
Abstract
Lung cancer is the commonest cause of cancer death in the world and carries a poor prognosis for most patients. While precision targeting of mutated proteins has given some successes for never- and light-smoking patients, there are no proven targeted therapies for the majority of smokers with the disease. Despite sequencing hundreds of lung cancers, known driver mutations are lacking for a majority of tumors. Distinguishing driver mutations from inconsequential passenger mutations in a given lung tumor is extremely challenging due to the high mutational burden of smoking-related cancers. Here we discuss the methods employed to identify driver mutations from these large datasets. We examine different approaches based on bioinformatics, in silico structural modeling and biological dependency screens and discuss the limitations of these approaches.
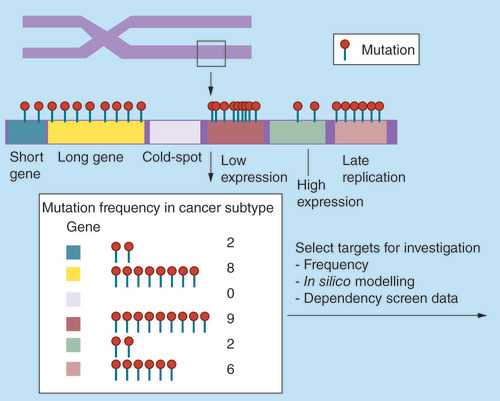
In areas of uniform mutational rates longer coding genes will demonstrate a higher mutational frequency if the data are not length corrected. Genes that are not expressed and those that replicate late in the cell cycle will have higher mutational rates. Genes with large GC-rich regions will have inadequate sequencing coverage and potential mutations will be missed leading to an underreporting of mutations in these genes.
Lung cancer is the most common cause of cancer death in the world; only 16.8% of patients survive to 5 years following a diagnosis of lung cancer [Citation1]. This is in stark contrast to prostate cancer (98.9% surviving to 5 years) and breast cancer (89.2% surviving to 5 years). A major reason for this disparity is that metastatic disease is diagnosed at presentation in the majority of lung cancer cases. In addition, the median age of lung cancer diagnosis is around 70 years, and patients have often smoked for a large period of their life, making successful treatment of lung cancer patients extremely challenging. As a consequence of smoking, many patients possess severe co-existing medical conditions that preclude them from receiving potentially toxic chemotherapeutic regimens. These patients cannot receive an active anticancer treatment and are only eligible for symptomatic palliation. Further, while some patients will benefit from palliative chemotherapy to extend survival, this is often short-lived and accompanied by toxic side effects. Therefore, the promise offered by targeted therapies, with their better-tolerated side effects, is of particular significance for lung cancer patients.
Most clinically effective targeted therapies rely on disruption of ‘oncogene addiction’ that occurs through genetic mutation or overexpression of genes conferring tumorigenic properties in line with the hallmarks of cancer [Citation2,Citation3]. The success of targeted precision therapies lies in identifying mutated genes that confer a growth or survival advantage (driver mutations) that can be subsequently targeted therapeutically. There have been some notable successes with this approach. EGF receptor (EGFR) inhibitors were first introduced into the clinic for the treatment of non small-cell lung cancer (NSCLC). The IPASS study compared the EGFR inhibitor gefitinib with a standard doublet chemotherapy regimen in patients from East Asia with first-line advanced lung adenocarcinoma [Citation4]. It showed superior progression-free survival (PFS) in the gefitinib arm as well as lower rates of severe toxicity. While the study did not stratify treatment based on EGFR mutation status, subgroup analysis showed that EGFR mutation positive patients had longer PFS with gefitinib while EGFR mutation negative patients had longer PFS with standard chemotherapy. The study population, consisting of East Asian never or light-smokers, was enriched for EGFR mutations (59.7%) compared with the heavy smoking population that forms the majority of Western lung adenocarcinoma cases (11% with EGFR mutation) [Citation5]. Subsequent trials of gefitinib, erlotinib and afatinib have demonstrated superior PFS in EGFR mutation positive patients when compared with standard chemotherapy leading to these agents being routinely used for treatment in EGFR mutation positive patients [Citation6–9]. More recently, ALK rearrangements have been identified in approximately 5% of NSCLC [Citation10]. Again patients with ALK rearrangements are more likely to be never/light smokers [Citation11]. Crizotinib, a small-molecule inhibitor of ALK (as well as MET and ROS1 kinases), has been shown to offer improved PFS, lower toxicity and better quality of life compared with standard chemotherapy [Citation12].
Other known oncogenes have been discovered to be mutated in a proportion of NSCLC cases and are currently being assessed in early phase clinical trials. The most commonly mutated gene is KRAS (approximately 25% depending on histology and more frequent in heavy smokers) [Citation13]. KRAS itself is not easily targetable and clinical trials have been developed using MEK inhibitors in combination with chemotherapy to block the downstream effects of oncogenic KRAS [Citation14]. This approach has seen some encouraging responses in KRAS mutation positive patients but phase 3 data are awaited and MEK inhibition on its own may not be sufficient in these patients given the multiple downstream effectors of KRAS. An additional downstream target of GOF mutant KRAS is PI3K (phosphoinosotide 3-kinase), where activation of PI3K leads to PIP3 (phosphoinosotide (3,4,5)-trisphosphate) mediated AKT activation to promote cancer cell survival [Citation15]. It would be expected that combination PI3K/MEK inhibitor therapy would promote tumor regression, however KRAS mutation positive colon cancer PDX models failed to demonstrate tumor regression highlighting the challenges in treating KRAS mutation positive cancers [Citation16]. BRAF is mutated in approximately 3% of patients (with half of cases being the V600E mutation that have been targeted to much success in melanoma) and early phase trials are taking place with BRAF inhibitors [Citation17]. However, the poor clinical response to V600E BRAF inhibition due to EGFR activation in colorectal tumors adds caution to any predictions of efficacy in lung cancer [Citation18]. HER2 amplification and activating mutations are seen in a proportion of NSCLC but clinical targeting with trastuzumab and lapatnib have not shown the efficacy seen in HER2 amplified breast cancer patients [Citation19,Citation20]. Other genetic alterations such as MET amplifications (8–10%), RET rearrangements (1–2%) and gain-of-function mutations in PIK3CA/AKT (2–5%) are being targeted in early phase clinical trials [Citation21].
