
Abstract
Type 2 diabetes (T2D) develops due to insulin resistance and impaired insulin secretion, predominantly in genetically predisposed subjects exposed to nongenetic risk factors like obesity, physical inactivity and ageing. Emerging data suggest that epigenetics also play a key role in the pathogenesis of T2D. Genome-wide studies have identified altered DNA methylation patterns in pancreatic islets, skeletal muscle and adipose tissue from subjects with T2D compared with nondiabetic controls. Environmental factors known to affect T2D, including obesity, exercise and diet, have also been found to alter the human epigenome. Additionally, ageing and the intrauterine environment are associated with differential DNA methylation. Together, these data highlight a key role for epigenetics and particularly DNA methylation in the growing incidence of T2D.
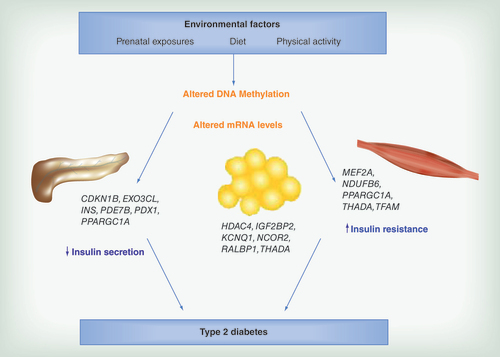
Altered DNA methylation and transcriptional activity of Type 2 diabetes candidate genes in human pancreatic islets may contribute to reduced insulin secretion. Additionally, environmental factors may induce insulin resistance in adipose tissue and skeletal muscle through epigenetic modifications.
Already in the early 1990s Hales and Barker suggested a role for epigenetics in the development of metabolic diseases, including Type 2 diabetes, when they described how early life events affected health outcome in adulthood [Citation1]. This first hypothesis was confirmed by a number of epidemiological studies, investigating altered fetal, early postnatal and childhood growth on the development of Type 2 diabetes or insulin resistance [Citation2–5]. Clearly this was an important beginning of a field that would start to be experimentally explored in humans many years later, with new laboratory technologies allowing for thorough investigations of epigenetic marks.
Type 2 diabetes is a complex disorder manifested by chronically elevated blood glucose levels, as a result of insufficient insulin secretion from the pancreatic β cells and decreased insulin sensitivity in target tissues, for example, the liver, skeletal muscle and adipose tissue. Genetic as well as environmental factors are involved in the development of Type 2 diabetes, with obesity as one of the main factors for the rapid increase of affected individuals worldwide [Citation6]. Interestingly, the interaction between genetic and environmental factors in disease development most likely involves epigenetic modifications [Citation7].
The most studied epigenetic mark is DNA methylation, the addition of a methyl group to the DNA nucleotide cytosine [Citation8]. A strictly regulated DNA methylation pattern is essential for normal development and has a key role throughout life in tissue specific gene regulation and transcription [Citation9]. Histone modifications are other important epigenetic marks, interacting with DNA methylation and tightly linked to the chromatin structure [Citation10,Citation11]. Also noncoding RNAs, with the ability to post transcriptionally regulate gene expression, is considered an epigenetic modification and implicated in the pathogenesis of Type 2 diabetes [Citation12]. In contrast to the stable DNA sequence, epigenetic modifications are dynamic and reversible and hence promising targets for pharmacological interventions.
This review will summarize published experimentally data supporting a role for epigenetic variation in the development of Type 2 diabetes, with a focus on findings from human studies, including both candidate gene approaches and genome-wide studies ().
Evidence of epigenetic alterations of genes involved in insulin secretion
The ability of pancreatic β cells to produce and secrete insulin in response to glucose is of utmost importance to maintain normoglycemia. During the development of Type 2 diabetes, the demands of insulin usually increase as a result of increased insulin resistance. Hence, mechanisms that affect β cell function, including cell viability, insulin production and secretion potential, are keys to find new treatment strategies for diabetes.
In a recent study, we investigated the genome-wide DNA methylation pattern in human pancreatic islets obtained from deceased donors [Citation13]. We found altered levels of DNA methylation in islets from Type 2 diabetic versus nondiabetic donors for a total of 1649 CpG sites, corresponding to 853 genes. These genes include known Type 2 diabetes loci such as TCF7L2, KCNQ1, THADA, FTO, IRS1 and PPARG, and are enriched in pathways involved in cancer, axon guidance and MAPK (mitogen-activated protein kinase) signaling. Furthermore, 102 of the genes showing differential methylation also displayed altered mRNA expression between islets from Type 2 diabetic versus nondiabetic donors, suggesting epigenetic regulation of transcriptional activity and a possible mechanism for how Type 2 diabetes risk genes contribute to disease development (). The majority of these genes showed increased DNA methylation and decreased gene expression in islets from diabetic versus nondiabetic donors. Functional analysis of some of those genes demonstrated, for example, impaired insulin secretion when Cdkn1a and Pde7b, respectively, were overexpressed in β cells, and also reduced β cell proliferation as a result of overexpression of Cdkn1a (p21). Additionally, silencing of Exoc3l, a gene encoding one component of the exocyst complex, in the β cells resulted in reduced exocytosis. Importantly, in this study there was no significant difference in the β cell content between diabetic and nondiabetic islets, showing that the identified epigenetic differences are not due to an altered cell type composition. Taken together, this study provides detailed information of the DNA methylome in human pancreatic islets and indeed highlights the importance of epigenetic regulation as a mechanism in the pathogenesis of Type 2 diabetes [Citation13].
