Abstract
FDA has been regulating diagnostic tests (including biomarkers) since passage of the Medical Device Amendments of 1976. Although always of interest as diagnostic tools, biomarkers (particularly genetic/genomic) have become of increased interest because of their potential impact on the development and personalized use of drugs. Unfortunately, there seem to be uncertainties among translational researchers as to the specific analytical and clinical measurement criteria needed for the approval of these novel biomarkers. This meeting presentation describes the current FDA perspective and major requirements and data for the validation/approval of an in vitro diagnostic device (IVD) based on a biomarker. The approval process for an IVD based on a biomarker used in the identification of a disease or condition (diagnosing, screening, monitoring) is well established, and is essentially identical to the process to generate sufficient analytical and clinical data for the approval of regular diagnostic devices. On the contrary, approvals for IVDs based on biomarker which may be designed to evaluate the efficacy or answer safety questions for new drug entities are less streamlined. The clinical studies are more complex, resulting in higher ethical standards, increased costs and requiring complex statistical evaluation. There is a small but growing literature on new models for co-development of drugs and diagnostics which will be discussed. Regulators like the FDA develop and bring a flexible regulatory toolbox to the table and are committed to assuring that scientific and regulatory thresholds are tempered to assure rapid access to new technologies while protecting public health.
Introduction to FDA/CDRH Device Classification
FDA regulations divide devices into one of three classes (class I, II, or III) [Citation1,Citation2]. In general, the classification of a diagnostic device depends on the intended use of the particular assay and its associated risk to the patient. An intended use with a higher risk will direct the classification of the device towards a high risk class III device, which requires a premarket application (PMA) to the FDA. Class III devices pose a significant risk of illness or injury to the patient or user or are important in preventing impairment of human health. Moderate-risk intended uses would allow a lower risk classification (class II), which in most cases requires a 510(k) submission. Class II devices are devices for which performance characteristics are well understood and predictable, carry moderate risk, or have associated special controls that mitigate possible harm to the patient. Class I devices carry a lower risk, offering little or no potential for the unreasonable risk of injury or illness. They are generally exempt from FDA premarket review. As many IVDs have moderate risk, the most common premarket regulatory pathway for IVDs is the 510(k), which is required for some class I and most class II devices. Information on cleared and approved IVDs is available online [Citation3].
For purposes of classifying IVDs, the physical risk to the patient from the testing process itself is generally small. The FDA's primary concern is the risk of harm derived from an incorrect result associated with in vitro diagnostics assays (i.e., false-negative and/or false-positive results). For example, what is the risk of an assay reporting a false-negative result when the patient actually has cancer? Would a false-positive result be followed by unnecessary surgery, a treatment with toxic side effects?
Types of biomarkers
Diagnosis: Diagnostic biomarkers are the most established biomarkers. They are results or measurements that provide information about a disease or condition e.g. cancer or the presence of an infection with an agent such as a virus. Diagnostic biomarkers are normally used in devices prescribed for patients with signs and symptoms of a specific disease/condition and not for the general population.
Early detection (screening): Screening biomarkers are biomarkers for the early detection of a disease/condition in a population of patients without specific signs and symptoms of a disease. The performance evaluation of screening biomarkers requires a clinical validation in a large intended use population. This extra requirement is necessary since the prevalence of any given/screened disease/condition is normally very low and a sufficient device performance evaluation for the positive and negative predictive value of the screening biomarker needs to be established.
Monitoring: Monitoring biomarkers are biomarkers for the follow-up of a previously diagnosed or established disease or condition. The performance requirements might be slightly less stringent for monitoring biomarkers in comparison to diagnostic biomarkers due to the fact that a possible one time false positive or negative result might not have such a negative impact on patient management since previous results are available e.g. viral load assays for hepatitis. Monitoring results, by themselves, are not therapy-directing. Claims for IVD safety and effectiveness for decisions to start or to change therapy are higher risk and require additional evidence. In addition, monitoring claims per se do not justify use of the IVD test results as a surrogate endpoint biomarker.
