
Abstract
There is no reference method that is generally acknowledged to be unbiased for the determination of the concentration of any protein in biological fluids. This is probably because mass spectrometry (MS) methods acknowledged as reference methods for determination of low molecular mass substances in biological fluids, e.g. creatinine, have been difficult to adapt for proteins. Here we suggest two tentative MS methods, which might be used as reference methods for the determination of protein concentrations in biological fluids. One is based upon the addition to the fluid of a non-proteome reference protein, very similar to the one to be measured, and analyzing the ratio between the corresponding peaks in a selected ion monitoring (SIM) chromatogram. We call this method LC-MS-NPRP (NPRP, Non-Proteome Reference Protein). The other method is based upon the classical standard addition assay for low molecular mass substances. The results of these assays for cystatin C in spinal fluid were compared to those obtained by an immunoassay. Both methods indicated lower concentration than the immunoassay. This was found to be due to the presence of a significant fraction of monohydroxylated cystatin C in spinal fluid. It turned out that the sum of the unhydroxylated and hydroxylated cystatin C concentrations, determined by either of the two MS methods, were close to the results obtained by the immunoassay. These MS-based methods analyze intact proteins and therefore seem more suitable for the determination of protein concentrations in biological fluids than other MS-based methods requiring proteolytic degradation with its inherent lack of precision.
Introduction
To secure agreeing and unbiased results for different assays measuring the concentration of a substance in a biological fluid two conditions must be fulfilled. One is the availability of an internationally accepted calibrator of the substance. The other is the availability of a ‘reference method’ for measurement of the substance concentration, i.e. a method that is internationally accepted as accurate (and without bias). Such calibrators and reference methods are not available for most of the substances used for diagnostic purposes in clinical medicine. While reference methods, often based upon mass spectrometry (MS), are available for a number of low molecular mass substances, e.g. glucose and creatinine, no such reference method is available for a single protein. So, although international calibrators are available for several proteins, the lack of reference methods means that it presently cannot be secured that the results for the concentrations of proteins in biological fluids obtained at different laboratories agree and are unbiased. The reason for the lack of reference methods for proteins is probably that it has been difficult to adapt MS methods for protein quantification. Although a multitude of MS methods for the analysis of proteins has been described, they are usually based upon proteolytic degradation of the proteins with several steps before the crucial mass analysis, which results in problems with reproducibility and precision. In the present work, we will suggest and test two non-degrading MS-based methods for the determination of protein concentrations in biological fluids in an effort to take a step towards MS-based reference methods for such determinations. One method is based upon the addition of a non-proteome reference protein, physicochemically very similar to the protein of interest, to the body fluid and using a liquid chromatography separation step before the MS (LC-MS-NPRP). The other is a classical standard addition method modified for intact proteins, also employing LC-MS. In our study we used cystatin C as the model protein, since the structure and mass of cystatin C is known [Citation1–3], recombinant production of the wild-type protein is available [Citation4], as well as an international primary calibrator [Citation5], and because cystatin C concentrations frequently are used in clinical medicine to assess e.g. kidney function [Citation6,Citation7]. We used spinal fluid as the biological fluid, since its concentration of cystatin C is relatively high, with cystatin C constituting about 1% of the proteins in the fluid [Citation8].
Methods
Materials
Recombinant native wild-type (non-hydroxylated) cystatin C (Mw 13343) and recombinant cystatin C, with four amino acid residues (Arg-8, Leu-9, Val-10, Trp-106) replaced by glycine (GGGG-cystatin C, Mw 13017), were produced as described [Citation4,Citation9] and lyophilized. Spinal fluid samples were obtained from the Department of Clinical Chemistry, University Hospital, Lund, Sweden. The protease inhibitor benzamidinium chloride was added to the samples to a final concentration of 1 mM to prevent protein degradation. Immunochemical determination of the cystatin C concentration in spinal fluid was performed as previously described [Citation10].
LC-MS
Liquid chromatography was performed with two Shimadzu LC 10AD-vp pumps configured in high-pressure gradient mixing mode, controlled by the integrated system controller in a SIL HTc autosampler. A Quattro II triple quadrupole mass spectrometer (Micromass) equipped with a Z-spray ion source and an electrospray probe was used for acquiring spectra and chromatograms. The m/z values specific for cystatin C, hydroxylated cystatin C and GGGG-cystatin C were used in selected ion monitoring (SIM) chromatograms.