DNA methylation in human pancreatic islets has also been investigated by Volkmar et al. [Citation14]. This study, covering less CpG sites and investigating less human islet donors as compared with the study by Dayeh et al., was still able to detect 276 CpG sites annotated to 254 gene promoters with differential DNA methylation in islets obtained from Type 2 diabetic versus nondiabetic donors. These genes are implicated in β cell function and survival and further support a role for DNA methylation in the pathogenesis of Type 2 diabetes. Interestingly, 27% of the CpG sites identified by Volkmar et al. was also differentially methylated in islets from Type 2 diabetics versus nondiabetic donors in our study [Citation13]. Moreover, both studies show that the CpG sites with significantly altered DNA methylation tend to have decreased DNA methylation in islets from Type 2 diabetic compared with nondiabetic donors (96 and 97%, respectively) [Citation13,Citation14]. As aberrant DNA methylation could be either a cause or arise as a consequence of disease, Volkmar et al. exposed pancreatic islets from nondiabetic donors to high glucose levels. They found no significant effect on DNA methylation for the 16 CpG sites tested, making the altered methylation pattern in islets from Type 2 diabetic patients unlikely to be a cause of hyperglycemia [Citation14]. However, it remains to be tested if prolonged hyperglycemia, in other words, more than 72 h as tested here, may have a different outcome as diabetes and its complications is known to develop for decades. To approach the same question from a different angle, we tested if the 1649 CpG sites with altered DNA methylation in islets from Type 2 diabetics versus nondiabetic donors were associated with age, BMI or HbA1c, all known risk factors for Type 2 diabetes, in nondiabetics [Citation13]. For HbA1c, 142 CpG sites (8.6%) were found to have significantly associated levels of DNA methylation in islets from nondiabetic donors, and for age and BMI we found 18 and 16 CpG sites, respectively. Importantly, 92% of those sites change in the same direction in islets from Type 2 diabetic donors as with increased age, BMI or HbA1c, hinting that some epigenetic alterations may be seen prior to onset of disease [Citation13].
We have also studied the genome-wide DNA methylation pattern in human pancreatic islets in response to palmitate treatment, to mimic the situation of elevated circulating free fatty acids often seen in Type 2 diabetes patients [Citation15]. Global DNA methylation, in other words, the average of all 483,844 CpG sites analyzed, was significantly higher in islets exposed to palmitate for 48 h compared with control islets. Additionally, exposure to palmitate altered the degree of DNA methylation of 46,977 sites at p < 0.05, which is approximately double the expected number and significantly more than expected by chance. However, after correction for multiple testing, no individual CpG site showed a significant difference in DNA methylation in response to palmitate treatment. Nevertheless, of the 1860 genes with significantly altered expression levels due to palmitate treatment, 290 genes (15.6%) had one or more CpG sites with a nominal difference in the level of DNA methylation in palmitate treated compared with control islets [Citation15], including Type 2 diabetes susceptibility genes like TCF7L2 [Citation16] and GLIS3 [Citation17]. Although this study suggests global epigenetic alterations in pancreatic islets due to palmitate exposure, further investigations are needed to determine if prolonged elevation of circulating free fatty acids, which is often seen in subjects with Type 2 diabetes, may have stronger effects on the DNA methylation pattern and to what extent the association with Type 2 diabetes is mediated by epigenetic factors [Citation15].
In an early study investigating DNA methylation of PPARGC1A as a candidate gene, we detected altered levels of methylation in a region upstream of the gene in human pancreatic islets from Type 2 diabetic versus nondiabetic donors [Citation18]. This increase in DNA methylation observed in islets from Type 2 diabetic donors was further accompanied by reduced PPARGC1A mRNA expression and reduced glucose-stimulated insulin secretion (). PPARGC1A is a master transcriptional regulator of genes participating in oxidative phosphorylation and hence decreased levels may lead to reduced ATP production from the mitochondria, impairing glucose-stimulated insulin secretion [Citation19]. The KCNQ1 imprinted locus is another candidate gene region associated with Type 2 diabetes, believed to act through impaired islet function [Citation20–22]. Using both adult human islets and fetal pancreas samples, Travers et al. were able to show that a Type 2 diabetes genetic risk variant in the KCNQ1 locus was associated with DNA methylation of a regulatory sequence in fetal samples, and with another region involving a PLAGL1 binding site in adult islets [Citation23]. However, no effect was observed on the level of gene expression. These results highlight the dynamic feature of DNA methylation, with different regulatory roles at different developmental stages.
An obvious target to investigate when studying Type 2 diabetes is the insulin gene (INS) and its regulation. As a matter of fact, two studies have shown that INS promoter DNA methylation is involved in regulating gene expression of insulin in human pancreatic islets and β cells, respectively [Citation24,Citation25] (). Kuroda et al. showed that the human INS promoter is demethylated specifically in β cells, and that methylation of this region in vitro suppresses INS gene expression [Citation24]. Furthermore, we have shown increased levels of DNA methylation in 4 CpG sites in the INS promoter region in human pancreatic islets obtained from Type 2 diabetic compared with nondiabetic donors [Citation25]. The methylation levels of these CpG sites, together with another 9 sites in the region, were negatively correlated with INS gene expression, indeed suggesting the insulin gene to be subject to epigenetic regulation. In further support of this hypothesis, we could show that hyperglycemia directly altered the degree of methylation of the insulin gene in clonal β cells cultured in vitro. Also, FACS (fluorescence-activated cell sorting) sorted human alpha cells had much higher degree of methylation of INS than β cells, supporting that INS methylation regulate cell specific gene expression. Additionally, the islets obtained from Type 2 diabetic donors displayed decreased insulin content and glucose-stimulated insulin secretion [Citation25]. A similar approach was undertaken to investigate PDX1 [Citation26], a transcription factor important for pancreas development and β cell maturation and function [Citation27]. We found three CpG sites located within the distal promoter and a set of seven CpG sites in the enhancer region of PDX1 that exhibit significantly increased DNA methylation in islets from Type 2 diabetic compared with nondiabetic donors. Interestingly, all nine analyzed CpG sites in the enhancer region correlated negatively with PDX1 mRNA expression levels in human islets, whereas this only was seen for one CpG site in the distal promoter and one in the proximal promoter, suggesting that epigenetic regulation of PDX1 is mediated mainly through the enhancer region. By the use of luciferase constructs, we could functionally demonstrate that methylation of the enhancer region of PDX1 decreases its transcriptional activity. Moreover, PDX1 mRNA levels were significantly correlated with INS mRNA expression and glucose-stimulated insulin secretion (). We also found a significant, positive association between PDX1 DNA methylation and HbA1c, suggesting that prolonged hyperglycemia may induce epigenetic alterations. This result was further experimentally verified by exposing clonal β cells to high glucose, resulting in increased DNA methylation and decreased mRNA expression of PDX1 [Citation26].