A biomarker-based precision medicine trial (known as the BATTLE trial) used tumor biopsies to stratify lung cancer patients into different treatment arms based on up-to-date mutational profiling, demonstrating early disease control for certain biomarker–drug combinations and highlighting that this approach is feasible [Citation22]. However, response rates were poor due to a heavily pretreated patient population combined with a lack of identifiable driver mutations and inhibitors for treatment. Overwhelmingly the most important factor in preventing this personalized approach for lung cancer is the lack of identifiable driver mutations. It was recently estimated that at least three driver mutations are required for the development of lung cancers [Citation23]. However, despite the whole exome sequencing of hundreds of lung cancer samples, approximately 50% of NSCLC have no identifiable activating mutations [Citation24]. In this review we shall discuss the different approaches and challenges to mining cancer genomics data to discover druggable driver mutations in lung cancer.
Online aggregated cancer genomics data
To enhance driver mutation discovery, large repositories of cancer genomics data have been published online. Aggregating the data from large numbers of sequenced cancers will aid in the discovery of commonly mutated genes for specific cancer subtypes. cBio is one of the most widely used databases, combining data from The Cancer Genome Atlas (TCGA) samples with other large studies including The Cancer Cell Line Encyclopedia (CCLE) [Citation25,Citation26]. Users can search by gene name to retrieve the frequency of mutation in different cancer subtypes and identify novel targets to evaluate further. The most straightforward approach is to seek genes that are commonly mutated in a large proportion of cancers so that effective and financially viable drug development can be undertaken for that target. However, this approach requires a strategy to distinguish somatic mutations that drive the oncogenic process (driver mutations) from somatic mutations that do not have a functional effect on the cell (passenger mutations) [Citation27]. In lung cancer, this is particularly challenging. Tobacco smoke contains a multitude of powerful carcinogens that form DNA adducts resulting in much higher mutational rates than a majority of other cancers [Citation28,Citation29]. The high mutational burden that yields large numbers of passenger mutations make it difficult to identify the driver mutations amongst this background of numerous inconsequential mutations [Citation30]. At the time of writing, the average number of protein coding mutations identified in lung squamous and lung adenocarcinoma TCGA samples was 319 and 280, respectively. This is 11–13-times greater than acute myeloid leukemia (AML), for which only 24 protein-coding mutations were reported per sample. In AML, the relatively low mutation burden has aided the discovery of more drivers. A study of 200 AML samples, for example, found that 99.5% possessed a nonsynonymous somatic mutation in a gene of biological significance [Citation31,Citation32]. This is in marked contrast to NSCLC in which only approximately 50% of cases have a known activating mutational driver [Citation24].
Given the relative failure to identify many common mutations in lung cancer it is likely that unknown cases are characterized by small groups of drivers, each accounting for 1–2% of the total. These drivers, affecting only a small proportion of patients can still be beneficial to pursue. This was evident in an expanded Phase 1 study of crizotinib, in which some patients with ROS1 rearrangements (only 1–2% of NSCLC patients) had dramatic responses to the drug with an overall longer median progression free survival than EML4-ALK patients receiving the same drug [Citation33,Citation34]. Given the high incidence of lung cancer, these targets that involve 1–2% of cases, represent a large global patient cohort.
Playing the numbers game
While cancer genomics data can be interrogated for genes with frequent mutations that segment according to histological type, it is not sufficient to assess statistical significance on the assumption that mutation rates for all genes are the same. Instead, more sophisticated models consider additional factors: first, gene-level mutation rates may be normalized according to length because when assuming a uniform background mutation rate, longer genes are more likely to acquire a mutation than shorter ones. Therefore, extremely long genes such as TTN have a high mutational frequency (52% in lung squamous TCGA data). lists the top 20 most frequently mutated genes in squamous lung cancer. Seven of these encode proteins in the top 20 longest proteins. The median protein length for the top 20 mutated is 4612 amino acids (mean for all proteins screened = 699 amino acids). While this effect is noted, it does not mean that very large proteins do not play a role in oncogenesis and indeed some propose that genes such as TTN may be an important driver of tumor progression [Citation35]. The length of these proteins obviously makes subsequent biological work more challenging and researchers may shy away from the associated technical challenges in search of lower hanging fruit.
Second, another issue that is particularly apparent in samples with a high mutation burden is that many mutations occur in genes that are not expressed as proteins in the given tumor. This is caused, at least in part by the differential effect of transcription-coupled repair; genes that are not expressed are less likely to be repaired and therefore mutations at these loci accumulate [Citation36,Citation37]. Following length correction the top 20 mutated genes () now contain a number of genes, such as olfactory receptors, that are not expressed in squamous lung cancer, and are unlikely to have a functional effect. The length corrected top 20 also contain very small proteins with a small number of incidental mutations as well as larger proteins with biological evidence of significance in lung cancer [Citation38,Citation39].
Third, replication timing is also important, since genes that replicate late will have a depleted pool of nucleotides available, and are therefore more likely to acquire mutations [Citation36]. This phenomenon has been used to explain increased germline variability and somatic mutations in late-replicating regions [Citation36,Citation40,Citation41]. This knowledge may help to explain the high density of mutations in a specific locus, but, as with issues of gene length, it does not rule out the possibility that a late-replication gene might play a significant role in cancer. Replication timing and expression have been used to develop the MutSigCV platform to better identify driver mutations using estimations of the background mutation rates in different cancer types using silent and noncoding mutations in a genetic region [Citation36]. However, it is acknowledged that larger amounts of next generation sequencing is required to get a better picture of local mutation rates and improve the method.