The spectra of isolated proteins were acquired during continuous infusion of a 10–15 mg/L protein solution in a 60/40 mixture of mobile phase A/B.
A 2.1 × 50 mm BEH300 C4 3.5μ column (Waters) was used for all chromatographic separations. Mobile phases were (A) water with 4 mM trifluoroacetic acid (VWR) and (B) acetonitrile (VWR). Samples were eluted with a binary gradient program from 28–38% B in 6 minutes, at a flow rate of 0.25 mL/min. The mass spectrometer was tuned to record data for the m/z corresponding to the + 12 ions for both cystatin C and GGGG-cystatin C. A post-column gate valve (Cheminert C2-1004 with electric actuator, Vici) was programmed to divert the eluate from the column to waste when no SIM data were recorded.
Preparation of samples and standards
Standards containing varying levels of cystatin C (0, 0.94, 2.1 and 4.7 mg/L) and 30 mg/L of GGGG-cystatin C (to inhibit unspecific binding of cystatin C in the chromatographic system) were prepared by weighing the pure lyophilized proteins and dissolving them in 30 mM formic acid in an 85/15 (v/v) mixture of water/acetonitrile. Aliquots of 70 microlitres of spinal fluid were mixed with 70 microlitres of each standard. Aliquots of 40 microlitres of the final mixture were injected in the chromatographic system, followed by integrating the resulting peak areas and evaluating the data for concentrations.
Note that samples and standards will mutually dilute each other by a factor of 2, so the actual concentrations in the injected samples will be half of the nominal concentrations.
Results
Mass spectrometry of isolated cystatin C and GGGG-cystatin C
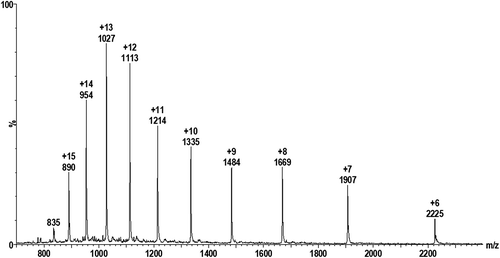
Mass spectra for pure cystatin C () and GGGG-cystatin C (not shown) exhibit the expected multiply-charged ion distributions. The multiply-charged ion z + 12 was selected for both cystatin C and GGGG-cystatin C and instrumental parameters were optimized for acquiring SIM data during chromatographic elution.
Figure 1. Electrospray mass spectrum for 15 mg/L cystatin C.
Adsorption of cystatin C and GGGG-cystatin C in the chromatographic system
Early experiments showed strong indications that pure cystatin C and GGGG-cystatin C displayed adsorption to vials and/or in the LC-chromatographic system used. Adsorption manifested mainly as non-linear calibration functions with negative intercepts, with the most pronounced effects when the protein concentrations were below 2–3 mg/L. It was discovered that the presence of GGGG-cystatin C in the cystatin C-standards reduced the adsorption of cystatin C and that a GGGG-cystatin-C concentration of 30 mg/L in the standards resulted in linear calibration functions for cystatin C. Analogously, the same concentration of cystatin C (30 mg/L) allowed linear calibration functions of GGGG-cystatin C. These results indicate that cystatin C and GGGG-cystatin C compete for the same adsorption sites of the chromatographic system.
The adsorption sites for cystatin C could evidently not be totally blocked by other proteins of spinal fluid, since addition of increasing amounts of pure cystatin C to spinal fluid samples (initial cystatin C concentrations of 2–5 mg/L, as determined by the immunochemical procedure) did not result in a linear relation between the area of the cystatin C-peak in LC-MS and the added amount of cystatin C. However, addition of increasing amounts of GGGG-cystatin C to the cystatin C-spiked spinal fluid samples resulted in less adsorption of cystatin C and at a final concentration of GGGG-cystatin C of 15 mg/L linear dose-response curves for cystatin C were obtained, at least to cystatin C-concentrations up to 10 mg/L.
Use of GGGG-cystatin C as a non-proteome reference protein in determination of cystatin C concentrations in biological fluids by non-degrading mass spectrometry
Attempting to establish a non-degrading LC-MS-based method for determination of protein concentrations in biological fluids, we choose GGGG-cystatin C as a reference protein to add to biological fluids for which we wanted to determine the level of cystatin C. The selection of GGGG-cystatin C was based upon the fact that it is not present in the human proteome, is very similar to cystatin C in physicochemical properties, and yet differs sufficiently from cystatin C in mass to allow a clear separation of the molecules by MS.