Incretin hormones are important for stimulating the release of insulin in response to a meal and its mimicking agents are used in Type 2 diabetes treatment [Citation28]. GLP1R encodes the receptor for one of the incretin hormones, glucagon-like peptide-1, and is expressed in pancreatic β cells [Citation29]. When investigating the epigenetic component of GLP1R gene regulation, we found a modest but significant difference, with increased DNA methylation in human pancreatic islets obtained from Type 2 diabetics compared with nondiabetic donors [Citation30]. Another CpG site in GLP1R, within a region containing SP1 and SP3 transcription factor binding sites, was negatively correlated with GLP1R expression and positively correlated with HbA1c and BMI [Citation30].
There is also evidence for altered levels of noncoding RNA in pancreatic islets from human Type 2 diabetic organ donors [Citation31]. Kameswaran et al. identified an imprinted locus with β cell specific expression of a cluster of miRNAs, which were downregulated in islets from Type 2 diabetic compared with nondiabetic donors. Interestingly, this was strongly correlated with promoter hypermethylation [Citation31]. This region, the DLK1-MEG3 locus, has previously been associated with different forms of cancer [Citation32]. This study also confirmed differential expression of miRNAs previously implicated in diabetes and β cell function, for example, miR-7 [Citation33,Citation34].
As epigenetics is known to regulate gene expression in a tissue specific manner, it is important to consider which tissue to investigate in relation to disease development. However, to be useful as a clinical biomarker, it is also essential to find markers that are reflected in readily accessible tissues or cells, such as blood samples. A few Type 2 diabetes case-control studies have investigated epigenetic alteration in Type 2 diabetes candidate genes in blood samples [Citation35–39]. Toperoff et al. detected differentially methylated regions (DMRs) in blood from patients with Type 2 diabetes compared with control individuals, including 6 DMRs in regions previous genetically associated with Type 2 diabetes (CENTD2, FTO, KCNJ11, TCF7L2 and WFS1) [Citation39]. Additionally, replication of individual CpG sites within the investigated DMRs found 13 CpG sites (intronic sites of THADA, JAZF1, TCF7L2, KCNQ1 and FTO, and 3′-UTR and downstream sites in SLC30A8) with a significant difference in DNA methylation between pooled cases and controls. They furthermore investigated DNA methylation in blood samples from a longitudinal study with healthy participants at inclusion, of which some progressed to impaired glucose metabolism. The results showed aberrant DNA methylation prior to disease, suggesting that DNA methylation patterns associated with Type 2 diabetes in tissues not directly involved in insulin secretion or action, like blood, is established early in development, and has a role as a predictive rather than a diagnostic marker [Citation39]. In peripheral blood leukocytes, del Rosario et al. demonstrated altered promoter methylation in offspring to mothers with diabetes during pregnancy [Citation36]. No individual genes were significantly different between offspring with (n = 14) and without (n = 14) diabetic mothers. However, differentially methylated genes were enriched for pathways involved in pancreas development and insulin secretion, including the pathways maturity onset diabetes of the young, Type 2 diabetes and Notch signaling. This suggests that the intrauterine environment, acting through epigenetic alterations, may exert an effect on future β cell function and risk of diabetes [Citation36]. DNA methylation of the TCF7L2 promoter was recently investigated in whole blood from patients newly diagnosed with Type 2 diabetes and controls matched for age and BMI [Citation35]. Here, altered levels of DNA methylation was detected in 13 of the 22 analyzed CpG sites in Type 2 diabetic patients compared with matched controls, after correction for multiple testing and confounding factors. Additionally, specific CpG sites were associated with metabolic traits including fasting glucose (four CpG sites) and total- and LDL-cholesterol (one CpG site). Importantly, the patients were only on diet without any pharmacological treatment affecting the results and this study suggests that DNA methylation alterations of a gene important for β cell function may be reflected in blood DNA [Citation35].
Epigenetic alterations & insulin sensitivity
Insulin resistance is a common feature of Type 2 diabetes, affecting the liver and peripheral tissues like skeletal muscle and adipose tissue. Impaired response to insulin in the peripheral tissues results in reduced capacity to clear the blood stream from glucose, and additionally, makes adipose tissue secrete adipokines which further negatively affect whole body glucose homeostasis. Insulin resistance in the liver results in failure to inhibit glucose output in response to high glucose levels, which adds to an increasing concentration of glucose in the circulation. Consequently, insulin resistance and the tissues involved in this process are, as well as insulin secretion and pancreatic β cells, a key to understand the pathogenesis of Type 2 diabetes.