Fourth, intratumoral mutation heterogeneity in NSCLC primary tumors has been demonstrated in two studies [Citation42,Citation43]. Zhang et al. showed that 76% of mutations were identified in all regions of individual tumors (including 20 out of 21 known cancer gene mutations) suggesting that analysis of primary tumor genomes will capture most driver mutations. However, it is not known if metastatic lesions that make up a large proportion of clinical presentations and have the most to gain from targeted systemic treatments, share this degree of homogeneity. While the TCGA dataset is comprised of primary tumors, cell lines are frequently derived from metastatic tissue; these differences need to be considered when pursuing candidate mutations in experimental systems based on tumor-derived cell lines. It is also important to consider whether previous lines of therapy have led to the selection of specific genetic clones. The fact that the primary tumors in Zhang et al. had especially good concordance for known cancer causing mutations suggests that selecting mutations with high allele frequency (reported in TCGA data) may be a beneficial strategy. However, uncertainties about the sampling and proportion of normal tissue contamination make this difficult.
Fifth, discrepancies between NGS datasets of the same samples highlight further challenges and opportunities with large-scale genomics data. Two prominent cancer genomics institutes (The Broad Institute [CCLE] and The Sanger Institute [COSMIC]) have published NGS data of commercially available cancer cell lines [Citation26,Citation44]. This work has been extremely beneficial to researchers around the world who have been able to use the data to select relevant cell lines in which to test their hypotheses. We had observed some inconsistencies between the two datasets, leading us to perform a formal comparison of the missense mutations reported by the two different institutes [Citation45]. We demonstrated marked discrepancies between the datasets with only 57% of the mutations reported across 568 cell lines being concordant. We found that one of the major reasons for this discordance was that GC-rich areas of the exome are still proving difficult to sequence by NGS, leading to over 400 significant areas of poor sequencing (cold-spots) in known cancer causing genes and kinases. A conservative estimate suggested that approximately three missense mutations occurring within known cancer census and kinase genes were being missed in each cell line (with many other mutations being missed at other loci). TCGA data are generally of a similar age and obtained with similar technologies, suggesting that these data may also suffer from cold-spots. GC-rich cold spots are more relevant in cancers, such as lung cancers, where mutations occurring more frequently in guanine nucleotides. It is likely, therefore, that mutations in genes with GC rich regions are under-reported in lung cancer, and suggests that these loci harbor additional common mutations that have yet to be identified.
Sixth, another source of disparity between the datasets we studied was poor consensus in the labeling of variants as either germline or somatic. The majority of cell lines do not have paired normal tissue for comparison, and therefore the somatic status of an observed variant was performed by matching to databases of known germline variants. The most common method employed simply removes all variants with an ‘rs’ oasis: entry in the dbSNP database [Citation46]. Unfortunately, dbSNP database is rapidly evolving and expanding, with the side effect that the date at which the filtering was performed can greatly affect the final output. Further, database submission is unrestricted leading to the occasional inclusion of a somatic mutation in error. Finally, high GC content can also lead to underreporting of germline variants in hard-to-sequence loci as well as bona fide mutations. As NGS technology improves, common germline SNPs in these regions can be uncovered, but since earlier germline sequencing approaches failed to identify them, they are not filtered against dbSNP leading them to be erroneously reported as rare or somatic.
From numbers to predictions: in silico analysis of mutations
Another method used to distinguish between driver and passenger mutations is to consider the structural impact of the resultant amino acid substitution. Tools such as mutationassessor.org, Polyphen2, Provean and SIFT are freely available online, and use information such as protein structure and sequence homology to predict whether a mutation might have a functional impact [Citation47–50]. They can be used to quickly analyze large batches of genomics data to allow researchers to assess whole exome data to select those mutations most likely to alter the function of the protein. A recent evaluation of these different tools demonstrated that they worked well to distinguish known pathogenic mutations from neutral ones, and that predictive power could be further enhanced by combining the outputs from multiple tools [Citation51]. From our experience, loss-of-function mutations (with their presumed greater structural disruption) are more likely to be identified than some more subtle activating oncogenes using these methods [Citation52]. Additional complexity arises because the majority of human protein-coding genes express more than one isoform, with the result that a missense mutation can have different effects according to the isoform it occurs in, and may not be present at all if it occurs in a spliced exon. Furthermore, proper interpretation of the data is difficult without access to protein expression data, since highly functional mutations will clearly not have an effect if that protein or mutant allele is not expressed. One useful source for these data is the Human Protein Atlas, which provides a valuable online resource in which immunohistochemistry data are used to catalogue the expression of proteins in different cancer types [Citation53].
The challenge of identifying driver mutations is well summarized by Tamborero and colleagues, who state that ‘The elucidation of cancer drivers relies on identifying the marks of positive selection that occur during the clonal evolution of tumors’ [Citation54]. These positive marks present themselves in various ways, from the clustering of mutations in a specific protein domain to the correlation of a mutated gene with a specific sub-phenotype present within the patient. Therefore state-of-the-art attempts at driver mutation identification combine a wide breadth of different data to identify the marks of positive selection. One such package is ‘MuSiC’ which combines statistical tests of mutational frequency and co-occurrence, clinical data, and information pertaining to the frequency of mutations in specific protein domains [Citation55]. Identifying mutations clustered at specific loci (whether using a focused approach based on known protein domain function or just identifying mutations in close proximity to another) can highlight potential mechanisms of positive selection. Another analysis focused on mutations only occurring in phosphosites of proteins to extract novel targets [Citation56]. Increasing understanding of protein domain function, from wet-lab studies, will provide further opportunities to create functionally relevant screens.