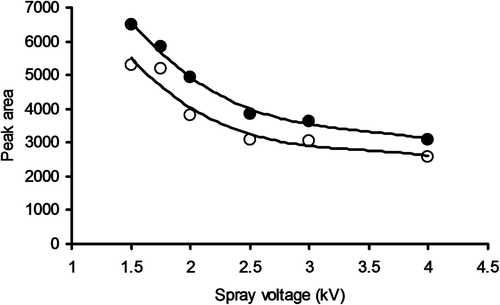
It was found that a spray voltage in the interval 3.0–3.5 kV gave the best response when acquiring spectra from continuous infusion of a 15 mg/L solution of pure cystatin C or GGGG-cystatin C. When acquiring SIM chromatograms from injecting 1 mg/L samples, the best response was achieved with lower spray voltages. It was also found that the use of a mobile phase degasser lowered the response (). Plausible explanations for these observations are found in the redox properties of the electrospray process [Citation11].
Figure 2. Peak area responses as a function of spray voltage when injecting 40 μL samples containing 1 mg/L cystatin C and acquiring SIM data for the z + 12 peak (●, without degasser; ○, with degasser).
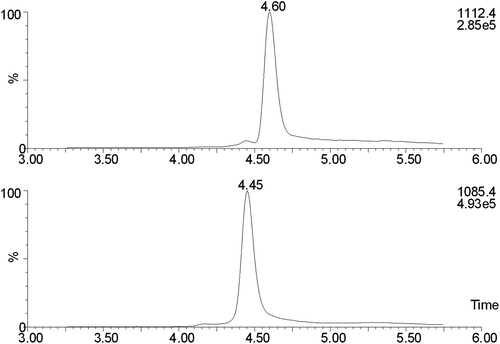
illustrates SIM chromatograms for native cystatin C and added GGGG cystatin C (15 mg/L) in a spinal fluid sample designed for determination of cystatin C concentrations (see below). It is evident that cystatin C and GGGG-cystatin C migrate very similarly in the chromatographic system.
Figure 3. SIM chromatograms for a spinal fluid sample with GGGG-cystatin C added to a concentration of 15 mg/L. The top trace is cystatin C and the bottom one is GGGG-cystatin C.
Two non-degrading LC-MS assays for determination of cystatin C concentration in spinal fluid
(A) Standard addition assay
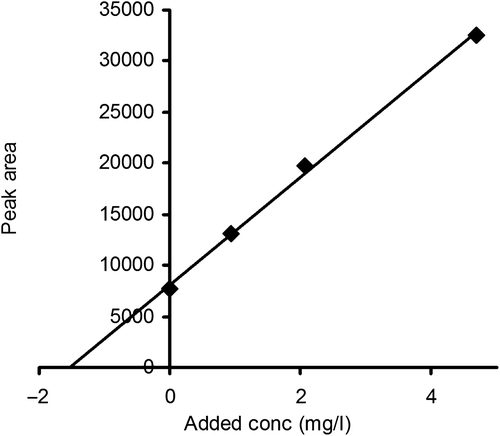
As the first non-degrading LC-MS-assay we chose to use the classical standard addition technique developed for low molecular mass substances considerably smaller than proteins. This assay was based upon the addition of increasing amounts of cystatin C to spinal fluid, which was then run in the LC-MS system described above. As mentioned in the Method section, all added cystatin C standards also contained 30 mg/L of GGGG-cystatin C to inhibit absorption of cystatin C. The peak areas for cystatin C were then plotted against the added amount of cystatin C (0, 0.94, 2.1 and 4.7 mg/L). A linear dose-response curve resulted and its extrapolation to a peak area of zero represents the cystatin C amount present in the spinal fluid before addition of further amounts of cystatin C. illustrates the linearity of this response for one sample of spinal fluid. To obtain an approximate estimate of the precision of the standard addition assay, we used a pool of spinal fluid and performed 12 replicate analyses of the pool. Here only two additions of cystatin C were analyzed, zero addition and addition of cystatin C corresponding to a concentration of 4.7 mg/L. When the peak areas were used to calculate the cystatin C level in the pool, a concentration of 1.2 mg/L and a CV of 4.3% was obtained.
