
Abstract
Since the first discovery that the bioactive lipid, lysophosphatidic acid (LPA) and LPA1 receptor signaling play a role in the initiation of neuropathic pain (NeuP), accumulated reports have supported the original findings and extended the study toward possible therapeutic applications. The present review describes beneficial roles of LPA receptor signaling in a variety of chronic pain, such as peripheral NeuP induced by nerve injury, chemotherapy and diabetes, central NeuP induced by cerebral ischemia with hemorrhage and spinal cord injury, and fibromyalgia-like wide spread pain induced by repeated cold, psychological and muscular acidic stress. Emerging mechanistic findings are the feed-forward amplification of LPA production through LPA1, LPA3 and microglia and the evidence for maintenance of chronic pain by LPA receptor signaling.
Repeated treatments with lysophosphatidic acid (LPA1/3) receptor antagonist cure the established chronic pain in mice models.
Target chronic pain includes peripheral and central neuropathic pain and fibromyalgia.
Feed-forward amplification of LPA production plays key roles in the mechanisms underlying LPA1/3 receptor antagonist-induced therapy of chronic pain memory.
Peripheral immune systems may play roles in reinforcement and maintenance of systemic feed-forward pain memory.
Pain could be classified into acute and chronic pain. Nociceptive (or physiological) and inflammatory pain are classified into acute pain and characterized as the reversibility by opioid analgesics and anti-inflammatory drugs. Tissue damage-induced inflammatory pain is reversible when underlying causes are rectified. Chronic pain, on the other hand, is in general refractory and lasts for long periods, even after the recovery from initial cause. Major types of chronic pain are represented by neuropathic pain (NeuP), which results from damage to components in pain transmission at the levels of primary afferent nerves, spinal cord and CNS. NeuP also occurs as a secondary symptom in diseases like diabetes, cancer, herpes zoster infection and stroke, and as a side effect of treatments with chemotherapeutics [Citation1–4]. The nature of NeuP is often characterized by stimulus-independent persistent pain, or allodynia and hyperalgesia, and the onset of NeuP is usually delayed after nerve damage, and pain remains even after the complete healing. Another newly proposed category of chronic and refractory pain is generalized wide-spread pain (WSP) disease, represented by fibromyalgia (FM), whose causes remain elusive, but it has been postulated to include intense physical and/or emotional stressful experiences [Citation5,Citation6]. As such, this type of pain could be called as ‘centralized pain’ [Citation7] and is discussed in relation to pain memory in the brain. This review introduces the experimental models for chronic pain, such as peripheral, central NeuP (CNP) and FM-like chronic WSP. In addition, the author introduces the experimental evidence for successful treatments with antagonists of lysophosphatidic acid (LPA) receptors, which play key roles in pathophysiology of these chronic pain models.
Characterization of peripheral nerve damage-induced NeuP models in rodents
The studies of NeuP using experimental animal models may provide some information to understand the pathophysiology in chronic pain patients. As the lack of verbal communication is a major drawback, however, the evaluation of pain in these models is based on empirical behavioral responses. The NeuP in experimental animal models is evaluated as allodynia or hyperalgesia, where the normally innocuous or mildly noxious stimuli induce a nociceptive withdrawal response. In peripheral NeuP models, damage may be due to compression, neurotoxins, infection, immune and metabolic diseases, and tumors. To simulate human peripheral NeuP conditions, nerve injury at or near sciatic nerves of rat or mouse has been used [Citation8,Citation9]. Partial injury to the sensory neurons is often used to induce NeuP behaviors. Such models include the chronic constriction injury of sciatic nerve model [Citation10], partial sciatic nerve ligation (pSNL) model [Citation11,Citation12] and the spared nerve injury model, in which two of the three terminal branches of sciatic nerve (tibial and common peroneal nerves) are tightly ligated or cut [Citation13].
In these peripheral nerve injury models, both thermal hyperalgesia and mechanical hyperalgesia/allodynia are observed at least for several weeks. These abnormal behaviors have been discussed by functional and molecular changes, such as upregulation of Nav1.3 in large (A-fiber) dorsal root ganglion (DRG) neurons [Citation14], TRPV1 and B1-type BK receptors in N52 (an A-fiber marker)-positive DRG neurons [Citation15,Citation16] or pituitary adenylyl cyclase activating polypeptide [Citation17,Citation18]. On the contrary, some molecules, such as substance P, Nav1.8 and Nav1.9 are down-regulated in C-fiber neurons [Citation19–22]. These negative signs in NeuP remained fully discussed, but they may underlie the unique hypoesthesia in patients with some types of NeuP [Citation23]. A few studies in the pSNL model provide some molecular-based mechanisms for such negative signs, in which nociceptive responses by intraplantar administration of type 1 peptidergic C-fiber stimulant, such as substance P, histamine or bradykinin were totally abolished in algogenic paw flexion test [Citation20,Citation24]. Similar findings were also observed in the study that paw withdrawal behaviors following an electric C-fiber stimulation of the foot at 5 Hz were markedly attenuated through an epigenetic gene silencing of Nav1.8 [Citation25].