Correlating mutational data with copy number deletions, immunohistochemistry, and considering the frequency of truncating mutations may assist in the prediction of loss-of-function mutations. However these cannot be solely relied upon given the effects of co-existing mutations and expression causing varying redundancy and unknowns regarding the presence or absence of a dominant negative effect [Citation57].
If greater computational resources are available, molecular dynamic (MD) simulations can provide more in depth in silico approaches with which to assess the effect of a given mutation. MD simulations model the movement of a protein over very short times scales (generally in the nanosecond range), making it possible to predict the structural variations that occur as a consequence of a mutation.
Initial MD simulations are kept relatively short, simulating up to 50 ns of time, due to the large computational burden required for such simulations. These typically focus on biochemically significant mutations that have either been published previously [Citation58,Citation59] or are analyzed in vitro/in vivo within the study itself [Citation60,Citation61]. These short simulations are generally utilized to confirm biochemical data and gain further understanding of the structural consequences of the identified mutation. As these simulations are only short, the information gained can range from as little as the position of the mutations in relation of other regions of the protein [Citation60] and movement of key regions of the structure [Citation61] up to changes in binding affinities of key substrates [Citation59]. All of this information can help to understand the potential impact the mutation is having on the protein in question.
More information can be gleaned from longer MD simulation studies. Many longer simulations focus on the alteration of key structural features (e.g., salt bridges, domain or feature orientation and drug binding) and how these affect the free energy landscape of the protein [Citation62–65]. These types of studies have provided information on the progression of these proteins into more active conformations following disease causing mutations [Citation64,Citation66] as well as critical information on the effect of mutations on drug resistance [Citation65,Citation66]. Such information is critical to understanding the structural effects occurring following mutation and providing the research community with this type of analysis could aid in drug development. However, in order to perform these MD simulations, a crystal structure of the protein is required. Furthermore, these more complex in silico methods require both a high level of computational knowledge and a large computational resource.
Genetic dependency screens
‘Oncogene addiction’ describes a phenomenon whereby cancer cells develop a dependency on a specific oncogene that has become either overexpressed or activated by mutation during the development of the cancer. This dependency leaves the cancer vulnerable should the activated oncogene be inhibited or suppressed. A number of mechanisms have been proposed to explain how dependency on a single oncogene occurs in tumors with a high burden of genetic mutations [Citation67]. This dependency creates a desirable differential between the normal and cancer cell that can be exploited and targeted for therapeutic intervention. Most clinically valuable targeted treatments owe their beneficial effect to disrupting the ‘oncogene addiction’ of a cancer cell to a mutated gene that drives cellular growth or survival. In fact it has been postulated that ‘most, if not all, dramatic responses of ’tumor shrinkage’ following molecularly targeted therapy result from the acute inactivation of an activated oncoprotein upon which the tumor cells became dependent’’ [Citation68]. Substantial effort is now being channeled into discovering more of these oncogenes against which small molecule inhibitors can be developed for cancer treatment.
Genetic dependency screens aim to exploit the phenomenon of oncogene addiction in a high-throughput manner using small interfering RNA against multiple targets and assessing the functional outcome in the cancer cell [Citation69]. Commercially available si/shRNA libraries mean the method is now widely used by groups investigating novel drivers of oncogenesis leading to novel target discovery [Citation70–75]. Project Achilles is a huge project from The Broad Institute initially undertaking the silencing of thousands of genes with shRNA in hundreds of cell lines [Citation76]. This has yielded some novel targets in different cancer subtypes [Citation77–79].
Rather than performing a genome-wide study, we developed a targeted approach using siRNA to knockdown only those genes harboring somatic mutations in specific lung cancer cell lines and assessed the effects on proliferation and cell survival [Citation61]. In three out of six of these cell lines we discovered novel gain of function mutations that were subsequently validated. One benefit of this approach is a clear endpoint, where the genes identified to harbor potential gain-of-function mutations, can then be generated in the laboratory and tested to determine if the mutation does in fact increase the catalytic activity of the protein. Alternatively the cancer mutant can be expressed in cells to determine if the mutant allele may promote increases in survival or proliferation by more subtle mechanisms such as altered cellular localization or differential substrate specificity. The mutant genes we identified (PAK5, FGFR4 and MAP3K9) all activated the MEK-ERK pathway and the respective cell lines had increased sensitivity to MEK inhibitors. Analyzing lung adenocarcinoma TCGA data reveals that the frequencies of cases with mutations in these genes are: PAK5 (11%), FGFR4 (5.2%) and MAP3K9 (4.7%). Inputting these three mutations into different online mutation assessors often predicts the FGFR4 and MAP3K9 mutations as unlikely to be pathogenic (). This highlights the benefit of using a targeted siRNA screen to identify novel drivers that would otherwise not be predicted to be pathogenic.
While the MAP3K9 mutation in the H2009 cell line was reported by CCLE, it was not reported by an earlier version of COSMIC that also sequenced the cell line. Therefore, using just COSMIC data would have missed this gain-of-function mutation. This demonstrates how a targeted genetic dependency screen relies on high quality genomics data to ensure all genes mutated in a sample are silenced. The issue is also important for nontargeted genetic dependency screens such as Project Achilles as well as pharmacogenomics screens. If the mutational data from these cell lines are not complete it hinders attempts to interpret the phenotypic response of the cell to the knockdown or inhibition of a specific gene.
Interestingly, siRNA knockdown screens of the remaining three cell lines in our study did not identify a stand out driver mutation in terms of cellular proliferation. It remains to be seen whether this is due to no mutational drivers present in the cell lines or that a mutation is present but being missed by inadequate sequencing of GC rich regions. Another consideration is that the driver mutations in these cell lines may exert their effect through loss-of-function mechanisms and a targeted screen like ours will not identify these. Synthetic lethality describes a mechanism by which tumor cells often become more dependent on a gene than a normal cell due to gain or loss of function of a different gene during the development of the cancer [Citation67,Citation80,Citation81]. Therefore, genome-wide knockdown, such as that used in the Achilles project, can be utilized to identify synthetically lethal genes that may be druggable. In addition, it is now possible to perform high-throughput screens for tumor suppressor genes using CRISPR/CAS technology [Citation82].