Figure 4. Peak areas for cystatin C in spinal fluid as a function of added amounts of cystatin C.
One operator, using the LC-MS equipment described in this work, can run about 100 samples per 8 h using the standard addition assay.
(B) LC-MS assay with the use of a non-proteome reference protein (LC-MS-NPRP)
The second non-degrading LC-MS assay was based upon the addition of a non-proteome reference protein to the samples and is, as far as we know, not described before. Step 1: Addition to the biological fluid of a selected amount of a reference protein, very similar in physicochemical properties to the protein to be analyzed, but not present in the proteome. Step 2: Comparison by LC-MS of the peak area for the reference protein with the peak area of the protein to be analyzed. Step 2 might require a chromatographic separation step before the analysis by MS. We suggest the designation LC-MS with a non-proteome reference protein (LC-MS-NPRP) for this type of assay. For determination of cystatin C in spinal fluid, we used GGGG-cystatin C as the non-proteome reference protein and the LC-MS system described above. We used a final concentration of GGGG-cystatin C in the spinal fluid of 15 mg/L. The ratio between the LC-MS peak areas for cystatin C and GGGG-cystatin C were used to calculate the cystatin C concentration in the sample, assuming identical, or very similar, MS dose response curves for cystatin C and GGGG-cystatin C. To test the linearity of the assay, increasing amounts of cystatin C were added to a spinal fluid sample and the ratios between the peak areas for cystatin C and GGGG-cystatin C were plotted against the added amount of cystatin C. As shown in , a linear relationship resulted.
Figure 5. The ratio of the peak areas for cystatin C and GGGG-cystatin C in spinal fluid as a function of the added amount of cystatin C. The GGGG-cystatin C (the non-proteome reference protein) concentration was 15 mg/L in all samples.
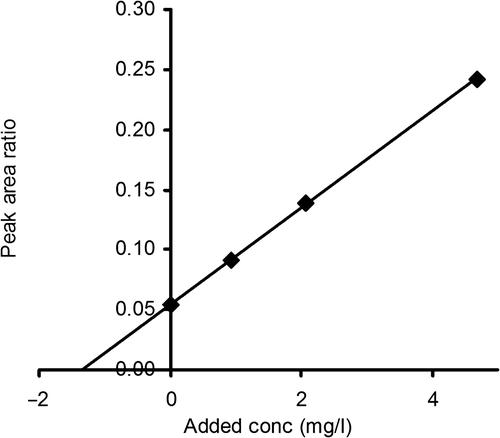
Like the standard addition assay, the CV of the LC-MS-NPRP assay at a concentration of 1.2 mg/L was 4–5%.
One operator, using the LC-MS equipment described in this work, can run about 150 samples per 8 h using the LC-MS-NPRP assay.
Assay comparisons. Demonstration of two physiological variants of cystatin C
As a preliminary test of the two LC-MS based assays, the cystatin C concentrations in four spinal fluid samples were determined using a commercially available immunoturbidimetric method [Citation10] and the samples then analyzed by the two LC-MS assays. The cystatin C concentrations measured by the LC-MS assays were only 30–40% of the concentrations obtained by the immunoturbidimetric assay ().
Table I. Cystatin C concentrations in four spinal fluid samples as analyzed by immunoturbidimetry and the LC-MS-based assays ‘standard addition’ and ‘LC-MS-with use of a non-proteome reference protein (LC-MS-NPRP). The percent columns refer to the results of the two LC-MS-based assays expressed as percentages of the immunochemical results.