LPA receptor-mediated mechanisms underlying pSNL-induced abnormal pain
In the course of searching key molecules to affect the function of myelinated A-fibers in the pSNL model, we took notice of LPA1 receptor-signaling, which causes the retraction of neuronal growth cones [Citation26] and morphological changes in Schwann cells [Citation27]. These functions seem to be related to a potential to cause C-fiber retraction [Citation20] and A-fiber demyelination and subsequent sprouting [Citation28], which may underlie a possible mechanism for allodynia [Citation29]. This speculation was supported by following experimental evidence, LPA1-mediated demyelination of dorsal root (DR) fibers following pSNL [Citation30], LPA-induced ex vivo demyelination of peripheral nerve fibers [Citation31], LPA-induced gene silencing of myelin protein in S16 Schwann cells [Citation32], LPA-induced demyelination and subsequent sprouting of myelinated fibers in a co-culture of separately isolated DRG neurons and Schwann cells [Citation24], LPA1-mediated functional cross-talk between Aβ-fibers and C- or Aδ-fibers following pSNL by measuring phosphorylation of ERK1/2 in dorsal horn neurons as a marker of pain transmission [Citation33]. Most importantly, pSNL-induced neuropathic hyperalgesia and allodynia were abolished in LPA1-knock-out (KO) mice. All these findings suggest that LPA1-signaling plays key roles in the pSNL-induced demyelination, a mechanism underlying synaptic reorganization or allodynia. Regarding the hypoesthesia following pSNL, the loss of substance P in the spinal dorsal horn and C-fiber hypoesthesia were reversed in LPA1 receptor-KO mice [Citation20] and partially by LPA1 receptor antagonist, respectively, suggesting that LPA1 receptor-signaling is also involved in these negative signs. Although the complete reversal of substance P expression in the dorsal horn may be explained by the LPA1-mediated neurite retraction [Citation34], the partial blockade of hypoesthesia remains to be clarified. Recent study demonstrates that intrathecal LPA causes an upregulation of Nav1.8 expression in the DRG [Citation35], which may counterbalance the negative (substance P loss) effects, following pSNL.
LPA1- & LPA3-mediated feed-forward amplification of LPA production
In the course of further studies of pSNL-induced demyelination, we found that LPA1-mediated demyelination was specifically observed in the DR, but not in sciatic or spinal nerves. As LPA causes demyelination in ex vivo preparations of sciatic, spinal and DR fibers [Citation36], the source of LPA for DR-demyelination following pSNL was speculated to be from the spinal cord. This speculation was evidenced by the pSNL-induced LPA production in the spinal dorsal horn by mass spectrometry. Stimulus-induced LPA production was also reproduced in experiments using ex vivo cultures of spinal cord slices, where the significant increase in LPA levels was observed by the simultaneous stimulation with substance P and NMDA, but not with substance P or NMDA alone [Citation37], indicating that LPA production and subsequent mechanisms underlying NeuP may require nonselective and intense stimulation of sensory neurons. From the pharmacological studies using phospholipase A2 (PLA2) inhibitors, enough amounts of production of lysophosphatidic choline (LPC), a precursor of LPA, require the activation of calcium-independent phospholipase A2 (iPLA2), which digests phospholipids containing a larger variety of fatty acids at the sn-2 position [Citation38], as well as cytosolic PLA2 (cPLA), which selectively digests the phospholipids with arachidonic acid [Citation38]. Thus, aliquots of LPC, which may be highly produced in neuronal cytosol, will be secreted afterward, which in turn will be converted to LPA by extracellular autotaxin (ATX). Most importantly, the addition of LPA to spinal cord slices in the presence of recombinant ATX increased the LPA production in a time-dependent manner, and the self-amplification of LPA production was lost in preparations from LPA3-KO mice [Citation39]. Further studies [Citation24] revealed that pSNL-induced LPA production in the dorsal horn is also self-amplified by LPA1- and LPA3-mediated microglial activation and cytokine (IL-1β) production, which in turn activates c/i-PLA2 in neurons [Citation40]. It is expected that produced LPA may go back to DR and DRG, where demyelination and Cavα2δ1 expression [Citation30] may cause mechanisms underlying allodynia and hyperalgesia, respectively. All these mechanisms appear to play key roles in the feed-forward system of LPA production (). The studies to examine whether the expression of LPA1 and LPA3 receptor at the late phase is changed would be necessary next subjects.
Peripheral nerve damage and following neuroinflammatory events cause intense nonphysiological pain signals to spinal dorsal horn neurons, in which cPLA2 and iPLA2 are activated, followed by a production of enough amounts of LPC. Some aliquots of LPC released from stimulated neurons are converted to LPA by autotaxin (lysophospholipase D), Thus, produced LPA in turn activates microglia and produces cytokines, such as IL-1β, which activates neuron and stimulates cPLA2 and iPLA2. and presumably also secretory sPLA2. Thus, the initial intense nonphysiological activation of spinal dorsal horn neurons may lead to a feed-forward LPA production, and possibly to a cause of chronic pain. LPA also goes back to DR fibers and DRG, where LPA1-mediated demyelination underlying allodynia and upregulation of Cavα2δ1 and ephrin B1 underlying hyperalgesia are caused. These mechanisms also contribute to the functional feed-forward system of pain transmission. On the other hand, LPA has an action of BDNF production in microglia through an activation of P2X4 receptor by secreted ATP. As BDNF is known to decrease neuronal plasma membrane KCC2 levels and increase cytosolic Cl- ion levels, resulting in a conversion of GABAA receptor function from inhibitory to excitatory one. All these mechanisms may play roles in the development of neuropathic pain [Citation24]. Astrocyte activation, on the other hand, may be also involved in the maintenance of neuropathic pain through a production of chemokines [Citation41].
DRG: Dorsal root ganglion; LPA: Lysophosphatidic acid; LPC: Lysophosphatidyl choline.