Conclusion & future perspective
We highlight the challenges of using cancer genomics data aggregated from a large number of samples to identify driver mutations. Given the urgent need for effective targeted therapies against lung cancer, it is important to develop solutions to tackle this problem. The first step is to identify commonly mutated signaling pathways against which to develop targeted therapies. The three clinically successful targets mentioned in this review (EGFR mutation, ALK rearrangements and ROS1 rearrangements) all occur predominantly in nonsmokers. Therefore smokers, often with multiple co-morbidities, lack targeted therapy options. Unfortunately, the mechanisms by which cigarette smoke causes cancer mean that smoking related tumors have a high mutational burden with many passenger mutations. illustrates the sources of potential bias in the analysis of aggregated genomics data. These issues become more problematic when mutational noise is increased. Gene length, expression level and replication timing all have the potential to distort mutation frequencies making it harder to identify driver mutations. We have shown how the ability to sequence difficult regions is improving with technological advances and that older data may be susceptible to bias due to sequencing cold-spots. It is obviously preferable to obtain matched normal tissue samples for comparison and, when this is not possible, inconsistencies in dbSNP reporting will impair the ability to identify driver mutations. Since germline data are obtained from a wide range of sources and situations, it can never be considered as reliable as matched normal tissue. A recent study has demonstrated that false-positive calling of actionable mutations is significantly increased without normal tissue control [Citation83]. Noncoding RNA and intronic mutations are not discussed here but present additional challenges.
Structural analysis has the potential to predict functional outcomes on a protein with a high degree of accuracy, but is currently unable to model how complex interactions between proteins within a cancer cell are disrupted as a consequence of co-occurring mutations, variability in gene expression and additional regulatory pathways involving, for example, miRNAs and other noncoding loci. The limitations of mutation assessors are illustrated by our siRNA screen data in which two of three activating mutations would be predicted to be neutral in most of the online mutation assessors. By contrast, potentially detrimental mutations supported by structural studies, are only of relevance if the protein is expressed in the tissue of interest.
Although the state of the art has advanced rapidly, biological confirmation of in silico results is still critical, and usually involves knockdown of the gene of interest with small interfering RNA (si/shRNA), combined with functional read-outs for proliferation, cell viability or apoptosis. It is possible to use these approaches to perform high-throughput studies, but limitations such as inadequate sequencing can make it hard in practice to associate observed sensitivity with mutation status, causing targets to be missed. Similarly large-scale inhibitor studies suffer the same limitations [Citation84]. In silico modeling is enhanced by the greater understanding of protein structure and function provided by wet-lab studies. Better characterization of protein domain function allows genomic data to be filtered for mutations by areas of functional importance. The biochemical validation of the effects of a mutation also provides valuable information with which to improve the training datasets used to develop these prediction tools. Driver mutation discovery is, therefore, enhanced if there is a virtuous circle in which existing genomics data are reanalyzed in the light of recent functional studies in order to identify further targets for evaluation at the bench – which then support additional rounds of progressive refinement and analysis.
NGS technology continues to progress rapidly, improving the coverage of hard to sequence regions and, as costs decrease, allowing genomes to be sequenced at higher depths. Together, these are contributing to substantial increases in the number of mutations that can be reliably detected. Unfortunately, since the technology itself is unable to distinguish between driver and passenger mutations, these advances come with the challenge of increased levels of mutational noise. The advances in genome profiling are, therefore, increasingly dependent on concomitant improvements in the techniques used to identify actionable mutations. While sequencing of cancer genomes will continue to accumulate, research efforts should shift to functional genomics to aid in the elucidation of novel drivers so that lung cancer patients can benefit from targeted therapies, likely in combination with immunotherapies. Pinpointing these drivers and targeting them with precision medicines will portend a future where lung cancer patients will be treated with therapies that extend survival while preserving quality of life.
Table 1A. Top 20 frequently mutated genes in squamous lung cancer with number of mutated cases and longest corresponding protein length.
Table 1B. Top 20 frequently mutated genes in squamous lung cancer normalized for length of longest protein and ranked by length corrected score.
Table 2. Mutation predictor data for the three pathogenic mutations discovered with a targeted siRNA screen [Citation61].
Online aggregated cancer genomics data
Large-scale cancer genomics programs such as TCGA, COSMIC and CCLE have been instrumental in discovering novel mutational drivers of cancer in many cancer subtypes.
In lung cancer there have been some notable clinical successes in targeting these drivers (EGFR mutations, ALK translocations, ROS translocations). However, the majority of lung cancer patients who benefit from these treatments are never/light smokers.
Lung cancer associated with smoking is characterized by a high mutational burden and the main hindrance to target discovery is identifying the few driver mutations from hundreds of inconsequential passenger mutations in each sample.
Playing the numbers game
Using mutational frequency in hundreds of cancer samples to select targets for further investigation is a powerful way to discover common mutations but certain considerations should be made.
Gene length will influence frequency results to over-represent longer proteins if a length correction is not made.
Genes with low expression and/or late replication timing will have a higher mutational rate.
Poor sequencing of GC-rich regions (sequencing cold-spots) will lead to under-reporting of mutations and these cold-spots may be hiding potential high frequency mutations.
Germline filtering without normal tissue comparison can introduce error.
From numbers to predictions: in silico analysis of mutations
Online mutation prediction programs can be used in a high-throughput manner to analyze whole exome data to select functional mutations.
Identifying marks of positive selection are fundamental to extracting the driver mutations. These marks are observed in a broad spectrum of data and combining different analyses will likely yield the most success.
Molecular dynamics simulations provide a more detailed analysis of the structural ramifications of a given mutation but require a known crystal structure and a large computational resource.