We considered if the relatively low values obtained by the two LC-MS-based assays could be due to the fact that cystatin C in biological fluids might consist of two, or more, variants, not recognized as different by the immunochemical procedure, but identified as different by the high-resolution capacity of LC-MS-based assays. A survey of the literature gave an indication that this could be the case, since, when the amino acid sequence of the single polypeptide chain of cystatin C, isolated from human urine, initially was determined in 1982, two phenylthiohydantoin derivatives, representing proline and hydroxyproline in approximately equal amounts, were obtained for residue number 3 [Citation1]. To test the possibility of this hydroxyproline-variant of cystatin C being present in spinal fluid, a separate SIM-trace was configured with the a priori assumption that hydroxylation will add 16 Da to the cystatin C, which at z + 12 translates to 16/12 = 1.33 in m/z difference between monohydroxylated and non-hydroxylated forms. The mass spectrometer was carefully tuned to maximize resolution and mass accuracy at the expense of some sensitivity. When injecting pure recombinant, non-hydroxylated, cystatin C, the SIM-trace for the assumed hydroxylated cystatin C was clean, while, when injecting spinal fluid samples, a chromatographic peak corresponding to the mass of monohydroxylated cystatin C was detected at the expected retention time (). The area of this peak was evaluated as if the hypothetic hydroxyproline variant of cystatin C would have the same MS response factor as the recombinant, nonhydroxylated, cystatin C. shows that the sums of the concentrations of hydroxylated and non-hydroxylated cystatin C, as determined by the two LC-MS assays, are much closer to 100% of the cystatin C concentrations as determined by immunoturbidimetry than when only nonhydroxylated cystatin C was measured by the two LC-MS assays.
Figure 6. SIM chromatograms of a standard of pure non-hydroxylated cystatin C (A) and of an intact spinal fluid sample (B). The trace for m/z 1112.7 represents non-hydroxylated cystatin C and 1114.1 represents the assumed hydroxylated cystatin C.
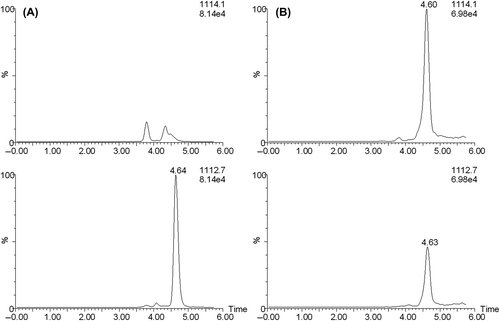
Table II. Cystatin C concentrations in four spinal fluid samples as analyzed by immunoturbidimetry and by the LC-MS-based assays ‘standard addition’ and ‘LC-MS-with use of a non-proteome reference protein (LC-MS-NPRP)’. The results of the MS-based assays are presented as concentrations of nonhydroxylated and monohydroxylated cystatin C and the corresponding sums. The percent columns refer to the results of the two LC-MS-based assays expressed as percentages of the immunochemical results.
Discussion
A fundamental problem in determining protein concentrations in biological samples by MS is the lack of blank sample matrices for preparing calibration samples and establishing calibration functions. Another difficulty is to compensate for individual variability between samples from different donors, which can cause different matrix effects and affect the MS response. A third problem is that different proteins display selective adsorption in the chromatographic system so that it is impossible to use a generally and easily available protein, e.g. albumin, to eliminate the adsorption of a specific protein. Although several MS methods for analysis of protein levels in biological fluids have been described, they are usually based upon proteolytic degradation of the proteins, require identification of one or more peptide fragments unique for the protein, and addition of internal standards with properties similar to the unique peptide(s), before chromatography and mass spectrometry. The variation in proteolytic efficiency, brought about by varying amounts of protease inhibitors in many biological fluids, and the varying proteolytic sensitivity of the proteins caused, e.g. by glycation or carbamylation of their amino groups as well as by other modifications of the proteins, contribute to the problems, when protein degradation is a crucial part of procedures to measure protein levels in biological fluids by mass spectrometry.
As far as we know, there is no reference method internationally accepted as accurate (i.e. without bias) for any protein in any biological fluid despite attempts to develop mass spectrometry-based procedures for such purposes. The method by Jeppson et al. to measure HbA1c has the status of a reference assay [Citation12]. However, it does not measure any protein concentration, but the proportion of glycated hemoglobin to total hemoglobin [Citation12]. The present investigation aimed at suggesting new mass spectrometry-based procedures, which might be developed into reference methods for protein quantification in biological fluids by omitting the degradation steps of presently used mass spectrometric assays. We chose to use the concentration of cystatin C in spinal fluid as a test of our attempts to develop such mass spectrometry-based assays, because of the relative simplicity of the task due to a relatively high concentration of cystatin C and the availability of some key reagents. It can be anticipated that the new, non-degrading, assays will be difficult to apply, without further development, to determine the concentrations of larger and/or glycosylated proteins in fluids, e.g. plasma, with higher total protein concentrations than spinal fluid.
