps owing to the relative small sample size.
As 15 of the genes subjected to CGI methylation analysis exhibited constant methylation intensities between paired primary and recurrent HCC, it was worthwhile to test if using just these 15 genes allowed determination of clonal origin in paired primary and recurrent HCCs. As shown in , the correlation between the primary tumor and the recurrent specimens reached statistical significance for Cases 1–8, while for Case 9 and two randomly chosen tumor specimens showed no correlation.
The expression of CSC biomarkers increased both in the primary & recurrent HCC tissues
It has been assumed that CSC expression levels in the primary tumor contribute to cancer recurrence [Citation11]. In a previous report, the expression of HCC CSC biomarkers, such as AFP, ALB, CD117,CD56, Nanog, OCT4 and ABCG2 were found to be increased in the recurrent HCC cell line Hep-12 compared with the primary Hep-11 [Citation30]. To investigate whether the expression of HCC CSC biomarkers also increased in the recurrent HCC tumors used in the present study, the mRNA expression levels of 22 CSC biomarkers in primary and recurrent HCC tumors were assayed by real time qPCR. As shown in , compared with the expression level in normal liver tissues, the mRNA levels of 7 (OCT4, LIN28A, SOX2, EpCAM, CK14, CD34 and AFP) out of the 22 CSC biomarkers were statistically higher in either primary or recurrent tumor tissues. However, none of the 22 CSC biomarkers showed a significant difference in expression between the primary and recurrent HCC tumors. To further compare the expression profile of these 22 CSC biomarkers in each patient, the relative expression levels of these genes in recurrent tumor tissue (normalized by that in the paired primary HCC tissue) were analyzed. As shown in , among the nine cases, increased expression of multiple CSC biomarkers was only seen in three cases (Cases 2, 7 and 8). By contrast, the expression of these tested CSC biomarkers in the other six cases showed a tendency to decrease. Taken together, these results suggested that the expression of some CSC biomarkers showed increased expression in either primary or recurrent HCC tumors and that the expression profiles of these 22 CSC biomarkers varied in different patients.
Discussion
That human cancer cells exhibit global DNA hypomethylation and certain promoter hypermethylation of tumor suppressor genes (TSGs), is a well-known marker for human malignancies. In this study, recurrent HCC tumors were also shown to exhibit hypermethylation of TSGs, furthermore their methylation pattern was associated with that found in the primary HCC. Such results first demonstrated that the constancy of methylation status can be used as a reliable biomarker for determining whether the primary and recurrent HCC share the same clonal origin.
HCC progression has been thought to be driven by CSCs through their capacity for self-renewal, production of heterogeneous lineages, resistance to chemotherapy and limitlessly division [Citation31]. Recently, CSCs shedding from the primary tumor to surrounding liver tissues and into the circulation before surgery have been linked to the recurrence of HCC [Citation11,Citation12]. However, there has not been enough evidence to support the proposition that the recurrent tumor shared clonality with the primary tumor. In this study for all seven monocentric primary HCC cases, the CGI methylation pattern in the recurrent HCC tumors remained the same as the primary tumors from that patient. These findings indicated that in addition to their well-known genetic similarity, recurrent HCC tumors might have the same epigenetic characteristics as the primary tumor. This study therefore provides a powerful evidence to support the CSC hypothesis. Interestingly, although the methylation pattern remained the same, the methylation intensity of some of the tested genes varied between the recurrent and the primary tumors, suggesting that during recurrent tumor development, the self-renewal division of CSCs in the primary HCC might be asymmetrical, which may also provide an explanation for the observed heterogeneity of HCC.