![Figure 1. Feed-forward LPA-mediated mechanisms in neuropathic pain following nerve damage or neuroinflammation.Peripheral nerve damage and following neuroinflammatory events cause intense nonphysiological pain signals to spinal dorsal horn neurons, in which cPLA2 and iPLA2 are activated, followed by a production of enough amounts of LPC. Some aliquots of LPC released from stimulated neurons are converted to LPA by autotaxin (lysophospholipase D), Thus, produced LPA in turn activates microglia and produces cytokines, such as IL-1β, which activates neuron and stimulates cPLA2 and iPLA2. and presumably also secretory sPLA2. Thus, the initial intense nonphysiological activation of spinal dorsal horn neurons may lead to a feed-forward LPA production, and possibly to a cause of chronic pain. LPA also goes back to DR fibers and DRG, where LPA1-mediated demyelination underlying allodynia and upregulation of Cavα2δ1 and ephrin B1 underlying hyperalgesia are caused. These mechanisms also contribute to the functional feed-forward system of pain transmission. On the other hand, LPA has an action of BDNF production in microglia through an activation of P2X4 receptor by secreted ATP. As BDNF is known to decrease neuronal plasma membrane KCC2 levels and increase cytosolic Cl- ion levels, resulting in a conversion of GABAA receptor function from inhibitory to excitatory one. All these mechanisms may play roles in the development of neuropathic pain [Citation24]. Astrocyte activation, on the other hand, may be also involved in the maintenance of neuropathic pain through a production of chemokines [Citation41].DRG: Dorsal root ganglion; LPA: Lysophosphatidic acid; LPC: Lysophosphatidyl choline.](/cms/asset/0bfc8f01-8dfd-4f31-926d-9b157bec052a/ipmt_a_12344353_f0001.jpg)
Roles of microglia & astrocytes in the maintenance of LPA signaling-mediated NeuP
From the view of pain management, it is more important to discuss whether the regulation of LPA receptor signaling could suppress the maintenance of NeuP. It is fortunate that a significant increase in LPA production at the dorsal horn was also observed at 2 and 3 weeks after pSNL [Citation41], suggesting that LPA receptor signaling may contribute to the maintenance of NeuP. This view was evidenced by the findings that intrathecal injection (it.) of siRNA for LPA1 or LPA3 at day 3 (P3) after pSNL significantly reversed the LPA production at P7 and hyperalgesia at P5 and P7 [Ueda et al., unpublished data]. In this study, the absolute contents (as a relative signal ratio to 17:0 LPA, an internal standard) of 18:1, 16:0 and 18:0 LPA were different, but their pSNL-induced changes were all approximately twofold, suggesting similar machineries may underlie the increase of these LPA species. Furthermore, it was also supported by the report that repeated intraperitoneal (ip.) treatments with LPA1/3 receptor antagonist Ki14625 from day 7 to day 13 reversed the basal pain threshold to the normal level and the beneficial effects lasted for another week after ceasing the treatment [Citation41] and by a finding that repeated it. treatments with LPA1/3 receptor antagonist VPC32183 [Citation42] also recovered the basal pain threshold [Ueda et al., unpublished data], it is evident that central LPA1 and LPA3 receptor signaling are responsible for the maintenance of NeuP, though this compound has inhibitory activities against LPA-degrading enzymes, such as lipid phosphate phosphatases. However, it still remains elusive which cell types are targets for LPA1/3 receptor siRNAs or antagonists. Further detailed studies revealed that microglia toxin, Mac1-saporin and astrocyte-specific toxin, L-α-aminoazipate inhibit the LPA production and hyperalgesia, respectively [Citation41]. The roles of astrocytes were evidenced by the findings that LPA1 receptor activation produces chemokines in cultured astrocytes, and it. injection of LPA-primed astrocytes causes potent LPA1 receptor-mediated hyperalgesia, possibly through an action of chemokines [Citation41].
Therapeutic roles of LPA receptor antagonists in a wide variety of chronic pain diseases
Chemotherapeutic agent-induced models
An animal model of chemotherapeutic agent-induced painful toxic neuropathy (CIPN) has been produced [Citation43,Citation44]. Paclitaxel (Taxol), which is an antineoplastic agent derived from the Pacific yew tree Taxus brevifolia [Citation45], promotes the microtubule stability and interferes with mitosis [Citation46]. Paclitaxel is often used to treat a variety of cancers including ovarian, breast and non-small-cell lung cancer [Citation47], but its beneficial anticancer effectiveness is limited by the highly incidental development of severe painful peripheral neuropathy, characterized by dysesthesia, numbness and burning pain [Citation48]. Although the CIPN trials are inconclusive regarding tricyclic antidepressants, gabapentin and ketamine [Citation49], the preventive treatment with gabapentin is effective for paclitaxel-induced NeuP [Citation50,Citation51], being consistent to an experimental study using mouse model [Citation52]. In four-times repeated paclitaxel treatment-induced NeuP model, the demyelination was observed at the DR proximal to the spinal cord and it was abolished in LPA1 or LPA3 receptor-KO mice, being consistent to the case with pSNL. Of most importance are the findings that paclitaxel-treatment causes LPA production in the dorsal horn of spinal cord, and the treatments of LPA1/3 receptor antagonist, Ki16425 at 30 min prior to each paclitaxel treatment abolished the CIPN [Citation53]. As CIPN is predicted before the start of treatments with chemotherapeutics, this type of prophylactic treatment with LPA1 or LPA3 receptor antagonist would be practically available in clinic, though it remains elusive whether the therapeutic treatment with LPA receptor antagonist may also cure the existing CIPN.
Diabetic NeuP
The NeuP commonly associated with diabetes mellitus (DM) is reproduced in experimental animal models. Streptozotocin (iv.)-induced type 1 DM model showed similar functional and structural abnormalities, as seen in human diabetic neuropathy [Citation54], and NeuP symptoms [Citation55,Citation56]. On the other hand, mice fed with high-fat diet and genetically obese mice deficient of leptin or leptin receptor (ob/ob or db/db mice) are also used as type 2 DM model, where allodynia and hyperalgesia are observed [Citation57]. Of interest is the finding that high-fat diet-fed mice and db/db mice also show C-fiber hypoesthesia as well as A-fiber hyperalgesia, as seen in the case with pSNL model. We have obtained that LPA1 and LPA3 receptor are also involved in the development and maintenance in such hyperalgesia and hypoesthesia by use of LPA1 and LPA3 receptor KO mice and antagonists [Ueda et al., unpublished data].