One of the two suggested procedures is the classical standard addition method used for low molecular mass substances modified for use with proteins. This procedure is based upon obtaining a linear relation between mass spectrometric response and added amount of standard substance. Usually the response is represented on the y-axis of a Cartesian coordinate system and the amount of standard added on the x-axis. The concentration of the substance in the original fluid is given by the intercept on the x-axis. Addition of pure cystatin C in increasing amounts to spinal fluid did not produce a linear response in our LC-MS system so our initial attempts to use the standard addition method did not work. However, addition of GGGG-cystatin C to a final concentration of 15 mg/L in all samples of spinal fluid resulted in a linear relation between mass spectrometric response and added amounts of pure cystatin C, thus suggesting that the standard addition method can be used for analysis of the cystatin C level in spinal fluid. The reason why addition of GGGG-cystatin C resulted in a linear relation is probably that GGGG-cystatin C efficiently blocks the adsorption sites for cystatin C in the analysis system. Since the high concentrations of other proteins in spinal fluid, e.g. albumin, did not block the binding of cystatin C as efficiently as GGGG-cystatin C at a much lower concentration, the adsorption sites for cystatin C probably display some specificity for cystatin C so that they require a molecule similar to cystatin C to be efficiently blocked. Since GGGG-cystatin C is not present in the proteome and has a molecular mass differing from cystatin C, it does not cause any problem in the analytical system used. Although the standard addition method theoretically should work for all substances, including all proteins, its practical usefulness is limited, at least as far as proteins are concerned, since it requires the availability of 100% pure proteins, structurally identical to those, the concentration of which are to be measured in the biological fluids. It also requires knowledge of the exact concentration of the solution of the pure protein to be added.
To expand the usefulness of non-degrading MS-based methods for the determination of protein concentrations in biological fluids, we suggest a procedure which does not require the availability of pure proteins identical to those whose concentrations are to be measured. The key element of this procedure is the addition to the biological fluid of a specific amount of a non-proteome reference protein (‘internal standard protein’) with properties as similar as possible to those of the protein to be analyzed, but with a small difference in mass. Such a reference protein has the potential to co-elute with the protein of interest in the chromatographic system used and to have similar, or identical, MS response per mole of protein. By using the ratio of the MS signals between the protein of interest and the selected non-proteome reference protein, an estimate of the concentration of the protein of interest should be possible to obtain. In the present study, a linear relation between the amount of added cystatin C and the ratio of the MS peak areas for cystatin C and the reference protein, GGGG-cystatin C, was obtained, which supports the hypothesis that the ratio between the peak areas is directly related to the concentration of the protein of interest and that the method thus not require addition of different amounts of the pure protein of interest. The non-proteome reference protein used in this study was GGGG-cystatin C, already available to the authors [Citation9], but although it is very similar in physicochemical properties to cystatin C, it is not the ideal reference protein to be used in LC-MS-NPRP assays for determination of cystatin C concentrations in biological fluids. A more ideal non-proteome reference protein would probably be, e.g. a cystatin C variant in which a leucine residue was replaced with a valine residue, or vice versa, because the only difference between cystatin C and such a cystatin C-variant would be a difference in molecular mass of 14 Da, which probably would secure identical migrations of the two proteins in the chromatographic system used and in similar, or identical, MS responses per mole of protein.
Alternative non-proteome reference proteins (‘internal standard proteins’) potentially useful in LC-MS-NPRP and in other types of MS-based assays of protein concentrations in biological fluids have been suggested. These are generally isotope-modified variants of the proteins of interest. Such internal standards have, however, several drawbacks. They require, for example, the availability of a system for (recombinant) production of the protein of interest and expensive isotopes like 2H and 13C. Moreover, such non-proteome reference proteins will generally not represent a homogeneous set of identical protein molecules, but a set of molecules with differing amounts of isotopes and thus masses [Citation13]. In contrast, in the LC-MS-NPRP assay described above, it is possible to use a homogeneous reference protein representing only one molecular mass.
As noted in the present study, the reference protein and the protein of interest might possess some specific adsorption, not completely blocked by unrelated proteins like albumin, in the chromatographic system used. If the adsorption of the reference protein and the protein of interest differs significantly, this would represent a problem in the LC-MS-NPRP assay, since the ratio of the responses between the reference protein and the protein of interest would change with the relative amounts of the proteins. However, since the reference protein is chosen to be as similar as possible to the protein of interest, the risk of a significant difference in adsorption is small.