Skeletal muscle is the main tissue responsible for glucose uptake, which is further stimulated by physical exercise involving muscle contractions. Additionally, individuals with a high genetic predisposition for Type 2 diabetes are characterized by reduced physical fitness [Citation40]. Clearly, exercise is beneficial to improve insulin sensitivity and prevent or delay the onset of Type 2 diabetes. Using an exercise intervention study, we investigated genome-wide DNA methylation in human skeletal muscle from individuals with or without a family history of Type 2 diabetes, in other words, a genetic predisposition for the disease [Citation41]. First, differential methylation at baseline between individuals with or without a family history of Type 2 diabetes was observed for genes involved in, for example, MAPK, insulin, Wnt and calcium signaling, as well as in genes important for muscle function like MAPK1, MYO18B, HOXC6 and PRKAB1. Of the genes differentially methylated due to a family history of Type 2 diabetes, 40% could be validated in skeletal muscle from monozygotic twin pairs discordant for the disease. Next, 134 genes were found to be differentially methylated in response to the six months exercise intervention, including MEF2A (exercise transcription factor), THADA (Type 2 diabetes candidate gene), NDUFC2 (mitochondrial function) and IL7 (cytokine). Several genes in this study also exhibited changes in both DNA methylation and gene expression levels (). Clearly, exercise induces genome-wide changes in DNA methylation in human skeletal muscle; however, the response may differ due to the genetic predisposition for Type 2 diabetes [Citation41]. Barres et al. also investigated DNA methylation in human skeletal muscle, and found a global decrease in DNA methylation in response to acute exercise, as well as decreases in promoter methylation of some candidate genes [Citation42]. The latter includes genes with increased expression in skeletal muscle after exercise and which also display altered levels of DNA methylation in Type 2 diabetes, in other words, PPARGC1A, TFAM, PPARD, PDK4 and CS [Citation43] ().
Also adipose tissue has a central role in glucose homeostasis, being an endocrine organ affecting many metabolic pathways [Citation44]. We recently investigated genome-wide DNA methylation and mRNA expression in adipose tissue from healthy individuals participating in a six months exercise intervention [Citation45,Citation46]. We found a change in global DNA methylation as well as in 17,975 individual CpG sites in response to exercise [Citation45]. Among the 39 genes with a validated association with Type 2 diabetes at that time [Citation47], 21 had one or more CpG sites with significantly altered levels of DNA methylation in adipose tissue before versus after exercise [Citation45]. KCNQ1 and TCF7L2 had most differentially methylated CpG sites, and for HHEX, IGF2BP2, JAZF1 and TCF7L2 increased DNA methylation in response to exercise was accompanied by decreased mRNA expression. To investigate the direct effect of DNA methylation on gene expression, a reporter gene construct of the human RALBP1 promoter was created where the number of CpG sites methylated can be modified. RALBP1 is implicated in metabolic disease and GLUT4 trafficking [Citation48,Citation49] and indeed increased DNA methylation of this promoter reduced its transcriptional activity. Furthermore, functional validation of genes (HDAC4 and NCOR2) that exhibit increased DNA methylation and decreased mRNA expression in adipose tissue after six months exercise resulted in increased insulin-stimulated lipogenesis when silenced in an adipocyte cell line [Citation45] (). We have also studied the genome-wide DNA methylation pattern in adipose tissue from subjects with Type 2 diabetes compared with nondiabetic controls [Citation50]. DNA methylation was altered in 15,627 individual CpG sites (7046 unique genes) with a false discovery rate of 15%. Replication in adipose tissue from monozygotic twins discordant for Type 2 diabetes showed a nominal association with disease for 1410 of these CpG sites. These include, for example, the Type 2 diabetes candidate genes KCNQ1, NOTCH2, TCF7L2 and THADA [Citation50] (). In adipose tissue from healthy twins, Grundberg et al. found 8638 CpG sites in 4133 unique genes where the shared common environment accounted for more than 30% of the methylation variance. A pathway analysis suggested these sites to be associated with metabolic diseases including Type 2 diabetes [Citation51], implying DNA methylation as a link between the environment and disease susceptibility.
Ribel-Madsen et al. analyzed genome-wide promoter DNA methylation in both skeletal muscle and adipose tissues from elderly MZ twin pairs discordant for Type 2 diabetes [Citation52]. After correction for multiple testing, only one CpG site in muscle (IL8) and seven in adipose tissue were significantly associated with disease, which may be due to the small sample size. However, analyzing Type 2 diabetes candidate genes separately further showed altered levels of DNA methylation in Type 2 diabetic versus control twins for HNF1A and CDKN2A in adipose tissue. Additionally, global DNA methylation (LINE1) did not associate with Type 2 diabetes status, but with the associated phenotypes BMI and 2 h plasma glucose [Citation52].
Also in DNA from peripheral blood leukocytes there is evidence for a relation between the level of DNA methylation and insulin resistance [Citation53]. Zhao et al. examined global DNA methylation (Alu repeats) in monozygotic twins and the relation to insulin resistance as measured by homeostasis model assessment. Each of the four CpG sites investigated was significantly associated with insulin resistance, after correcting for confounding factors including several known risk factors of insulin resistance. However, the mechanism behind this association remains unknown, although the authors speculate that it may be a result of genomic instability due to the altered methylation of the Alu elements [Citation53]. Simar et al. went one step further and investigated global DNA methylation in individual cell types in blood [Citation54]. Using this approach, they found increased global DNA methylation in natural killer and B cells from patients with Type 2 diabetes, which correlated positively with insulin resistance. This highlights the need for not only tissue specific but also cell type specific epigenetic analyses and the results potentiate a role for DNA methylation in immune function and metabolic disorders [Citation54].
Low levels of circulating insulin-like growth factor binding protein-1 (IGFBP1) is associated with insulin resistance and Type 2 diabetes [Citation55], and is also a predictor for development of Type 2 diabetes [Citation56]. To investigate how this gene is regulated, Gu et al. analyzed IGFBP1 DNA methylation in blood from patients with Type 2 diabetes and age-matched controls [Citation38]. While the level of DNA methylation was higher, IGFBP1 serum levels were lower in Type 2 diabetic patients compared with controls. Additionally, among individuals newly diagnosed with Type 2 diabetes, a family history of diabetes was associated with higher levels of IGFBP1 DNA methylation [Citation38], excluding the results to be caused by treatment and suggesting altered methylation of this gene to be a predictive marker of disease. Also IGFBP7 has been linked to insulin resistance and Type 2 diabetes, and is further able to interact both with insulin and insulin-like growth factor-1. In a similar approach as above, Gu et al. found increased IGFBP7 DNA methylation in men newly diagnosed with Type 2 diabetes, however, no correlation with serum IGFBP7 protein levels was observed [Citation37].