Central post stroke pain
Cerebral ischemic stroke leads to irreversible neurological damage with dysfunctions of motor skill, cognition and sensory perception [Citation58,Citation59], but there are limited strategies to restore the blood flow. Although tissue plasminogen activator (tPA) has been used for acute treatment, there is a limitation of clinical use due to short time frame for safe intervention. If the reperfusion therapies with tPA are applied at the time of 4.5 h and/or later, the risk to cause cerebral hemorrhage increases [Citation60]. Central post stroke pain (CPSP) or stroke-induced headache is one of representative cerebral hemorrhage-related toxic events in survivors [Citation61,Citation62].
As post stroke pain may include multiple pain conditions associated with CNS disorders, such as headache, painful spasms, hemiplegic shoulder pain and musculoskeletal pain, CNP or CPSP should be defined to be the pain arising as a direct consequence of a lesion or disease affecting the central somatosensory system [Citation63]. From the careful study, the ratio of CPSP was estimated as only 10% of all types of chronic pain after stroke and the prevalence of CPSP in patients with stroke is between 1 and 12% [Citation61]. In the experimental animal study, the transient middle cerebral artery occlusion for 1 h causes heavy motor dysfunction and thereby makes it difficult to evaluate reliable pain behaviors. The short period (15 min) of transient middle cerebral artery occlusion, which does not cause the motor dysfunction, successfully shows weak abnormal pain behavior in sensitive nociception tests using electrical stimulation to the foot [Citation64]. However, this protocol does not show stable thermal or mechanical hyperalgesia. Similar weak hyperalgesia was also observed in the case with lacunar infarction-induced stroke by use of photochemically induced thrombosis (PIT) in mice [Citation65]. When tPA was given to mice at 6 h after PIT-treatment, however, sporadic hemorrhage clots were observed at cortex, striatum and thalamus on the ipsilateral side, but these treatments were not lethal. In this model, stable and long-lasting CNP was observed on both sides in mechanical and thermal nociception tests [Citation65]. The bilateral hyperalgesia was abolished in LPA1 or LPA3 receptor-KO mice and by repeated i.c.v. treatments with AM966, an LPA1/3 receptor antagonist. Similar beneficial effects were also observed with systemic and repeated administrations (per os) of brain penetrable LPA1 receptor antagonist [Ueda et al., unpublished data]. Although details of bilateral hyperalgesia following one side PIT treatment remain elusive, it may be related to the LPA production in mediodorsal thalamus [Citation65], and insula [Ueda et al., unpublished data], both brain areas which are related to the emotional pain and/or bilateral commissure connection, respectively.
Spinal cord injury-induced NeuP
The global incidence of traumatic spinal cord injury (SCI) is estimated as 10.5 cases per 100,000 persons per year [Citation66]. Patients with SCI often experience chronic pain, which sometimes exceed the impact due to other SCI induced consequences. NeuP is present in 40–50% of patients with pain, and tends to become chronic [Citation67–69]. During over the last 20 years, many therapeutic targets have emerged, but only gabapentinoids retained their status as first-line treatments, though their mechanism of actions remain elusive [Citation70]. Therefore, it is still necessary to discover new pharmacotherapeutic targets toward CNP using experimental animal models. Important concerns in developing SCI-induced CNP are to avoid motor dysfunction or wait for the recovery from the dysfunction, which makes it difficult to appropriately evaluate the nociceptive withdrawal behaviors. For this purpose, female C57BL/6J mice (20–22 g) were anesthetized and given a reliable contusion injury by rapidly applying a force-defined and weaker impact (40 kdyn) with a stainless steel-tipped impounder using a commercially available SCI device (IH impactor, Precision Systems and Instrumentation, KY, USA), as described previously [Citation71,Citation72]. The motor dysfunction evaluated by Rota-rod treadmill test was worst at day 1 after the SCI, but gradually improved throughout the day. Complete recovery was found at day 7 to 14 (2 weeks). Therefore, the relatively reliable nociceptive threshold without influence of motor effects could be obtained at 2 weeks after the SCI. The SCI-induced mechanical hyperalgesia was constantly observed for more than 12 weeks, and they were abolished in LPA1-KO mice and ATX-heterozygous KO mice, but not in LPA2-KO mice [Ueda et al., unpublished data]. In this study, milder impact was used for SCI to make complete recovery from the motor dysfunction within 1–2 weeks, and nociception test was performed at 2 weeks and later. Therefore, the milder SCI-induced hyperalgesia through LPA1 receptor may not be influenced by the LPA1-mediated demyelination and motor dysfunction through microglial activation, in which stronger SCI used in the previous report [Citation73].
FM-like chronic WSP
Following the nerve damage, NeuP is caused to the side closely associated in the pain pathway. In contrast, FM is a WSP disease, which is poorly characterized in terms of causes, primary and secondary loci. FM is known to comprise an approximate 2% population ratio in developed countries [Citation5,Citation74], and is less responsive to classic and commonly used analgesics, such as nonsteroidal anti-inflammatory drugs and opioids. Many studies demonstrated that FM is closely associated to brain mechanisms [Citation6], though there is also an increasing body of evidence supporting a role for peripheral small fiber neuropathy [Citation5,Citation75,Citation76]. Therefore, it is expected that basic studies using animal models, which mimic pathophysiological symptoms and pharmacotherapeutic sensitivities seen in FM patients would be expected for better diagnosis or therapy. Following several pioneering studies with vagotomized animals [Citation77] and acid saline-induced muscle (AcMP) pain model [Citation78], intermittent cold stress (ICS) or intermittent psychological stress (IPS)-induced chronic WSP models have been recently developed [Citation79,Citation80]. In AcMP-model to induce generalized chronic muscle pain, 20 μl of pH 4.0 sterile saline was injected into the right gastrocnemius muscle of mouse twice, 5 days apart. The mouse ICS-model used a type of autonomic stress by alternating environmental temperatures between 24 and 4°C every 30 min from 10:00 am to 4:30 pm. The stress by alternating temperatures was given for 2 days. The mouse IPS-model, on the other hand, used intermittent psychological stress or empathy, using the communication box with divided transparent plastic walls. Mice were exposed to psychological stress by seeing, hearing and smelling the foot-shocked mice. The stress was given once per day for 5 days and called the poststress day 1 as P1.