Genetic-dependency screens
si/shRNA screens exploit the oncogene addiction of cancer cells on a high-throughput scale to compare the functional effects of gene knockdown.
We used a targeted screen to knockdown all mutated genes in specific lung cancer cell lines and discovered three novel mutational drivers of lung cancer (PAK5, MAP3K9, FGFR4).
Incomplete genomics data (including sequencing cold-spots) and potential loss-of-function mutational drivers may explain why three of the cell lines tested did not have an identifiable driver mutations using the targeted siRNA screen.
Conclusion & future perspective
Identifying driver mutations in lung cancer genomics data remains a large challenge and there is much opportunity to identify targetable mutations for the benefit of patients.
The different methods detailed in this review have specific strengths and weaknesses and a combination of approaches is required to capture all driver mutations.
As sequencing technology improves and becomes cheaper, the scale of mutational data will increase but this will also increase the amount of mutational noise.
An increased focus on functional genomics is required to develop clinically effective precision medicines from the large-scale data.
Financial & competing interests disclosure
This work was solely funded by Cancer Research, UK. The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Seer. Surveillance, epidemiology, and end results (seer) program: Seer 18 regs research data Nov 13 sub ( 2000–2011 ).
- Weinstein IB . Addiction to oncogenes – the Achilles heel of cancer . Science297 ( 5578 ), 63 – 64 ( 2002 ).
- Hanahan D , WeinbergRA . The hallmarks of cancer . Cell100 ( 1 ), 57 – 70 ( 2000 ).
- Mok TS , WuYL , ThongprasertSet al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma . N. Engl. J. Med.361 ( 10 ), 947 – 957 ( 2009 ).
- Dogan S , ShenR , AngDCet al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers . Clin. Cancer Res.18 ( 22 ), 6169 – 6177 ( 2012 ).
- Sequist LV , YangJC , YamamotoNet al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations . J. Clin. Oncol.31 ( 27 ), 3327 – 3334 ( 2013 ).
- Maemondo M , InoueA , KobayashiKet al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR . N. Engl. J. Med.362 ( 25 ), 2380 – 2388 ( 2010 ).
- Rosell R , CarcerenyE , GervaisRet al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced egfr mutation-positive non-small-cell lung cancer (eurtac): a multicentre, open-label, randomised Phase 3 trial . The Lancet Oncology13 ( 3 ), 239 – 246 ( 2012 ).
- Mitsudomi T , MoritaS , YatabeYet al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (wjtog3405): an open label, randomised Phase 3 trial . The Lancet Oncology11 ( 2 ), 121 – 128 ( 2010 ).
- Soda M , ChoiYL , EnomotoMet al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer . Nature448 ( 7153 ), 561 – 566 ( 2007 ).
- Shaw AT , YeapBY , Mino-KenudsonMet al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK . J. Clin. Oncol.27 ( 26 ), 4247 – 4253 ( 2009 ).
- Solomon BJ , MokT , KimDWet al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer . N. Engl. J. Med.371 ( 23 ), 2167 – 2177 ( 2014 ).
- Dearden S , StevensJ , WuYL , BlowersD . Mutation incidence and coincidence in non small-cell lung cancer: Meta-analyses by ethnicity and histology (mutmap) . Ann. Oncol.24 ( 9 ), 2371 – 2376 ( 2013 ).
- Janne PA , ShawAT , PereiraJRet al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study . The Lancet Oncology14 ( 1 ), 38 – 47 ( 2013 ).
- Luo J , ManningBD , CantleyLC . Targeting the pi3k-akt pathway in human cancer: rationale and promise . Cancer Cell4 ( 4 ), 257 – 262 ( 2003 ).
- Migliardi G , SassiF , TortiDet al. Inhibition of MEK and PI3K/MTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas . Clin. Cancer Res.18 ( 9 ), 2515 – 2525 ( 2012 ).
- Paik PK , ArcilaME , FaraMet al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations . J. Clin. Oncol.29 ( 15 ), 2046 – 2051 ( 2011 ).
- Prahallad A , SunC , HuangSet al. Unresponsiveness of colon cancer to BRAF(v600e) inhibition through feedback activation of EGFR . Nature483 ( 7387 ), 100 – 103 ( 2012 ).
- Gatzemeier U , GrothG , ButtsCet al. Randomized Phase ii trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer . Ann. Oncol.15 ( 1 ), 19 – 27 ( 2004 ).
- Ross HJ , BlumenscheinGRJr , AisnerJet al. Randomized Phase ii multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer . Clin. Cancer Res.16 ( 6 ), 1938 – 1949 ( 2010 ).
- Califano R , AbidinA , TariqNU , EconomopoulouP , MetroG , MountziosG . Beyond EGFR and ALK inhibition: unravelling and exploiting novel genetic alterations in advanced non small-cell lung cancer . Cancer Treat Rev. ( 2015 ).
- Kim ES , HerbstRS , WistubaIIet al. The battle trial: personalizing therapy for lung cancer . Cancer Discov.1 ( 1 ), 44 – 53 ( 2011 ).
- Tomasetti C , MarchionniL , NowakMA , ParmigianiG , VogelsteinB . Only three driver gene mutations are required for the development of lung and colorectal cancers . Proc. Natl Acad. Sci. USA112 ( 1 ), 118 – 123 ( 2015 ).
- Pao W , GirardN . New driver mutations in non-small-cell lung cancer . The Lancet. Oncology12 ( 2 ), 175 – 180 ( 2011 ).
- Cerami E , GaoJ , DogrusozUet al. The CBIO cancer genomics portal: an open platform for exploring multidimensional cancer genomics data . Cancer Discov.2 ( 5 ), 401 – 404 ( 2012 ).
- Barretina J , CaponigroG , StranskyNet al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity . Nature483 ( 7391 ), 603 – 607 ( 2012 ).