Prognosis: Prognostic biomarkers inform about the outcome of already-diagnosed disease, independent of a specific treatment. They are associated with the likelihood of an outcome (survival, response, recurrence) in a population that is untreated or on a standard (non-targeted) treatment. Prognostic biomarkers predict the probability of disease recurrence, progression, etc. or categorize an individual into risk classes for the disease. Tests intended to estimate the risk that a patient will develop disease in the future often involve testing of larger populations and may carry higher risk than do tests for prognosis of already-diagnosed disease.
Safety: Safety biomarkers inform about the risk of an adverse event and are specifically dependent on the biomarker. For example the FDA cleared biomarker UGT1A1, which can help in assessing the risk of neutropenia in patients taking irinotecan for colorectal cancer.
Efficacy prediction: Efficacy biomarkers can predict the differential effect of a particular treatment (e.g. compared to the effect of no treatment or an alternative treatment) on an outcome (such as survival, response, or recurrence). Predictive claims may be therapy directing. Statistically, there is an interaction (with respect to outcome) between the biomarker and the particular treatment, and it is important to include the phrase “predictive for __x__ therapy”. A predictive biomarker needs to show (for example) that a therapy works better for test positive patients than for test negative ones. It is a single trait or signature of traits that separates different populations with respect to the outcome of interest in response to a particular (targeted) treatment.
Prognostic-, safety- and predictive efficacy biomarkers are attempting to foresee the future.
It is important to note that endpoints for Prognostic and Predictive Markers: (1) need to be defined in advance of a confirmatory trial (e.g., recurrence free survival, time to metastases, overall survival), (2) can be measured in time to event or survival times. Clinical trial designs for such biomarkers are challenges as described in more detail later.
Efforts to support biomarker qualification
Beginning in March 2005 and still relevant at the current time, most pharmacogenomic data are of an exploratory or research nature, and FDA regulations do not require that these data be submitted. However, voluntary submissions can benefit both the industry and FDA in a general way by providing a means for sponsors to ensure that regulatory scientists are familiar with and prepared to appropriately evaluate future genomic submissions. Therefore, FDA is requesting that sponsors conducting such programs consider providing pharmacogenomic data to the Agency voluntarily, when such data are not otherwise required under the regulations.
In February 2006 the Food and Drug Administration (FDA), the National Cancer Institute (NCI), and the Centers for Medicare & Medicaid Services (CMS) kicked off the Oncology Biomarker Qualification Initiative (OBQI), an agreement to collaborate on improving the development of cancer therapies and the outcomes for cancer patients through biomarker development and evaluation. This initiative is the first time these three Department of Health and Human Services (HHS) agencies have focused together on biomarkers as a way of speeding the development and evaluation of cancer therapies. The goal of OBQI is to validate particular biomarkers so that they can be used to evaluate new, promising technologies in a manner that will shorten clinical trials, reduce the time and resources spent during the drug development process, improve the linkage between drug approval and drug coverage, and increase the safety and appropriateness of drug choices for cancer patients.
Critical path initiative
In addition to the previously mentioned programs the FDA started its Critical Path Initiative to “Qualify Biomarkers” [Citation4] for regulatory use via public-private partnership consortia to overcome the identified obstacles.
This initiative has clear objectives towards Advancing Innovative Trial Designs [Citation5]. Numerous projects have been identified and sponsored such as project #34: Design of Active Controlled Trials, #35: Enrichment Designs, #36: Use of Prior Experience or Accumulated Information in Trial Design, #37: Development of Best Practices for Handling Missing Data, #39: Analysis of Multiple Endpoints.