Although long-lasting mechanical hyperalgesia from day 1 after the second acid saline injection is observed for 2–3 weeks in AcMP-model, but no thermal hyperalgesia is observed. The mechanical hyperalgesia is suppressed by pregabalin [Citation81] or antidepressants, as seen in clinics. Unlike the case in the clinic, AcMP-induced hyperalgesia is also suppressed by morphine [Citation82]. On the other hand, ICS- and IPS-models show mechanical and thermal hyperalgesia, and antimuscle pain activity [Citation79,Citation80]. There is a female-predominant sex difference, and it is more evident when the ICS or IPS is given 3 weeks after the gonadectomy [Citation79,Citation80]. In addition to the pathophysiological features, there were clear pharmacotherapeutic similarity to the cases in clinic for FM patients. Both ICS- and IPS-models show good sensitivities to gabapentinoids and antidepressants, but not to morphine [Citation79,Citation80,Citation83,Citation84].
The involvement of LPA1 receptor in the development of FM-like chronic WSP was also clarified. Experimental evidence was reported with ICS, IPS and AcMP models by use of LPA1 receptor-KO mice, which completely abolished the WSP in all three models [Citation80]. The repeated treatments with AM966, an LPA1/3 receptor antagonist largely blocked the established pain in the IPS model, while it has no acute or instant actions against hyperalgesia [Citation80].
Other important components in NeuP mechanisms
Besides LPA1 and LPA3 receptors, there are some studies that suggest the LPA5 receptor is also involved in the mechanisms underlying NeuP in rodents by use of LPA5 receptor-KO mice and LPA5 receptor-specific antagonist [Citation85,Citation86]. There are a number of studies that show the activation of peripheral neuroinflammatory system plays a key role in the mechanisms for NeuP as well as inflammatory pain [Citation87]. As chronic pain status is also largely abolished in genetic deficiency of responsible key molecules, the author proposes the hypothesis that peripheral immune and central LPA receptor mechanisms work for the development and maintenance of chronic pain in a tandem and feed-forward manner.
Clinical evidence for LPA production in NeuP patients
Regarding the LPA levels in pain patients, McDougall et al. [Citation88] reported that LPA levels in synovial fluid of patients with knee osteoarthritis increased significantly with disease severity. In this report, they also demonstrated that intraarticular injection of LPA in the rat causes joint pain and joint nerve demyelination in a Ki16425-reversible manner. They suggest that LPA is a candidate molecule for the development of osteoarthritis nerve damage and the origin of joint NeuP. Most recently, there are two important reports that LPA levels in the cerebrospinal fluid (CSF) in NeuP patients are associated with the degree of pain [Citation89], and the CSF levels in patients with lumbar spinal stenosis are associated with the severity of lumbar spinal stenosis [Citation90].
Conclusion
Since the first demonstration that LPA1 receptor signaling initiates the nerve injury-induced NeuP and underlying mechanisms through demyelination and Cavα2δ1 upregulation in DRG, much advances have been unveiled. They are the evidence of feed-forward amplification of LPA production in the spinal cord through microglial actions, and the evidence for the roles of LPA1 and LPA3 receptor signaling for the maintenance of NeuP. Of further importance are the findings that LPA receptor signaling play roles in many other chronic pain diseases, such as peripheral CIPN, diabetic NeuP, CNP CPSP and FM-like chronic WSP.
Future perspective
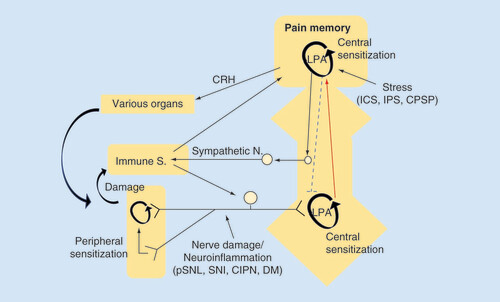
Emerging evidence supports the roles of LPA receptor signaling in chronic pain patients. Considering a number of studies of involvement of peripheral neuroinflammatory system in NeuP, I would like to propose the hypothesis that LPA receptor-mediated central pain memory is developed with and reinforced by the peripheral immune system (). FM is now classified as chronic primary pain due to lack of knowledge of cause. One of most important clues is the fact that LPA1 receptor-KO mice abolish the abnormal pain in FM-like mouse models. On the analogy of the study that LPA levels in the cerebrospinal fluid of NeuP patients are increased as the pain degree, the study of LPA measurement in FM patients would be very important. In addition, it is also interesting to further examine how LPA1 receptor system is involved in the development and maintenance of generalized pain. From the view of diagnosis and therapy, the studies to clarify the related peripheral immune system in NeuP or FM patients, and their animal models may lead to a discovery of biomarkers (for diagnosis) or key molecules underlying maintenance (for therapy).
In the peripheral NeuP model, nerve damage or neuroinflammation causes LPA production in the spinal dorsal horn, where LPA production is self-amplified and LPA-mediated pain memory is developed and maintained in a feed-forward manner. In the centralized pain, represented by fibromyalgia, various types of intense stress experience develop and maintain the LPA receptor-mediated pain memory. Although conclusive data, including LPA-involvement, are not yet available, the brain plasticity related to pain memory may attenuate the descending pain-inhibitory system and activate peripheral immune system, which in turn reinforces the brain pain memory through the peripheral pain pathway.