CSC biomarkers exhibit an increased expression in HCC, which has been related to poor prognosis [Citation19,Citation32–34]. Consistent with these findings, seven of the 22 CSC biomarkers analyzed in this study were upregulated in either the primary or recurrent HCC as compared with the levels seen in normal liver tissue. However, no differences in expression were seen for these CSC biomarkers between the paired primary and recurrent HCC tumors. As the stem cell phenotype was only compared with normal liver tissue and not with other HCCs that have not displayed potency to recur, whether the recurrent tumor exhibited more CSC properties could not be determined in this study. However, as CSCs comprise only a minor subpopulation of malignant cells in solid tumors, measurement of mRNA levels for specific genes in total HCC tissues may not faithfully reflect their real expression levels in CSCs. Consequently, to provide additional evidence in support of the CSC properties of the recurrent tumor, the establishment of more primary culture model-systems made from the same patient’s primary and recurrent HCC tissues are needed for characterizing the precise nature of the cells related to HCC recurrence. Interestingly, the upregulated or downregulated tendency of the 22 CSC biomarkers in recurrent tumors was found to vary in different patients. This is consistent with asymmetrical self-renewal division of CSCs. In addition, the majority of the HCC patients recruited in this study are infected with HBV, which itself results in more complicated and heterogenous tumor characteristics being seen. To date, only two pioneering reports have explored the potential involvement of CSC properties in HBV related HCC and the over expression of EpCAM in HCC has also been reported to be linked to HBV infection [Citation35,Citation36]. Thus, further studies are needed to investigate the potential correlation between HBV infection and the properties of CSCs at both the cellular and molecular levels.
In this study the recurrent tumors were always found in the same or nearby liver segment. A possible explanation to this is that, hepatitis and systemic inflammatory response induced by major hemorrhage, viral infection, blood transfusion and postoperative complications might produce a favorable microenvironment for CSCs to develop into a new tumor nodule [Citation12]. The interval between two operations of these nine patients ranges from 3.3 to 48.2 months. Although it is not possible to exclude the possibility that the recurrent tumor came from residual cancer cells from the primary HCC, since the longest interval between surgery was more than 4 years, it is believable, at least in part, that the recurrent HCC might have developed from dormant CSCs. The possible presence of such ‘dormant CSCs’ strongly suggests that in addition to removal of the tumor block during surgery, efforts in the future should focus on the eradication of the CSCs shed away from the tumor to avoid tumor recurrence.
The simultaneous occurrence of multiple HCCs may result from either the dissemination of malignant cells from a single primary tumor (intrahepatic metastasis), or multiple independent tumors. These two possible mechanisms of multiple HCCs development have a different impact on clinical treatment and prognosis [Citation37,Citation38]. Multiple HCCs developing from intrahepatic metastasis might be poorly differentiated and more aggressive. Therefore, it is valuable to investigate whether the multiple nodules have the same clonal origin, which will provide clinical evidence for the tumor treatment regime proscribed. Critically, since the result of MSRE-qPCR is obtained through a statistical analysis, it was much more precise and objective. Consequently in comparison to other methods, the CGI methylation assay described here could be a simpler and time-saving method for determining clonal origin of HCC tumors.
The major limitation of this study is the low case number of HCC tissues. The paired primary and recurrent HCC nodules were hard to collect and therefore are very precious. Furthermore, the results obtained from these specimens were highly accordant. Together with the validation by HBV integration assay, the results obtained suggested that the majority of the recurrent and primary tumors have the same origin, which somewhat supports the CSC hypothesis. This is the first study in which that analysis of DNA methylation profiles has been shown to be useful for discriminating the clonal origin of different nodules, found in recurrent HCC.
Conclusion & future perspective
In summary, our findings demonstrated that the recurrent HCC tumors exhibited similar global-level methylation to that in the primary HCC, indicating that the majority of recurrent HCC might have developed from the CSCs originated from the primary tumors. The methylation assay could be used to determine whether different tumor nodules have the same clonal origin.
Table 1. Clinicopathological parameters of the nine patients with primary and recurrent hepatocellular carcinoma.
Table 2. The HBV DNA integration status in host cellular genome of six patients with primary and recurrent hepatocellular carcinoma.
Table 3. Correlation of every gene’s methylation between the eight paired primary and recurrent tumors.