Challenges
General challenges: The challenges for biomarker development, qualification and eventual approval of an IVD are complex and plentiful. Due to the fact that there is still limited information available regarding the best and most effective way for co development of biomarkers and therapies, the research community and FDA have identified the need for transparency on how to get a biomarker validated/approved as an IVD. FDA promotes transparency and the Office of In Vitro Diagnostic Device Evaluation and Safety (OIVD) has invested a significant amount of resources to make information such as IVD review documentation available. Another challenge is the fact that regulatory goals are often different from research and exploratory goals. Regulatory scientists have to assure that a test, device, or biomarker is safe and effective and can be measured and used robustly and reproducibly in its established final form. Basic researchers often have the desire to continuously improve their tests, a well appreciated desire, but hindering approach for the validation of a test, device, or biomarker as major changes require revalidation of analytical and clinical performances. Finally, this new and developing field also requires new paradigms. Previously established clinical study designs do not always work due to ethical, prevalence, economical or other reasons, the development of new approaches is essential. Some have already been used and established in oncology. Prospective studies are not always feasible, so it is common sense to try to use other types of studies to generate the required data. Retrospective and/or banked samples are sometimes an option, however it is of extreme importance to consider some general issues: (1) these samples need to be representative of the intended use population, (2) should be of a prospective nature, meaning being an “all-comers” study, where all the eligible patients/samples were prospectively enrolled/collected therefore generating a set of consecutive cases/patient samples that meet the predetermined inclusion/exclusion criteria, (3) storage conditions are addressed and it is important to assure that they do not have any impact on assay results.
Methods, results and discussion
Analytical validation – quality of measurement. The analytical verification of any assay/device including biomarker assays has been well established and includes a list of parameters that need to be evaluated. These parameters include: (1) Precision (repeatability, reproducibility), (2) Trueness, (3) Performance around the cut-off, (4) Linearity for a quantitative test, (5) Limit of Detection, (6) Specificity (reactivity, interference, cross-reactivity, interference, cross-reactivity), (7) Sample type/matrix, (8) Sample preparation/preanalytical validation, (9) Platform/instrument – preparation, purification, detection, (10) Potential for carryover, cross-hybridization. Briefly, Precision intends to capture total test variability, including performance of the entire device, which means all steps from specimen preparation (preanalytical steps, e.g., extraction method) to final result. The experiments for the precision should demonstrate that the intended users can get reliable results, and should demonstrate reproducibility of the test at external sites. Major sources of variability should be determined. Precision needs to be established for each marker or the final score if applicable. Accuracy is defined by CLSI as the closeness of agreement between a measured quantity value and a true quantity value of a measurand. It is aimed to compare results from real clinical samples to a reference method.
Performance around the cut-off describes the performance of a qualitative biomarker/device at the decision point of being reported as positive or negative. The cut-off should be optimized to minimize false positive and negative results by testing a representative sampling of specimens containing, and not containing, the target organism. It should be established analytically and confirmed using the results of specimens from the clinical trial.
Linearity describes the ability (within a given range) to provide results that are directly proportional to the concentration (amount) of the analyte in the test sample [Citation6].
Limit of detection represents the lowest (and highest) concentration of input sample that yields a reliable and accurate result. The limit of detection (LoD) should be determined by using limited dilutions of biomarker/analyte.
Specificity (reactivity, interference, cross-reactivity). Reactivity demonstrates that the test can detect different versions or strains of the biomarker/analyte that represents temporal and geographical diversity. Interference evaluates whether endogenous and exogenous common substances are efficiently removed by sample preparation procedures and do not negatively influence the biomarker/analyte. Interference studies should be performed at the assay cut-off for each biomarker/analytes and for each of the interfering substances. For cross-reactivity, medically relevant levels of analytes similar, but different, from the intended analyte need to be evaluated. In general, it is important to identify the scenarios under which the biomarker/analyte will be used and the relevant interferences that could occur.
Sample preparation and preanalytical validation are increasingly important aspects in evaluating the performance characteristics of a device. Some biomarkers/analytes are labile or unstable and therefore performance data on these unstable measurands should be provided that includes the evaluation of the early preparation steps in a clinical setting.
Potential for carryover, cross-hybridization addressed the increased risks of carryover due to the amplification methodologies utilized in molecular testing.
Standards for evaluating tests
As previously mentioned, a large body of information from many resources is available which also includes standards for evaluating tests. These standards include: (1) the Medical Devices Standards Database [Citation7] (2) the Clinical and Laboratory Standards Institute (CLSI), this organization develops global consensus standards and guidelines for healthcare testing [Citation8]: (3) the International Standards Organization (ISO).