Mitochondrial dysfunction likely has a role not only in impairing insulin secretion from the pancreatic β cells, but also in insulin resistance [Citation57]. Interestingly, differential DNA methylation of the promoter of mitochondrial transcription factor A (TFAM) in blood samples has been shown to be associated with insulin resistance [Citation58]. Ageing is also associated with insulin resistance and an increased risk for Type 2 diabetes. We have previously found increased DNA methylation in the promoter of two candidate genes for the disease, NDUFB6 and COX7A1, in skeletal muscle from elderly compared with young subjects [Citation59,Citation60]. The increased methylation was further associated with decreased gene expression. While NDUFB6 encodes a protein which is part of complex one of the mitochondrial respiratory chain, COX7A1 encodes a member of complex four. Additionally, the expression of both of these genes is decreased in muscle from subjects with Type 2 diabetes compared with nondiabetic controls [Citation61] ().
Finally, energy rich diets are associated with obesity, insulin resistance and an increased risk for Type 2 diabetes. Interestingly, a five days high fat diet altered the DNA methylation pattern in skeletal muscle from young healthy men [Citation62]. The 6508 genes with altered levels of DNA methylation were enriched in pathways affecting inflammation, cancer and reproduction. The diet was also associated with metabolic alterations such as hepatic insulin resistance and reduced nonesterified fatty acid levels [Citation62].
Interaction between genetic & epigenetic factors in Type 2 diabetes pathogenesis
There may be an interaction between genetic and epigenetic factors, together affecting physiological processes and disease pathogenesis. Already in 2007, we described the interaction of genetic and epigenetic factors in human skeletal muscle [Citation59]. The investigated gene, NDUFB6, is involved in oxidative phosphorylation, and is known to be decreased in insulin resistant and Type 2 diabetic subjects [Citation61]. We found a genetic variant in NDUFB6 that introduces a CpG site subject to DNA methylation, with the ability to regulate gene expression and glucose uptake in an age dependent matter [Citation59]. Based on this finding, and to reveal a possible disease mechanism, we continued to investigate if genetic variants associated with Type 2 diabetes introduce or remove possible DNA methylation sites [Citation63]. Surpassing our expectations, 19 of the 40 known single nucleotide polymorphisms (SNPs) associated with Type 2 diabetes at that time [Citation47] indeed altered the DNA sequence to introduce or remove a CpG site (also known as CpG-SNPs). We hence analyzed 16 of these CpG-SNPs in human pancreatic islets and found that all were associated with altered levels of DNA methylation in the SNP site. In six of the loci (CDKN2A, TCF7L2, HMGA2, KCNQ1, ADCY5 and WFS1), not only the DNA methylation site directly affected by the SNP, but also surrounding CpG sites, showed altered levels of DNA methylation significantly associated with the CpG-SNP. Furthermore, two CpG-SNPs were additionally associated with altered pancreatic islets mRNA expression, four with alternative splicing events (SLC30A8, WFS1, CDKAL1 and TCF7L2), and four with islet function as measured by insulin secretion and content and glucagon secretion. These findings suggest a mechanism where genetic and epigenetic factors interact to affect gene function and possible risk of disease [Citation63].
As methods for large scale studies improved, we further extended our analyses of genetic and epigenetic interactions by investigating genome-wide SNP (n = 574,533) and DNA methylation (n = 468,787) data in human pancreatic islets [Citation64]. The methylation quantitative trait loci (mQTL) analysis identified 67,438 SNP-CpG pairs in cis and 2562 in trans, including 11,735 and 383 individual CpG sites, respectively, where genetic variation is significantly associated with altered DNA methylation in human islets. Represented are many previous reported Type 2 diabetes loci, such as ADCY5, KCNJ11, INS, PDX1 and GRB10. A causality inference test further suggested DNA methylation to mediate the effect of genotype on pancreatic islet mRNA expression levels and insulin secretion. Functional validation of candidate genes with genetic variation associated with both DNA methylation and mRNA expression, showed that silencing of Gpx7 or Gstt1 results in increased cell death signaling, while silencing of Snx19 increases β cell proliferation in a β cell line [Citation64]. Indeed, there is evidence for a strong interaction between genetic and epigenetic factors, leading to altered tissue specific gene expression and eventually disease phenotypes.
Using DNA from blood, Bell et al. also performed an integrated analysis of interaction between genotypes and epigenotypes in the context of Type 2 diabetes [Citation65]. The method applied in this study investigates the average degree of DNA methylation over larger genomic regions, here exemplified by LD blocks of known Type 2 diabetes associated loci, and not the methylation level of individual CpG sites. Using this approach, they identified haplotype-specific DNA methylation in FTO, a locus previously associated with Type 2 diabetes and obesity [Citation66], but not for any of the other investigated Type 2 diabetes regions. Further evaluation of the FTO methylation signal pointed to a putative enhancer region, with CpG-creating SNPs affecting the ability for DNA methylation [Citation65], in agreement with enrichment of H3K4me1 in this region as previously described [Citation67,Citation68]. Tobi et al. showed that SNPs in IGF2 and INS, respectively, were associated with the level of DNA methylation in blood in gene regulatory regions [Citation69]. There was further an independent and additive effect of prenatal famine on the DNA methylation pattern of these genes.
Conclusion
As methods for analysis of epigenetic modifications have dramatically improved during the last years, maps describing the epigenome in various tissues in health and disease have evolved. In this review, we have specifically described differences in the epigenomes observed in Type 2 diabetic patients compared with nondiabetic control subjects, in tissues relevant for the disease, in other words, pancreatic islets, skeletal muscle, adipose tissue and blood. We have also included associations between DNA methylation and phenotypes or environmental factors relevant for Type 2 diabetes, for example, insulin secretion and action, BMI, HbA1c and exercise.