Table 4. The correlation between the paired primary and recurrent cell lines and tumor tissues by using the 15 constant methylated genes.
Aims
To investigate if the DNA methylation assay could identify the primary tumor cell origin of recurrent hepatocellular carcinoma (HCC).
Materials & methods
The methylation status of 24 well-known hypermethylated genes was measured by MSRE-qPCR in the primary and recurrent HCC tumor tissues from a total of ten HCC patients.
HBV DNA integration pattern was measured by Alu-PCR in those HBV-associated HCC specimens.
The mRNA levels of cancer stem cell (CSC) biomarkers were measured by real time RT-qPCR.
Determination of the clonal origin by methylation pattern & HBV integration pattern in paired primary and recurrent HCC cell lines & tumor tissues
The cell lines Hep-11 and Hep-12 showed a similar methylation pattern and the same integration pattern.
Among the nine pairs of tumor tissues, similar methylation patterns between the primary and recurrent tumors were seen in eight pairs.
In five cases with HBV infection, the same HBV DNA integration was identified both in the paired primary and recurrent tumor specimens.
The methylation pattern and integration pattern in the same patient was consistent.
The CGI methylation intensity of some genes varied between the primary & the recurrent tumors
Although methylation level of 15 genes showed statistical association between the primary and its recurrent tumor, AKAP12, CDKN2A and GSTP1 methylation was obviously different.
Using the 15 methylation-constant genes would be a better combo to identify the clonal origin of tumor nodules.
The expression of CSC biomarkers increased both in the primary & recurrent HCC tissues
The mRNA levels of seven CSCs biomarkers were significantly higher in either primary or recurrent tumor tissues compared with normal liver tissues, while no difference was found between these two groups of tumors.
Discussion & conclusion
The majority of recurrent HCC might have developed from the CSCs from the primary tumors.
Tumor methylation assay could determine whether different tumor nodules have the same clonal origin.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Supplemental Figure 1
Download MS Word (446.5 KB)Acknowledgements
We appreciate the excellent work done by MA McCrae for his carefully English editing on this manuscript. We also thank Z Zhang for providing the DNA of HCC cell lines Hep-11 and Hep-12.
Financial & competing interests disclosure
This work was supported by the 973 Program (2015CB554000), the National S & T Major Project for Infectious Diseases (2012ZX10004–904), the Project of Beijing Municipal Science and Technology Commission (Z111107067311027), the National Natural Science Foundation of China (81372603) and the 111 Project (B07001). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Vivarelli M , MontaltiR , RisalitiA . Multimodal treatment of hepatocellular carcinoma on cirrhosis: an update . World J. Gastroenterol.19 ( 42 ), 7316 – 7326 ( 2013 ).
- Yazici C , NiemeyerDJ , IannittiDA , RussoMW . Hepatocellular carcinoma and cholangiocarcinoma: an update. Expert Rev . Gastroenterol. Hepatol.8 ( 1 ), 63 – 82 ( 2014 ).
- Dupont-Bierre E , CompagnonP , RaoulJL , FayetG , de Lajarte-ThirouardAS , BoudjemaK . Resection of hepatocellular carcinoma in noncirrhotic liver: analysis of risk factors for survival . J. Am. Coll. Surg.201 ( 5 ), 663 – 670 ( 2005 ).
- Lim KC , ChowPK , AllenJC , SiddiquiFJ , ChanES , TanSB . Systematic review of outcomes of liver resection for early hepatocellular carcinoma within the Milan criteria . Br. J. Surg.99 ( 12 ), 1622 – 1629 ( 2012 ).
- Dhir M , LydenER , SmithLM , AreC . Comparison of outcomes of transplantation and resection in patients with early hepatocellular carcinoma: a meta-analysis . HPB (Oxford)14 ( 9 ), 635 – 645 ( 2012 ).
- Arii S , TeramotoK , KawamuraTet al. Characteristics of recurrent hepatocellular carcinoma in Japan and our surgical experience . J. Hepatobiliary Pancreat. Surg.8 ( 5 ), 397 – 403 ( 2001 ).