If the analytical verification of tests/devices/biomarkers is so well established, one may ask the question: Why is analytical data insufficient? The reasons vary and depend on the type of technology the assay/device uses. For example, most important of all is how well the assay/device works with real clinical samples at different locations in the hands of intended or end users. Also, how well does the assay work at the extremes i.e. the cut-off or at the medical decision points? One may need to characterize performance at low levels of the analyte and possibly across the entire measurement interval. If the medical test interpretation combines the results of multiple probes, analytes or biomarkers into a single result or score then the device falls into the category of an In Vitro Diagnostic Multivariate Index Assay (IVDMIA) which requires a “locked down” set of biomarkers or classifiers that need to be validated with an Independent Validation Data Set:. This means a confirmatory studies performed on an appropriate number of specimens to ensure that the biomarker/assay/score developed with a training set of a few hundred patients actually works post-market in a large number of patients [Citation9].
Clinical validation and study trial design for predictive claims
When biomarker information emerges as part of exploratory analyses, the most informative way to move forward is with a prospectively designed randomized controlled trial. This approach assures several study strengths that are questionable when instead retrospectively mining of previously banked trial specimens is used. First, with the prospective trial, the relevant biomarker is pre-specified and there is no concern about inflating test performance through multiple comparisons used to identify the biomarker. During specimen accrual, specimen collection, preservation and storage can be optimized for the biomarker assay. With a prospective design, specimen accountability may be substantially increased and non-random loss of specimens, with its attendant risk of bias, can be actively minimized. In executing the new trial, there is the opportunity to stratify based on the biomarker status (preferably, as assessed using a fully specified and analytically verified assay) before randomizing into treatment groups. This helps assure maximum power in the study design and removes the confounding effect of other variables that might be correlated with the biomarker. In designing the new trial, one has the opportunity to manage the allocation of Type 1 statistical error, or to maximize trial efficiency through adaptive design. New (prospective) randomized controlled trials (RCT) are hard or impossible to beat.
Clinical validation: Common types of clinical studies:
Completely randomized design: One study design uses an untargeted or completely randomized approach. All patients are randomized to drug or control, and the diagnostic test/biomarker interaction could be determined later. Such a study design generates data that addresses all parameters with the biomarker and the drug. It is a study in which there is at least some descriptive data available to allow investigators to see drug action (independent of biomarker), the biomarker action (independent of drug), and then the intersection between the two.
The study is designed to determine: (1) Sensitivity (SENS) – fraction of responders who test positive, (2) Specificity (SPEC) – fraction of non-responders who test negative, (3) Positive Predictive Value (PPV) – fraction of test positives who respond, and (4) Negative Predictive Value (NPV) – fraction of test negatives who do not respond. A study like this is depicted in . It will test if the drug works in the entire population and will assure a mixture of marker positive and negative drug effects.
Figure 1. Complete randomized design.

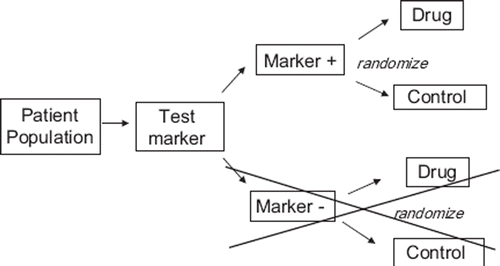
Randomized block design: All patients are tested with a diagnostic assay and then randomized within each of two blocks, test positive and test negative, see . One might think that the completely randomized design and randomized block design will generate an identical final data set, however if the accrual rate is not equal for the marker positive and marker negative individuals the study could be enriched with test positives patient samples. In such a case the SENS and SPEC will be incorrectly determined due to bias in the study population but PPV and NPV can be established correctly.
Figure 2. Randomized block design.