- Haber DA , SettlemanJ . Cancer: drivers and passengers . Nature446 ( 7132 ), 145 – 146 ( 2007 ).
- Hecht SS . Tobacco smoke carcinogens and lung cancer . J. Natl Cancer Inst.91 ( 14 ), 1194 – 1210 ( 1999 ).
- Pleasance ED , StephensPJ , O’mearaSet al. A small-cell lung cancer genome with complex signatures of tobacco exposure . Nature463 ( 7278 ), 184 – 190 ( 2010 ).
- Alexandrov LB , Nik-ZainalS , WedgeDCet al. Signatures of mutational processes in human cancer . Nature500 ( 7463 ), 415 – 421 ( 2013 ).
- Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia . N. Engl. J. Med.368 ( 22 ), 2059 – 2074 ( 2013 ).
- Chen SJ , ShenY , ChenZ . A panoramic view of acute myeloid leukemia . Nat. Genet.45 ( 6 ), 586 – 587 ( 2013 ).
- Shaw AT , OuSH , BangYJet al. Crizotinib in ros1-rearranged non-small-cell lung cancer . N. Engl. J. Med.371 ( 21 ), 1963 – 1971 ( 2014 ).
- Gold KA . ROS1– targeting the one percent in lung cancer . N. Engl. J. Med.371 ( 21 ), 2030 – 2031 ( 2014 ).
- Hofree M , ShenJP , CarterH , GrossA , IdekerT . Network-based stratification of tumor mutations . Nat. Methods10 ( 11 ), 1108 – 1115 ( 2013 ).
- Lawrence MS , StojanovP , PolakPet al. Mutational heterogeneity in cancer and the search for new cancer-associated genes . Nature499 ( 7457 ), 214 – 218 ( 2013 ).
- Hanawalt PC , SpivakG . Transcription-coupled DNA repair: two decades of progress and surprises . Nat. Rev. Mol. Cell Biol.9 ( 12 ), 958 – 970 ( 2008 ).
- Andujar P , WangJ , DescathaAet al. P16ink4a inactivation mechanisms in non-small-cell lung cancer patients occupationally exposed to asbestos . Lung Cancer67 ( 1 ), 23 – 30 ( 2010 ).
- Singh A , Boldin-AdamskyS , ThimmulappaRKet al. RNAI-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy . Cancer Res.68 ( 19 ), 7975 – 7984 ( 2008 ).
- Koren A , PolakP , NemeshJet al. Differential relationship of DNA replication timing to different forms of human mutation and variation . Am. J. Hum. Genet.91 ( 6 ), 1033 – 1040 ( 2012 ).
- Stamatoyannopoulos JA , AdzhubeiI , ThurmanRE , KryukovGV , MirkinSM , SunyaevSR . Human mutation rate associated with DNA replication timing . Nat. Genet.41 ( 4 ), 393 – 395 ( 2009 ).
- Zhang J , FujimotoJ , WedgeDCet al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing . Science346 ( 6206 ), 256 – 259 ( 2014 ).
- De Bruin EC , McgranahanN , MitterRet al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution . Science346 ( 6206 ), 251 – 256 ( 2014 ).
- Forbes SA , BindalN , BamfordSet al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer . Nucleic Acids Res.39 ( Database issue ), D945 – 950 ( 2011 ).
- Hudson AM , YatesT , LiYet al. Discrepancies in cancer genomic sequencing highlight opportunities for driver mutation discovery . Cancer Res.74 ( 22 ), 6390 – 6396 ( 2014 ).
- Sherry ST , WardMH , KholodovMet al. DBSNP: the NCBI database of genetic variation . Nucleic Acids Res.29 ( 1 ), 308 – 311 ( 2001 ).
- Reva B , AntipinY , SanderC . Predicting the functional impact of protein mutations: application to cancer genomics . Nucleic Acids Res.39 ( 17 ), e118 ( 2011 ).
- Adzhubei IA , SchmidtS , PeshkinLet al. A method and server for predicting damaging missense mutations . Nat. Methods7 ( 4 ), 248 – 249 ( 2010 ).
- Choi Y , SimsGE , MurphyS , MillerJR , ChanAP . Predicting the functional effect of amino acid substitutions and indels . PLoS One7 ( 10 ), e46688 ( 2012 ).
- Kumar P , HenikoffS , NgPC . Predicting the effects of coding non-synonymous variants on protein function using the sift algorithm . Nat. Protoc.4 ( 7 ), 1073 – 1081 ( 2009 ).
- Dong C , WeiP , JianXet al. Comparison and integration of deleteriousness prediction methods for nonsynonymous snvs in whole exome sequencing studies . Hum Mol. Genet. ( 2014 ).
- Brognard J , ZhangYW , PutoLA , HunterT . Cancer-associated loss-of-function mutations implicate dapk3 as a tumor-suppressing kinase . Cancer Res.71 ( 8 ), 3152 – 3161 ( 2011 ).
- Uhlen M , FagerbergL , HallstromBMet al. Proteomics. Tissue-based map of the human proteome . Science347 ( 6220 ), 1260419 ( 2015 ).
- Tamborero D , Gonzalez-PerezA , Lopez-BigasN . Oncodriveclust: exploiting the positional clustering of somatic mutations to identify cancer genes . Bioinformatics29 ( 18 ), 2238 – 2244 ( 2013 ).
- Dees ND , ZhangQ , KandothCet al. MuSiC: identifying mutational significance in cancer genomes . Genome Res.22 ( 8 ), 1589 – 1598 ( 2012 ).
- Reimand J , BaderGD . Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers . Mol. Syst. Biol.9 , 637 ( 2013 ).
- Herskowitz I . Functional inactivation of genes by dominant negative mutations . Nature329 ( 6136 ), 219 – 222 ( 1987 ).
- D’ursi P , OrroA , MorraGet al. Molecular dynamics and docking simulation of a natural variant of activated protein C with impaired protease activity: implications for integrin-mediated antiseptic function . J. Biomol. Struct. Dyn.33 ( 1 ), 85 – 92 ( 2015 ).