CIPN: Chemotherapy-induced peripheral neuropathy; CPSP: Central post-stroke pain; CRH: Corticotropin releasing hormone; DM: Diabetes mellitus; ICS: Intermittent cold stress; IPS: Intermittent psychological stress; LPA: Lysophosphatidic acid; pSNL: Partial sciatic nerve ligation; SNI: Spare nerve injury.
Financial & competing interests disclosure
This work was supported by the MEXT KAKENHI (grant number 26253077 to H Ueda) and the Platform for Drug Discovery, Informatics and Structural Life Science (16am0101012j0005) (H Ueda) from the Japan Agency for Medical Research and Development (AMED). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Bridges D , ThompsonSW , RiceAS. Mechanisms of neuropathic pain. Br. J. Anaesth.87(1), 12–26 (2001).
- Henry JL , LallooC , YashpalK. Central poststroke pain: an abstruse outcome. Pain Res. Manag.13(1), 41–49 (2008).
- Sah DW , OssipoMH , PorrecaF. Neurotrophic factors as novel therapeutics for neuropathic pain. Nat. Rev. Drug Discov2(6), 460–472 (2003).
- Woolf CJ , MannionRJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet353(9168), 1959–1964 (1999).
- Clauw DJ . Fibromyalgia: a clinical review. JAMA311(15), 1547–1555 (2014).
- Clauw DJ , UedaH. Summary of the fibromyalgia research symposium 2016 in Nagasaki. Pain Rep.2(1), e582 (2017).
- Eller-Smith OC , NicolAL , ChristiansonJA. Potential mechanisms underlying centralized pain and emerging therapeutic interventions. Front. Cell Neurosci.12, 35 (2018).
- Hogan Q . Animal pain models. Reg. Anesth. Pain Med.27(4), 385–401 (2002).
- Wang LX , WangZJ. Animal and cellular models of chronic pain. Adv. Drug Deliv. Rev.55(8), 949–965 (2003).
- Bennett GJ , XieYK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain33(1), 87–107 (1988).
- Malmberg AB , BasbaumAI. Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain76(1–2), 215–222 (1998).
- Seltzer Z , DubnerR , ShirY. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain43(2), 205–218 (1990).
- Decosterd I , WoolfCJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain87(2), 149–158 (2000).
- Kim CH , OhY , ChungJM , ChungK. The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res. Mol. Brain Res.95(1–2), 153–161 (2001).
- Rashid MH , InoueM , KondoS , KawashimaT , BakoshiS , UedaH. Novel expression of vanilloid receptor 1 on capsaicin-insensitive fibers accounts for the analgesic effect of capsaicin cream in neuropathic pain. J. Pharmacol. Exp. Ther.304(3), 940–948 (2003).
- Rashid MH , InoueM , MatsumotoM , UedaH. Switching of bradykinin-mediated nociception following partial sciatic nerve injury in mice. J. Pharmacol. Exp. Ther.308(3), 1158–1164 (2004).
- Mabuchi T , ShintaniN , MatsumuraSet al. Pituitary adenylate cyclase-activating polypeptide is required for the development of spinal sensitization and induction of neuropathic pain. J. Neurosci.24(33), 7283–7291 (2004).
- Zhang Q , ShiTJ , JiRRet al. Expression of pituitary adenylate cyclase-activating polypeptide in dorsal root ganglia following axotomy: time course and coexistence. Brain Res.705(1–2), 149–158 (1995).
- Dib-Hajj SD , FjellJ , CumminsTRet al. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain83(3), 591–600 (1999).
- Inoue M , YamaguchiA , KawakamiM , ChunJ , UedaH. Loss of spinal substance P pain transmission under the condition of LPA1 receptor-mediated neuropathic pain. Mol. Pain2, 25 (2006).
- Ruscheweyh R , ForsthuberL , SchoffneggerD , SandkuhlerJ. Modification of classical neurochemical markers in identified primary afferent neurons with Abeta-, Adelta-, and C-fibers after chronic constriction injury in mice. J. Comp. Neurol.502(2), 325–336 (2007).
- Sleeper AA , CumminsTR , Dib-HajjSDet al. Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J. Neurosci.20(19), 7279–7289 (2000).
- Maier C , BaronR , TolleTRet al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): somatosensory abnormalities in 1236 patients with different neuropathic pain syndromes. Pain150(3), 439–450 (2010).
- Ueda H . Lysophosphatidic acid signaling is the definitive mechanism underlying neuropathic pain. Pain158(Suppl. 1), S55–S65 (2017).
- Uchida H , MaL , UedaH. Epigenetic gene silencing underlies C-fiber dysfunctions in neuropathic pain. J. Neurosci.30(13), 4806–4814 (2010).
- Campbell DS , HoltCE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron32(6), 1013–1026 (2001).
- Weiner JA , FukushimaN , ContosJJ , SchererSS , ChunJ. Regulation of Schwann cell morphology and adhesion by receptor-mediated lysophosphatidic acid signaling. J. Neurosci.21(18), 7069–7078 (2001).
- Ueda H . Lysophosphatidic acid as the initiator of neuropathic pain. Biol. Pharm. Bull.34(8), 1154–1158 (2011).
- Ueda H , MatsunagaH , OlaposiOI , NagaiJ. Lysophosphatidic acid: chemical signature of neuropathic pain. Biochim. Biophys. Acta1831(1), 61–73 (2013).
- Inoue M , RashidMH , FujitaR , ContosJJ , ChunJ , UedaH. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat. Med.10(7), 712–718 (2004).
- Fujita R , KiguchiN , UedaH. LPA-mediated demyelination in ex vivo culture of dorsal root. Neurochem. Int.50(2), 351–355 (2007).
- Tsukahara R , UedaH. Myelin-related gene silencing mediated by LPA1 – Rho/ROCK signaling is correlated to acetylation of NFkappaB in S16 Schwann cells. J. Pharmacol. Sci.132(2), 162–165 (2016).