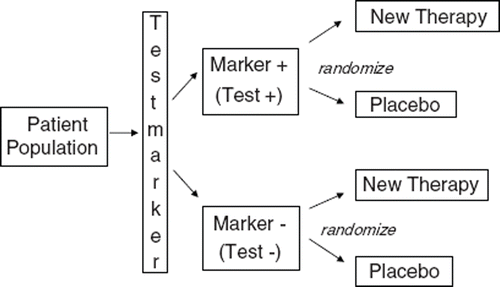
Targeted (enriched) design: Treatment response (e.g., clinical recovery rate) stratified by test outcome, see . The drug will be evaluated in one study arm only. The targeted design can answer the question whether the drug works in the test positive group or not. However, it can not answer whether the drug works in the test negative group. It is important to determine whether the diagnostic test is different from a random test for selecting subjects with a drug response. Also it is significant to note that this design does not establish any predictive claim for the diagnostic device. These types of studies are the more usual studies – one in which there is information on drug response versus placebo in biomarker positive patients only. The result that can be obtained from such a targeted design study is PPV, not sensitivity, not specificity and not NPV. An established pre-specified analysis plan is essential and it should be clearly stated whether the use of test is for safety or effectiveness and what type of hypotheses is evaluated, superiority or non-inferiority.
Figure 3. Targeted (enriched) design.
Ideal and real live scenarios for the Rx-Dx co-development process/pipeline
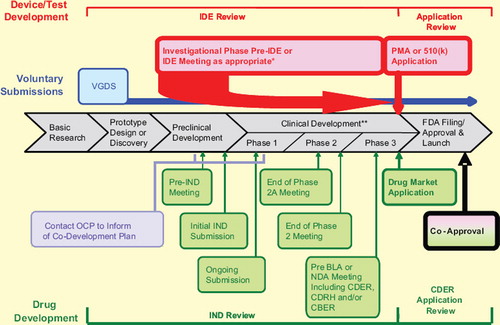
depicts the idealized pathway for test development which is at the same time as the drug is being developed, including ample time for early analytical validation of the test. An ideal scenario is one in which the relationship of the biomarker to potential action of the drug is recognized very early. In this setting, many milestones for development of the biomarker assay might be reached in an orderly way. The identity of the biomarker should be established early, along with a reliable means to measure its concentration. If the biomarker has an impact on the natural course of disease, such a prognostic relationship might be elucidated. Through pre-clinical studies and early clinical trials, support might grow for applicability of the biomarker as an indicator of drug effect. This is the point at which formulation of a specific intended use for the biomarker might emerge, and resources are committed to complete the analytical validation of a fully specified biomarker test. When a pivotal trial of the drug is undertaken, its design should incorporate a validation of the complete test, so that firm conclusions can be drawn concerning both the safety and efficacy of the drug and the safety and effectiveness of the biomarker test for informing use of the drug [Citation10,Citation11].
Figure 4. Drug-device co-development process: Formal industry-FDA interactions (noncombination product example).
For various practical reasons, such well choreographed development of a new biomarker test along with a new drug is a rarity so far. For example the idealized concurrent development of a new drug and a new biomarker test may be upset by late emergence of critical information (for example, late recognition of the biomarker or its putative relevance to drug effect).
When previously studied drugs are evaluated in new contexts and in light of new biomarker information, analytical verification of the new test may be very late in the development process, and it is tempting for clinical validation plans to rely on the analysis of specimens that were retained from ongoing or already completed clinical trials. Such data may not be complete and issues regarding missing data need to be addressed or questions of bias may cloud the real performance evaluation of the test. A different situation is when a biomarker that has been previously well characterized, and reduced to a test approved for clinical use, seems relevant to the development of a new drug. Here the marketed test “validated” for one intended use may not imply being validated for another intended use, due to possible differences in patient population, assay cut-off values, interferences with respect to intended patient population and specimen type. It is also feasible that a previously studied drug will be re-evaluated in the context of a previously studied IVD test, for example when the drug is reviewed for a new indication.