DNA methylation is highly relevant to serve as a diagnostic tool for Type 2 diabetes, as many epigenetic changes are seen prior to disease development, for example, associated with increased BMI or HbA1c in healthy individuals or detected in prospective studies. The finding of altered DNA methylation in genes relevant for Type 2 diabetes also in blood suggests that at least some of these epigenetic alterations are established in early development. Epigenetics also holds promises for therapeutic advances, with numerous studies of, for instance, HDAC inhibitors clearly demonstrating beneficial effects on diabetic phenotypes. However, there is a significant need to translate the research from cultured cell lines or animals into human clinical investigations.
The results presented in this review show that epigenetics is of major significance for Type 2 diabetes and improves our understanding of the disease, and will guide the development of future drugs.
Future perspective
The arrays investigating genome-wide DNA methylation have greatly contributed to improved knowledge and understanding of the human epigenome, however, whole genome bisulfite sequencing will take it to the next level by providing details of DNA methylation on single base resolution throughout the complete DNA sequence. As more genome-wide epigenetic data becomes available, there will also be a better understanding of the complex interplay between DNA methylation and the various histone modifications, acting together with genetic variation in gene regulation and establishment of the chromatin structure. Together, these data will help identify and develop novel diagnostic and therapeutic agents that can be used in the battle against Type 2 diabetes.
The DNA methylation pattern differs between patients with Type 2 diabetes and matched controls in target tissues for the disease, in other words, human pancreatic islets, skeletal muscle and adipose tissue.
Both insulin secretion and insulin resistance show associations with altered DNA methylation in multiple human tissues.
Alterations in the level of DNA methylation are widespread across the entire genome, but also present in numerous candidate genes for Type 2 diabetes.
Epigenetic variations are present prior to disease development and are found to be associated with risk factors for Type 2 diabetes, including age, BMI and HbA1c.
Patients with Type 2 diabetes display altered levels of DNA methylation of genes implicated in the disease also in blood samples, providing an opportunity to be used as clinical biomarkers.
As epigenetic changes are dynamic and potentially reversible, targeting drugs, for example, histone deacetylase (HDAC) inhibitors, are suggested for disease prevention or in the treatment of Type 2 diabetes.
Financial & competing interests disclosure
This work was supported by grants from the Swedish Research Council, Region Skåne, Lund University Diabetes Centre (LUDC), EXODIAB, Linné grant (B31 5631/2006), Knut & Alice Wallenberg Foundation, Fredrik & Ingrid Thurings Foundation, Kungliga Fysiografiska Society, Tore Nilssons Foundation, Påhlsson Foundation, Novo Nordisk Foundation, ALF, The Swedish Diabetes Foundation and Söderberg Foundation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Hales CN , BarkerDJ . Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis . Diabetologia35 ( 7 ), 595 – 601 ( 1992 ).
- Barker DJ , HalesCN , FallCH , OsmondC , PhippsK , ClarkPM . Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth . Diabetologia36 ( 1 ), 62 – 67 ( 1993 ).
- Eriksson JG , ForsenT , TuomilehtoJ , JaddoeVW , OsmondC , BarkerDJ . Effects of size at birth and childhood growth on the insulin resistance syndrome in elderly individuals . Diabetologia45 ( 3 ), 342 – 348 ( 2002 ).
- Hales CN , BarkerDJ , ClarkPMet al. Fetal and infant growth and impaired glucose tolerance at age 64 . BMJ303 ( 6809 ), 1019 – 1022 ( 1991 ).
- Lithell HO , MckeiguePM , BerglundL , MohsenR , LithellUB , LeonDA . Relation of size at birth to non-insulin dependent diabetes and insulin concentrations in men aged 50–60 years . BMJ312 ( 7028 ), 406 – 410 ( 1996 ).
- Wu Y , DingY , TanakaY , ZhangW . Risk factors contributing to Type 2 diabetes and recent advances in the treatment and prevention . Int. J. Med. Sci.11 ( 11 ), 1185 – 1200 ( 2014 ).
- Ling C , GroopL . Epigenetics: a molecular link between environmental factors and Type 2 diabetes . Diabetes58 ( 12 ), 2718 – 2725 ( 2009 ).
- Laird PW . Principles and challenges of genomewide DNA methylation analysis . Nat. Rev. Genet.11 ( 3 ), 191 – 203 ( 2010 ).
- Jones PA . Functions of DNA methylation: islands, start sites, gene bodies and beyond . Nat. Rev. Genet.13 ( 7 ), 484 – 492 ( 2012 ).
- Cheng X , BlumenthalRM . Coordinated chromatin control: structural and functional linkage of DNA and histone methylation . Biochemistry49 ( 14 ), 2999 – 3008 ( 2010 ).
- Hashimoto H , VertinoPM , ChengX . Molecular coupling of DNA methylation and histone methylation . Epigenomics2 ( 5 ), 657 – 669 ( 2010 ).
- Muhonen P , HolthoferH . Epigenetic and microRNA-mediated regulation in diabetes . Nephrol. Dial. Transplant.24 ( 4 ), 1088 – 1096 ( 2009 ).
- Dayeh T , VolkovP , SaloSet al. Genome-wide DNA methylation analysis of human pancreatic islets from Type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion . PLoS Genet.10 ( 3 ), e1004160 ( 2014 ).
- Volkmar M , DedeurwaerderS , CunhaDAet al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from Type 2 diabetic patients . EMBO J.31 ( 6 ), 1405 – 1426 ( 2012 ).
- Hall E , VolkovP , DayehTet al. Effects of palmitate on genome-wide mRNA expression and DNA methylation patterns in human pancreatic islets . BMC Med.12 , 103 ( 2014 ).