- Xie W , MatsumotoM , ChunJ , UedaH. Involvement of LPA1 receptor signaling in the reorganization of spinal input through Abeta-fibers in mice with partial sciatic nerve injury. Mol. Pain4, 46 (2008).
- Fukushima N , WeinerJA , KaushalDet al. Lysophosphatidic acid influences the morphology and motility of young, postmitotic cortical neurons. Mol. Cell. Neurosci.20(2), 271–282 (2002).
- Pan HL , LiuBL , LinW , ZhangYQ. Modulation of Nav1.8 by lysophosphatidic acid in the induction of bone cancer pain. Neurosci. Bull.32(5), 445–454 (2016).
- Ueda H . Molecular mechanisms of neuropathic pain – phenotypic switch and initiation mechanisms. Pharmacol. Ther.109(1–2), 57–77 (2006).
- Inoue M , MaL , AokiJ , UedaH. Simultaneous stimulation of spinal NK1 and NMDA receptors produces LPC which undergoes ATX-mediated conversion to LPA, an initiator of neuropathic pain. J. Neurochem.107(6), 1556–1565 (2008).
- Mouchlis VD , DennisEA. Phospholipase A2 catalysis and lipid mediator lipidomics. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids1864(6), 766–771 (2019).
- Ma L , UchidaH , NagaiJet al. Lysophosphatidic acid-3 receptor-mediated feed-forward production of lysophosphatidic acid: an initiator of nerve injury-induced neuropathic pain. Mol. Pain5, 64 (2009).
- Ma L , NagaiJ , UedaH. Microglial activation mediates de novo lysophosphatidic acid production in a model of neuropathic pain. J. Neurochem.115(3), 643–653 (2010).
- Ueda H , NeyamaH , NagaiJ , MatsushitaY , TsukaharaT , TsukaharaR. Involvement of lysophosphatidic acid-induced astrocyte activation underlying the maintenance of partial sciatic nerve injury-induced neuropathic pain. Pain159(11), 2170–2178 (2018).
- Heasley BH , JaroszR , LynchKR , MacdonaldTL. Initial structure-activity relationships of lysophosphatidic acid receptor antagonists: discovery of a high-affinity LPA1/LPA3 receptor antagonist. Bioorg. Med. Chem. Lett.14(11), 2735–2740 (2004).
- Cavaletti G , TrediciG , BragaM , TazzariS. Experimental peripheral neuropathy induced in adult rats by repeated intraperitoneal administration of taxol. Exp. Neurol.133(1), 64–72 (1995).
- Hama A , TakamatsuH. Chemotherapy-induced peripheral neuropathic pain and rodent models. CNS Neurol. Disord. Drug Targets15(1), 7–19 (2016).
- Rowinsky EK . The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu. Rev. Med.48, 353–374 (1997).
- Rowinsky EK , DonehowerRC. Paclitaxel (Taxol). N. Engl. J. Med.332(15), 1004–1014 (1995).
- Mekhail TM , MarkmanM. Paclitaxel in cancer therapy. Expert Opin. Pharmacother.3(6), 755–766 (2002).
- Velasco R , BrunaJ. Taxane-induced peripheral neurotoxicity. Toxics3(2), 152–169 (2015).
- Hershman DL , LacchettiC , DworkinRHet al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol.32(18), 1941–1967 (2014).
- Aghili M , ZareM , MousaviNet al. Efficacy of gabapentin for the prevention of paclitaxel induced peripheral neuropathy: a randomized placebo controlled clinical trial. Breast J.25(2), 226–231 (2019).
- Brewer JR , MorrisonG , DolanME , FlemingGF. Chemotherapy-induced peripheral neuropathy: current status and progress. Gynecol. Oncol.140(1), 176–183 (2016).
- Matsumoto M , InoueM , HaldA , YamaguchiA , UedaH. Characterization of three different sensory fibers by use of neonatal capsaicin treatment, spinal antagonism and a novel electrical stimulation-induced paw flexion test. Mol. Pain2, 16 (2006).
- Uchida H , NagaiJ , UedaH. Lysophosphatidic acid and its receptors LPA1 and LPA3 mediate paclitaxel-induced neuropathic pain in mice. Mol. Pain10, 71 (2014).
- Sima AA , SugimotoK. Experimental diabetic neuropathy: an update. Diabetologia42(7), 773–788 (1999).
- Courteix C , EschalierA , LavarenneJ. Streptozocin-induced diabetic rats: behavioural evidence for a model of chronic pain. Pain53(1), 81–88 (1993).
- Rashid MH , InoueM , BakoshiS , UedaH. Increased expression of vanilloid receptor 1 on myelinated primary afferent neurons contributes to the antihyperalgesic effect of capsaicin cream in diabetic neuropathic pain in mice. J. Pharmacol. Exp. Ther.306(2), 709–717 (2003).
- Obrosova IG . Diabetic painful and insensate neuropathy: pathogenesis and potential treatments. Neurotherapeutics6(4), 638–647 (2009).
- De Vloo P , MorlionB , van LoonJ , NuttinB. Animal models for central poststroke pain: a critical comprehensive review. Pain158(1), 17–29 (2017).
- Vestergaard K , NielsenJ , AndersenG , Ingeman-NielsenM , Arendt-NielsenL , JensenTS. Sensory abnormalities in consecutive, unselected patients with central post-stroke pain. Pain61(2), 177–186 (1995).
- Woo D , BroderickJP , KothariRUet al. Does the National Institutes of Health Stroke Scale favor left hemisphere strokes? NINDS t-PA Stroke Study Group. Stroke30(11), 2355–2359 (1999).
- Klit H , FinnerupNB , JensenTS. Central post-stroke pain: clinical characteristics, pathophysiology, and management. Lancet Neurol.8(9), 857–868 (2009).