Safeguards to ensure success
As previously mentioned, it is highly beneficial to start thinking about co-development of a biomarker test early in drug development and one should plan for the possibility that a test may be needed. FDA strongly recommends that sufficient patient samples are collected, clinical trial samples are appropriately stored and most important informed consent is obtained that allows all future possible use of the clinical samples. These three safe guards are of particular importance if there is no validated diagnostic test available at the beginning of the phase III study. On the data analysis side, statistical methods should be evaluated that may allow for adaptive trial design if a drug is not effective in the general population. If possible, utilize bridging studies as these may be necessary in certain situations such as where the test used in drug trials is not the marketed version or if a platform change from a higher complexity and more expensive to a faster and cheaper test is desirable. Both marker positive and marker negative samples from Phase 2 and 3 studies should be stored for use in any bridging studies that might be necessary in the future. These banked trial specimens will also be extremely useful for validation of future versions of a biomarker test. In general, it is strongly advised to talk to the FDA, as early as possible about any potential co-development programs and plans on how to execute them.
Conclusions
Biomarker validation-approval still is a challenging field with minimal tolerance for mistakes. One has to get it right the first time, since the ethical and financial circumstances often prevent repeating a study. The objectives that need to be covered to facilitate clearance/approval of a biomarker device are (1) finding, designing and translating a biomarker/analyte/device into a functional product usable in a clinical decision making environment, (2) obtaining a clinical trial designer, medical experts in the underlying disease and statisticians that understand practical challenges coming from the use of a not perfectly designed statistical trial, (3) organizing stakeholders from all scientific disciplines including physicians, scientists, and regulators to come together and provide concepts and outlines and alert the community about already determined pitfalls. The process to find a pathway has begun, but is clearly not complete. The FDA with its outreach program of guidance documents has summarized concepts and new updates and several others are in the pipeline. The path is worthwhile but challenging, and it is recommended to get an appropriate team together since the stakes are too high and the pitfalls too plentiful.
The late emergence of critical information, causing re-evaluation of a well-studied drug in light of a new biomarker, seems to be common. Also, the analytical verification of the new biomarker test may be very late in the development process, but it is essential to verify it before testing clinical trial specimens. Problems in the assessment of assay performance characteristics can occur when one device version is used for patient accrual and another version is used for final clinical validation. The extent to which revision of the drug indication and clinical validation of the IVD test can be based on retrospective analyses of retained specimens requires scrutiny. The methodology is well-developed to assess the informativeness of a biomarker test but may not be well-known in therapeutic circles, and it is important to assess whether the molecular diagnostic really adds anything to what is already known. Studies to demonstrate informativeness of a biomarker can be quite difficult to design, conduct and analyze.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Food and Drug Administration. 21 USC 360 c (a)(1). Classification of devices intended for human use; device classes. http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/FDCActChapterVDrugsandDevices/ucm110188.htm (accessed 2010-04-27).
- Food and Drug Administration. Overview of IVD regulation. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/IVDRegulatoryAssistance/ucm123682.htm#4b: (accessed 2010-04-27). Within section “Medical devices.”
- Food and Drug Administration. Information on cleared and approved IVDs. http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/LabTest/ucm126189.htm (accessed 2010-04-27).
- Food and Drug Administration. Critical Path Initiative. http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/default.htm
- Food and Drug Administration. Critical Path Initiative. http://www.fda.gov/oc/initiatives/criticalpath/reports/opp_list.pdf (accessed 2010-04-27).
- CLSI, previous NCCLS. Evaluation of the linearity of quantitative measurements procedures: A statistical approach; Approved Guideline NCCLS document EP6-A [ISBN 1-56238-498] Wayne PA USA, 2003.
- Medical Devices Standards Database http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Standards/default.htm (accessed 2010-04-27).
- Clinical and Laboratory Standards Institute (CLSI). www.clsi.org (accessed 2010-04-27).
- Russek-Cohen E, Tezak Z, Philip R, Pastel M (2007) Multivariate assays reporting a composite score. JSM Proceedings CD-Rom, 2007.
- Food and Drug Administration. Preliminary concept paper: Drug-diagnostic co-development concept paper. http://www.fda.gov/downloads/Drugs/ScienceResearch/Research Areas/Pharmacogenetics/UCM116689.pdf (accessed 2010-04-27).
- Taube SE, Clark GM, Dancey JE, McShane LM, Sigman CC, Gutman SI. A perspective on challenges and issues in biomarker development and drug and biomarker codevelopment. J Natl Cancer Inst 2009;101:1453–63.