- Choi GH , KimDH , KangCMet al. Prognostic factors and optimal treatment strategy for intrahepatic nodular recurrence after curative resection of hepatocellular carcinoma . Ann. Surg. Oncol.15 ( 2 ), 618 – 629 ( 2008 ).
- Reya T , MorrisonSJ , ClarkeMF , WeissmanIL . Stem cells, cancer, and cancer stem cells . Nature414 ( 6859 ), 105 – 111 ( 2001 ).
- Visvader JE , LindemanGJ . Cancer stem cells in solid tumours: accumulating evidence and unresolved questions . Nat. Rev. Cancer8 ( 10 ), 755 – 768 ( 2008 ).
- Kyo S , MaidaY , InoueM . Stem cells in endometrium and endometrial cancer: accumulating evidence and unresolved questions . Cancer Lett.308 ( 2 ), 123 – 133 ( 2011 ).
- Pantel K , Alix-PanabieresC , RiethdorfS . Cancer micrometastases . Nat. Rev. Clin. Oncol.6 ( 6 ), 339 – 351 ( 2009 ).
- Fan ST , YangZF , HoDW , NgMN , YuWC , WongJ . Prediction of posthepatectomy recurrence of hepatocellular carcinoma by circulating cancer stem cells: a prospective study . Ann. Surg.254 ( 4 ), 569 – 576 ( 2011 ).
- Yamamoto T , KajinoK , KudoM , SasakiY , ArakawaY , HinoO . Determination of the clonal origin of multiple human hepatocellular carcinomas by cloning and polymerase chain reaction of the integrated hepatitis B virus DNA . Hepatology29 ( 5 ), 1446 – 1452 ( 1999 ).
- Ng IO , GuanXY , PoonRT , FanST , LeeJM . Determination of the molecular relationship between multiple tumour nodules in hepatocellular carcinoma differentiates multicentric origin from intrahepatic metastasis . J. Pathol.199 ( 3 ), 345 – 353 ( 2003 ).
- He B , TangZY , LiuKD , ZhouG . Analysis of the cellular origin of hepatocellular carcinoma by p53 genotype . J. Cancer Res. Clin. Oncol.122 ( 12 ), 763 – 766 ( 1996 ).
- Xu X , XingB , HuMet al. Recurrent hepatocellular carcinoma cells with stem cell-like properties: possible targets for immunotherapy . Cytotherapy12 ( 2 ), 190 – 200 ( 2010 ).
- Wang Y , ChengJ , XuCet al. Quantitative methylation analysis reveals gender and age differences in p16INK4a hypermethylation in hepatitis B virus-related hepatocellular carcinoma . Liver Int.32 ( 3 ), 420 – 428 ( 2012 ).
- Jiang S , YangZ , LiWet al. Re-evaluation of the carcinogenic significance of hepatitis B virus integration in hepatocarcinogenesis . PLoS ONE7 ( 9 ), e40363 ( 2012 ).
- Yang XR , XuY , YuBet al. High expression levels of putative hepatic stem/progenitor cell biomarkers related to tumour angiogenesis and poor prognosis of hepatocellular carcinoma . Gut59 ( 7 ), 953 – 962 ( 2010 ).
- Goeppert B , SchmezerP , DutruelCet al. Down-regulation of tumor suppressor A kinase anchor protein 12 in human hepatocarcinogenesis by epigenetic mechanisms . Hepatology52 ( 6 ), 2023 – 2033 ( 2010 ).
- Moribe T , IizukaN , MiuraTet al. Identification of novel aberrant methylation of BASP1 and SRD5A2 for early diagnosis of hepatocellular carcinoma by genome-wide search . Int. J. Oncol.33 ( 5 ), 949 – 958 ( 2008 ).