- Tversky S , LibmanRB , SchlossERet al. Catastrophic intracranial hemorrhages after IV tPA in a patient with insidious onset of fever and back pain. J. Stroke Cerebrovasc. Dis.25(5), e69–e70 (2016).
- Treede RD , JensenTS , CampbellJNet al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology70(18), 1630–1635 (2008).
- Halder SK , YanoR , ChunJ , UedaH. Involvement of LPA1 receptor signaling in cerebral ischemia-induced neuropathic pain. Neuroscience235, 10–15 (2013).
- Ueda H , NeyamaH , SasakiK , MiyamaC , IwamotoR. Lysophosphatidic acid LPA1 and LPA3 receptors play roles in the maintenance of late tissue plasminogen activator-induced central poststroke pain in mice. Neurobiol. Pain5, 100020 (2019).
- Kumar R , LimJ , MekaryRAet al. Traumatic spinal injury: global epidemiology and worldwide volume. World Neurosurg.113, e345–e363 (2018).
- Baastrup C , FinnerupNB. Pain in spinal cord injury. Pain Manag.2(1), 87–94 (2012).
- Finnerup NB . Pain in patients with spinal cord injury. Pain154(Suppl. 1), S71–S76 (2013).
- Siddall PJ , McClellandJM , RutkowskiSB , CousinsMJ. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain103(3), 249–257 (2003).
- Alles SRA , SmithPA. Etiology and pharmacology of neuropathic pain. Pharmacol. Rev.70(2), 315–347 (2018).
- Ma L , NagaiJ , SekinoY , GotoY , NakahiraS , UedaH. Single application of A2 NTX, a botulinum toxin A2 subunit, prevents chronic pain over long periods in both diabetic and spinal cord injury-induced neuropathic pain models. J. Pharmacol. Sci.119(3), 282–286 (2012).
- Scheff SW , RabchevskyAG , FugacciaI , MainJA , LumppJEJr. Experimental modeling of spinal cord injury: characterization of a force-defined injury device. J. Neurotrauma20(2), 179–193 (2003).
- Santos-Nogueira E , Lopez-SerranoC , HernandezJet al. Activation of lysophosphatidic acid receptor Type 1 contributes to pathophysiology of spinal cord injury. J. Neurosci.35(28), 10224–10235 (2015).
- Russell IJ , VaeroyH , JavorsM , NybergF. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum.35(5), 550–556 (1992).
- Doppler K , RittnerHL , DeckartM , SommerC. Reduced dermal nerve fiber diameter in skin biopsies of patients with fibromyalgia. Pain156(11), 2319–2325 (2015).
- Ramirez M , Martinez-MartinezLA , Hernandez-QuintelaE , Velazco-CasapiaJ , VargasA , Martinez-LavinM. Small fiber neuropathy in women with fibromyalgia. An in vivo assessment using corneal confocal bio-microscopy. Semin. Arthritis Rheum.45(2), 214–219 (2015).
- Khasar SG , MiaoFJ , JanigW , LevineJD. Vagotomy-induced enhancement of mechanical hyperalgesia in the rat is sympathoadrenal-mediated. J. Neurosci.18(8), 3043–3049 (1998).
- Sluka KA , KalraA , MooreSA. Unilateral intramuscular injections of acidic saline produce a bilateral, long-lasting hyperalgesia. Muscle Nerve24(1), 37–46 (2001).
- Nishiyori M , UedaH. Prolonged gabapentin analgesia in an experimental mouse model of fibromyalgia. Mol. Pain4, 52 (2008).
- Ueda H , NeyamaH. LPA1 receptor involvement in fibromyalgia-like pain induced by intermittent psychological stress, empathy. Neurobiol. Pain1, 16–25 (2017).
- Yokoyama T , MaedaY , AudetteKM , SlukaKA. Pregabalin reduces muscle and cutaneous hyperalgesia in two models of chronic muscle pain in rats. J. Pain8(5), 422–429 (2007).
- Sluka KA , RohlwingJJ , BusseyRA , EikenberrySA , WilkenJM. Chronic muscle pain induced by repeated acid injection is reversed by spinally administered mu- and delta-, but not kappa-, opioid receptor agonists. J. Pharmacol. Exp. Ther.302(3), 1146–1150 (2002).
- Nishiyori M , NagaiJ , NakazawaT , UedaH. Absence of morphine analgesia and its underlying descending serotonergic activation in an experimental mouse model of fibromyalgia. Neurosci. Lett.472(3), 184–187 (2010).
- Nishiyori M , UchidaH , NagaiJet al. Permanent relief from intermittent cold stress-induced fibromyalgia-like abnormal pain by repeated intrathecal administration of antidepressants. Mol. Pain7, 69 (2011).
- Lin ME , RiveraRR , ChunJ. Targeted deletion of LPA5 identifies novel roles for lysophosphatidic acid signaling in development of neuropathic pain. J. Biol. Chem.287(21), 17608–17617 (2012).
- Murai N , HiyamaH , KisoTet al. Analgesic effects of novel lysophosphatidic acid receptor 5 antagonist AS2717638 in rodents. Neuropharmacology126, 97–107 (2017).
- Sommer C , LeindersM , UceylerN. Inflammation in the pathophysiology of neuropathic pain. Pain159(3), 595–602 (2018).
- McDougall JJ , AlbaceteS , SchuelertNet al. Lysophosphatidic acid provides a missing link between osteoarthritis and joint neuropathic pain. Osteoarthritis Cartilage25(6), 926–934 (2017).
- Kuwajima K , SumitaniM , KuranoMet al. Lysophosphatidic acid is associated with neuropathic pain intensity in humans: an exploratory study. PLoS ONE13(11), e0207310 (2018).
- Hayakawa K , KuranoM , OhyaJet al. Lysophosphatidic acids and their substrate lysophospholipids in cerebrospinal fluid as objective biomarkers for evaluating the severity of lumbar spinal stenosis. Sci. Rep.9(1), 9144 (2019).