- Grant SF , ThorleifssonG , ReynisdottirIet al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of Type 2 diabetes . Nat. Genet.38 ( 3 ), 320 – 323 ( 2006 ).
- Nogueira TC , PaulaFM , VillateOet al. GLIS3, a susceptibility gene for Type 1 and Type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim . PLoS Genet.9 ( 5 ), e1003532 ( 2013 ).
- Ling C , Del GuerraS , LupiRet al. Epigenetic regulation of PPARGC1A in human Type 2 diabetic islets and effect on insulin secretion . Diabetologia51 ( 4 ), 615 – 622 ( 2008 ).
- Lin J , HandschinC , SpiegelmanBM . Metabolic control through the PGC-1 family of transcription coactivators . Cell Metab.1 ( 6 ), 361 – 370 ( 2005 ).
- Jonsson A , IsomaaB , TuomiTet al. A variant in the KCNQ1 gene predicts future Type 2 diabetes and mediates impaired insulin secretion . Diabetes58 ( 10 ), 2409 – 2413 ( 2009 ).
- Unoki H , TakahashiA , KawaguchiTet al. SNPs in KCNQ1 are associated with susceptibility to Type 2 diabetes in East Asian and European populations . Nat. Genet.40 ( 9 ), 1098 – 1102 ( 2008 ).
- Yasuda K , MiyakeK , HorikawaYet al. Variants in KCNQ1 are associated with susceptibility to Type 2 diabetes mellitus . Nat. Genet.40 ( 9 ), 1092 – 1097 ( 2008 ).
- Travers ME , MackayDJ , Dekker NitertMet al. Insights into the molecular mechanism for Type 2 diabetes susceptibility at the KCNQ1 locus from temporal changes in imprinting status in human islets . Diabetes62 ( 3 ), 987 – 992 ( 2013 ).
- Kuroda A , RauchTA , TodorovIet al. Insulin gene expression is regulated by DNA methylation . PLoS ONE4 ( 9 ), e6953 ( 2009 ).
- Yang BT , DayehTA , KirkpatrickCLet al. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA(1c) levels in human pancreatic islets . Diabetologia54 ( 2 ), 360 – 367 ( 2011 ).
- Yang BT , DayehTA , VolkovPAet al. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with Type 2 diabetes . Mol. Endocrinol.26 ( 7 ), 1203 – 1212 ( 2012 ).
- Kaneto H , MiyatsukaT , KawamoriDet al. PDX-1 and MafA play a crucial role in pancreatic beta-cell differentiation and maintenance of mature beta-cell function . Endocr. J.55 ( 2 ), 235 – 252 ( 2008 ).
- Drucker DJ . The biology of incretin hormones . Cell Metab.3 ( 3 ), 153 – 165 ( 2006 ).
- Tornehave D , KristensenP , RomerJ , KnudsenLB , HellerRS . Expression of the GLP-1 receptor in mouse, rat, and human pancreas . J. Histochem. Cytochem.56 ( 9 ), 841 – 851 ( 2008 ).
- Hall E , DayehT , KirkpatrickCL , WollheimCB , Dekker NitertM , LingC . DNA methylation of the glucagon-like peptide 1 receptor (GLP1R) in human pancreatic islets . BMC Med. Genet.14 , 76 ( 2013 ).
- Kameswaran V , BramswigNC , MckennaLBet al. Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human Type 2 diabetic islets . Cell Metab.19 ( 1 ), 135 – 145 ( 2014 ).
- Benetatos L , HatzimichaelE , LondinEet al. The microRNAs within the DLK1-DIO3 genomic region: involvement in disease pathogenesis . Cell Mol. Life Sci.70 ( 5 ), 795 – 814 ( 2013 ).
- Correa-Medina M , Bravo-EganaV , RoseroSet al. MicroRNA miR-7 is preferentially expressed in endocrine cells of the developing and adult human pancreas . Gene Expr. Patterns9 ( 4 ), 193 – 199 ( 2009 ).
- Wang Y , LiuJ , LiuC , NajiA , StoffersDA . MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic beta-cells . Diabetes62 ( 3 ), 887 – 895 ( 2013 ).
- Canivell S , RuanoEG , Siso-AlmirallAet al. Differential methylation of TCF7L2 promoter in peripheral blood DNA in newly diagnosed, drug-naive patients with Type 2 diabetes . PLoS ONE9 ( 6 ), e99310 ( 2014 ).
- Del Rosario MC , OssowskiV , KnowlerWC , BogardusC , BaierLJ , HansonRL . Potential epigenetic dysregulation of genes associated with MODY and Type 2 diabetes in humans exposed to a diabetic intrauterine environment: an analysis of genome-wide DNA methylation . Metabolism63 ( 5 ), 654 – 660 ( 2014 ).
- Gu HF , GuT , HildingAet al. Evaluation of IGFBP-7 DNA methylation changes and serum protein variation in Swedish subjects with and without Type 2 diabetes . Clin. Epigenetics5 ( 1 ), 20 ( 2013 ).
- Gu T , GuHF , HildingAet al. Increased DNA methylation levels of the insulin-like growth factor binding protein 1 gene are associated with Type 2 diabetes in Swedish men . Clin Epigenetics5 ( 1 ), 21 ( 2013 ).
- Toperoff G , AranD , KarkJDet al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood . Hum. Mol. Genet.21 ( 2 ), 371 – 383 ( 2012 ).
- Isomaa B , ForsenB , LahtiKet al. A family history of diabetes is associated with reduced physical fitness in the Prevalence, Prediction and Prevention of Diabetes (PPP)-Botnia study . Diabetologia53 ( 8 ), 1709 – 1713 ( 2010 ).
- Nitert MD , DayehT , VolkovPet al. Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with Type 2 diabetes . Diabetes61 ( 12 ), 3322 – 3332 ( 2012 ).