- Kumar A , RajendranV , SethumadhavanR , PurohitR . Relationship between a point mutation s97c in ck1delta protein and its affect on ATP-binding affinity . J. Biomol. Struct. Dyn.32 ( 3 ), 394 – 405 ( 2014 ).
- Liu HC , LinTM , EngHL , LinYT , ShenMC . Functional characterization of a novel missense mutation, HIS147ARG, in A1 domain of FV protein causing type ii deficiency . Thromb. Res.134 ( 1 ), 153 – 159 ( 2014 ).
- Fawdar S , TrotterEW , LiYet al. Targeted genetic dependency screen facilitates identification of actionable mutations in FGFR4, MAP3K9, and PAK5 in lung cancer . Proc. Natl Acad. Sci. USA110 ( 30 ), 12426 – 12431 ( 2013 ).
- Zhu Y , WuY , LuoY , ZouY , MaB , ZhangQ . R102q mutation shifts the salt-bridge network and reduces the structural flexibility of human neuronal calcium sensor-1 protein . J. Phys. Chem. B118 ( 46 ), 13112 – 13122 ( 2014 ).
- Corbi-Verge C , MarinelliF , Zafra-RuanoA , Ruiz-SanzJ , LuqueI , Faraldo-GomezJD . Two-state dynamics of the SH3-SH2 tandem of ABL kinase and the allosteric role of the n-cap . Proc. Natl Acad. Sci. USA110 ( 36 ), E3372 – 3380 ( 2013 ).
- Sutto L , GervasioFL . Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase . Proc. Natl Acad. Sci. USA110 ( 26 ), 10616 – 10621 ( 2013 ).
- Sun H , LiY , TianS , WangJ , HouT . P-loop conformation governed crizotinib resistance in g2032r-mutated ROS1 tyrosine kinase: clues from free energy landscape . PLoS Comput. Biol.10 ( 7 ), e1003729 ( 2014 ).
- Doss GP , RajithB , ChakrabortyC , NagasundaramN , AliSK , ZhuH . Structural signature of the g719s-t790m double mutation in the EGFR kinase domain and its response to inhibitors . Sci. Rep.4 , 5868 ( 2014 ).
- Torti D , TrusolinoL . Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils . EMBO Mol. Med.3 ( 11 ), 623 – 636 ( 2011 ).
- Sharma SV , SettlemanJ . Oncogene addiction: setting the stage for molecularly targeted cancer therapy . Genes Dev.21 ( 24 ), 3214 – 3231 ( 2007 ).
- Sachse C , EcheverriCJ . Oncology studies using sirna libraries: the dawn of RNAI-based genomics . Oncogene23 ( 51 ), 8384 – 8391 ( 2004 ).
- Swanton C , MaraniM , PardoOet al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs . Cancer Cell11 ( 6 ), 498 – 512 ( 2007 ).
- Henderson MC , GonzalesIM , AroraSet al. High-throughput rnai screening identifies a role for TNK1 in growth and survival of pancreatic cancer cells . Mol. Cancer Res.9 ( 6 ), 724 – 732 ( 2011 ).
- Hu K , LeeC , QiuDet al. Small interfering rna library screen of human kinases and phosphatases identifies polo-like kinase 1 as a promising new target for the treatment of pediatric rhabdomyosarcomas . Mol. Cancer Ther.8 ( 11 ), 3024 – 3035 ( 2009 ).
- Tiedemann RE , ZhuYX , SchmidtJet al. Identification of molecular vulnerabilities in human multiple myeloma cells by rna interference lethality screening of the druggable genome . Cancer Res.72 ( 3 ), 757 – 768 ( 2012 ).
- Morgan-Lappe SE , TuckerLA , HuangXet al. Identification of RAS-related nuclear protein, targeting protein for xenopus kinesin-like protein 2, and stearoyl-coa desaturase 1 as promising cancer targets from an RNAI-based screen . Cancer Res.67 ( 9 ), 4390 – 4398 ( 2007 ).
- Thaker NG , ZhangF , McdonaldPRet al. Identification of survival genes in human glioblastoma cells by small interfering RNA screening . Mol. Pharmacol.76 ( 6 ), 1246 – 1255 ( 2009 ).
- Cowley G , WeirB , VazquezFet al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies . Scientific Data1 , Article number: 140035 ( 2014 ).
- Cheung HW , CowleyGS , WeirBAet al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer . Proc. Natl Acad. Sci. USA108 ( 30 ), 12372 – 12377 ( 2011 ).
- Helming KC , WangX , WilsonBGet al. Arid1b is a specific vulnerability in ARID1A-mutant cancers . Nat. Med.20 ( 3 ), 251 – 254 ( 2014 ).
- Rosenbluh J , NijhawanD , CoxAGet al. Beta-catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis . Cell151 ( 7 ), 1457 – 1473 ( 2012 ).
- Weinstein IB , JoeA . Oncogene addiction . Cancer Res.68 ( 9 ), 3077 – 3080 discussion 3080 ( 2008 ).
- Kaelin WG Jr . The concept of synthetic lethality in the context of anticancer therapy . Nat. Rev. Cancer5 ( 9 ), 689 – 698 ( 2005 ).
- Wang T , WeiJJ , SabatiniDM , LanderES . Genetic screens in human cells using the CRISPR-CAS9 system . Science343 ( 6166 ), 80 – 84 ( 2014 ).
- Jones S , AnagnostouV , LytleKet al. Personalized genomic analyses for cancer mutation discovery and interpretation . Sci. Transl. Med.7 ( 283 ), 283ra253 ( 2015 ).
- Haibe-Kains B , El-HachemN , BirkbakNJet al. Inconsistency in large pharmacogenomic studies . Nature504 ( 7480 ), 389 – 393 ( 2013 ).