- Moribe T , IizukaN , MiuraTet al. Methylation of multiple genes as molecular markers for diagnosis of a small, well-differentiated hepatocellular carcinoma . Int. J. Cancer125 ( 2 ), 388 – 397 ( 2009 ).
- Nishida N , KudoM , NagasakaT , IkaiI , GoelA . Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma . Hepatology56 ( 3 ), 994 – 1003 ( 2012 ).
- Yu J , TaoQ , CheungKFet al. Epigenetic identification of ubiquitin carboxyl-terminal hydrolase L1 as a functional tumor suppressor and biomarker for hepatocellular carcinoma and other digestive tumors . Hepatology48 ( 2 ), 508 – 518 ( 2008 ).
- Xie B , ZhouJ , YuanLet al. Epigenetic silencing of Klotho expression correlates with poor prognosis of human hepatocellular carcinoma . Hum. Pathol.44 ( 5 ), 795 – 801 ( 2013 ).
- Ammerpohl O , PratschkeJ , SchafmayerCet al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma . Int. J. Cancer.130 ( 6 ), 1319 – 1328 ( 2012 ).
- Ren J , HeW , ZhangRet al. RASSF2A promoter methylation in hepatitis B virus-related hepatocellular carcinogenesis and its correlation with elevated serum alpha-fetoprotein level . J Huazhong Univ Sci Technolog Med Sci.29 ( 3 ), 309 – 312 ( 2009 ).
- Kuo CC , LinCY , ShihYLet al. Frequent methylation of HOXA9 gene in tumor tissues and plasma samples from human hepatocellular carcinomas . Clin. Chem. Lab. Med.52 ( 8 ), 1235 – 1245 ( 2014 ).
- Tao R , LiJ , XinJet al. Methylation profile of single hepatocytes derived from hepatitis B virus-related hepatocellular carcinoma . PLoS ONE6 ( 5 ), e19862 ( 2011 ).
- Xu XL , XingBC , HanHBet al. The properties of tumor-initiating cells from a hepatocellular carcinoma patient’s primary and recurrent tumor . Carcinogenesis31 ( 2 ), 167 – 174 ( 2010 ).
- Oishi N , YamashitaT , KanekoS . Molecular biology of liver cancer stem cells . Liver Cancer3 ( 2 ), 71 – 84 ( 2014 ).
- Guo Z , LiLQ , JiangJH , OuC , ZengLX , XiangBD . Cancer stem cell markers correlate with early recurrence and survival in hepatocellular carcinoma . World J. Gastroenterol.20 ( 8 ), 2098 – 2106 ( 2014 ).
- Yin X , LiYW , JinJJet al. The clinical and prognostic implications of pluripotent stem cell gene expression in hepatocellular carcinoma . Oncol. Lett.5 ( 4 ), 1155 – 1162 ( 2013 ).
- Yin X , LiYW , ZhangBHet al. Coexpression of stemness factors Oct4 and Nanog predict liver resection . Ann. Surg. Oncol.19 ( 9 ), 2877 – 2887 ( 2012 ).
- Arzumanyan A , FriedmanT , NgIO , ClaytonMM , LianZ , FeitelsonMA . Does the hepatitis B antigen HBx promote the appearance of liver cancer stem cells?Cancer Res.71 ( 10 ), 3701 – 3708 ( 2011 ).
- Kimura O , KondoY , KogureTet al. Expression of EpCAM increases in the hepatitis B related and the treatment-resistant hepatocellular carcinoma . Biomed. Res. Int.2014 , 172913 ( 2014 ).
- Yasui M , HaradaA , NonamiTet al. Potentially multicentric hepatocellular carcinoma: clinicopathologic characteristics and postoperative prognosis . World J. Surg.21 ( 8 ), 860 – 864 , 864 – 865 ( 1997 ).
- Kaibori M , MatsuiY , SaitoT , KamiyamaY . Risk factors for different patterns of recurrence after resection of hepatocellular carcinoma . Anticancer Res.27 ( 4C ), 2809 – 2816 ( 2007 ).