- Barres R , YanJ , EganBet al. Acute exercise remodels promoter methylation in human skeletal muscle . Cell Metab.15 ( 3 ), 405 – 411 ( 2012 ).
- Barres R , OslerME , YanJet al. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density . Cell Metab.10 ( 3 ), 189 – 198 ( 2009 ).
- Ronti T , LupattelliG , MannarinoE . The endocrine function of adipose tissue: an update . Clin. Endocrinol. (Oxf)64 ( 4 ), 355 – 365 ( 2006 ).
- Ronn T , VolkovP , DavegardhCet al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue . PLoS Genet.9 ( 6 ), e1003572 ( 2013 ).
- Ronn T , VolkovP , TornbergAet al. Extensive changes in the transcriptional profile of human adipose tissue including genes involved in oxidative phosphorylation after a 6-month exercise intervention . Acta Physiol. (Oxf)211 ( 1 ), 188 – 200 ( 2014 ).
- Mccarthy MI . Genomics, Type 2 diabetes and obesity . N. Engl. J. Med.363 ( 24 ), 2339 – 2350 ( 2010 ).
- Chen XW , LetoD , ChiangSH , WangQ , SaltielAR . Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c . Dev. Cell.13 ( 3 ), 391 – 404 ( 2007 ).
- Singhal J , NagaprashanthaL , VatsyayanR , AwasthiS , SinghalSS . RLIP76, a glutathione-conjugate transporter, plays a major role in the pathogenesis of metabolic syndrome . PLoS ONE6 ( 9 ), e24688 ( 2011 ).
- Nilsson E , JanssonPA , PerfilyevAet al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes . Diabetes63 ( 9 ), 2962 – 2976 ( 2014 ).
- Grundberg E , MeduriE , SandlingJKet al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements . Am. J. Hum. Genet.93 ( 5 ), 876 – 890 ( 2013 ).
- Ribel-Madsen R , FragaMF , JacobsenSet al. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for Type 2 diabetes . PLoS ONE7 ( 12 ), e51302 ( 2012 ).
- Zhao J , GoldbergJ , BremnerJD , VaccarinoV . Global DNA methylation is associated with insulin resistance: a monozygotic twin study . Diabetes61 ( 2 ), 542 – 546 ( 2012 ).
- Simar D , VersteyheS , DonkinIet al. DNA methylation is altered in B and NK lymphocytes in obese and Type 2 diabetic human . Metabolism63 ( 9 ), 1188 – 1197 ( 2014 ).
- Gokulakrishnan K , VelmuruganK , GanesanS , MohanV . Circulating levels of insulin-like growth factor binding protein-1 in relation to insulin resistance, Type 2 diabetes mellitus, and metabolic syndrome (Chennai Urban Rural Epidemiology Study 118) . Metabolism61 ( 1 ), 43 – 46 ( 2012 ).
- Petersson U , OstgrenCJ , BrudinL , BrismarK , NilssonPM . Low levels of insulin-like growth-factor-binding protein-1 (IGFBP-1) are prospectively associated with the incidence of type 2 diabetes and impaired glucose tolerance (IGT): the Soderakra Cardiovascular Risk Factor Study . Diabetes Metab.35 ( 3 ), 198 – 205 ( 2009 ).
- Kim JA , WeiY , SowersJR . Role of mitochondrial dysfunction in insulin resistance . Circ. Res.102 ( 4 ), 401 – 414 ( 2008 ).
- Gemma C , SookoianS , DieuzeideGet al. Methylation of TFAM gene promoter in peripheral white blood cells is associated with insulin resistance in adolescents . Mol. Genet. Metab.100 ( 1 ), 83 – 87 ( 2010 ).
- Ling C , PoulsenP , SimonssonSet al. Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle . J. Clin. Invest.117 ( 11 ), 3427 – 3435 ( 2007 ).
- Ronn T , PoulsenP , HanssonOet al. Age influences DNA methylation and gene expression of COX7A1 in human skeletal muscle . Diabetologia51 ( 7 ), 1159 – 1168 ( 2008 ).
- Mootha VK , LindgrenCM , ErikssonKFet al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes . Nat. Genet.34 ( 3 ), 267 – 273 ( 2003 ).
- Jacobsen SC , BronsC , Bork-JensenJet al. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men . Diabetologia55 ( 12 ), 3341 – 3349 ( 2012 ).
- Dayeh TA , OlssonAH , VolkovP , AlmgrenP , RonnT , LingC . Identification of CpG-SNPs associated with Type 2 diabetes and differential DNA methylation in human pancreatic islets . Diabetologia56 ( 5 ), 1036 – 1046 ( 2013 ).
- Olsson AH , VolkovP , BacosKet al. Genome-wide associations between genetic and epigenetic variation influence mRNA expression and insulin secretion in human pancreatic islets . PLoS Genet.10 ( 11 ), e1004735 ( 2014 ).
- Bell CG , FinerS , LindgrenCMet al. Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO Type 2 diabetes and obesity susceptibility locus . PLoS ONE5 ( 11 ), e14040 ( 2010 ).
- Frayling TM , TimpsonNJ , WeedonMNet al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity . Science316 ( 5826 ), 889 – 894 ( 2007 ).
- Bhandare R , SchugJ , Le LayJet al. Genome-wide analysis of histone modifications in human pancreatic islets . Genome Res.20 ( 4 ), 428 – 433 ( 2010 ).
- Heintzman ND , HonGC , HawkinsRDet al. Histone modifications at human enhancers reflect global cell-type-specific gene expression . Nature459 ( 7243 ), 108 – 112 ( 2009 ).
- Tobi EW , SlagboomPE , Van DongenJet al. Prenatal famine and genetic variation are independently and additively associated with DNA methylation at regulatory loci within IGF2/H19 . PLoS ONE7 ( 5 ), e37933 ( 2012 ).