Both the standard addition and the LC-MS-NPRP assays, described here for determination of protein levels in biological fluids, avoid proteolytic degradation of the protein of interest. This avoids not only the known problems concerning the varying yields of peptides in different samples, but also the requirement to find a peptide unique for the protein of interest and the addition of an internal standard peptide with properties similar to the unique peptide. Interestingly, an assay similar to the LC-MS-NPRP assay described here for proteins, has been described for hepcidin, a 25-amino acid peptide [Citation14].
Determination of protein concentrations in biological fluids is generally done by immunochemical methods. However, no immunochemical method is considered to be a proven ‘reference method’ (i.e. without bias) for any substance, including proteins. Therefore, the procedure used when the concentration of a primary reference calibrator (usually a pure protein in a solution with its concentration determined by dry-mass analysis), is going to be used to determine the protein concentration in a secondary calibrator with a relevant biological matrix, e.g. serum, several different immunological methods are used and the mean value accepted as the concentration of the protein in the secondary calibrator. This was recently illustrated by the production of the international cystatin C calibrator ERM-DA471/IFCC [Citation15,Citation16]. Hoofnagle and Wener have recently discussed fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry [Citation17].
When the presently described LC-MS assays, designated standard addition and LC-MS-NPRP, were used to determine the cystatin C concentration in four human spinal fluid samples, the values obtained were considerably lower than those obtained by an immunochemical assay using the international cystatin C calibrator. The mean values by the two LC-MS assays were only 31 and 41 %, respectively, of those obtained by the immunochemical assay (). These discrepancies might, of course, be due to errors/problems in the LC-MS assays, but another possibility could be that two, or more, cystatin C variants are present in biological fluids and not recognized as different by the immunochemical procedure, but identified as separate molecules by the high-resolution capacity of LC-MS-based assays. The possibility of two different, but closely related, variants of cystatin C in biological fluids was indicated by the amino acid sequence analysis of cystatin C isolated from urine 30 years ago, which showed two phenylthiohydantoin derivatives, representing proline and hydroxyproline, for residue number 3 [Citation1]. There is also a 20-year-old report that a mass spectrum of cystatin C isolated from urine showed not only a peak indicating the expected mass (13343 Da), but also a peak indicating a mass of 13359 Da, compatible with the presence of monohydroxylated cystatin C [Citation2]. We therefore wanted to study if hydroxylated cystatin C is present in spinal fluid and chose a m/z-value specific for the molecular mass of monohydroxylated cystatin C (13343 + 16Da) and used this to produce SIM-chromatograms of recombinant (non-hydroxylated) cystatin C and of spinal fluid. No response was produced by nonhydroxylated cystatin C, but the response from spinal fluid indicated the presence of hydroxylated cystatin C in this fluid. Although the identity as hydroxylated cystatin C of the molecule producing the response must be confirmed, e.g. by time-of-flight or orbitrap mass spectrometry, we used a MS response factor identical to that for nonhydroxylated cystatin C to calculate the concentration of hydroxylated cystatin C in spinal fluid according to both the standard addition and the LC-MS-NPRP assays with GGGG-cystatin C as the reference protein. The mean values for the concentrations of (nonhydroxylated) cystatin C in the four spinal fluid samples using the standard addition and LC-MS-NPRP assays were 31 and 41%, respectively, of those determined by the immunochemical assay (). In contrast, the sum of nonhydroxylated and hydroxylated cystatin C, as determined by the two MS-based assays, were much closer to the results of the immunochemical assay (). This indicates that the immunochemical assay, indeed, co-determines nonhydroxylated and monohydroxylated cystatin C. The difference between the results of the standard addition and LC-MS-NPRP assays indicates that further work has to be performed to optimize the procedures. Such efforts must include studies using the international primary cystatin C calibrator, an improved non-proteome reference protein, e.g. a cystatin C variant with one leucine residue replaced with a valine residue, or vice versa, and actual determination of the MS response per mole of cystatin C and per mole of the selected non-proteome reference protein.
The possibility to measure both hydroxylated and nonhydroxylated cystatin C in biological fluids might be of biomedical importance, since the ratio of hydroxylated and nonhydroxylated cystatin C will be an indicator of the enzyme system performing hydroxylation of proteins with the target sequence X-Pro-Gly, not only present in the amino-terminal end (Ser-Ser-Pro-Gly) of the cystatin C polypeptide chain, but also in many of the major proteins of the body, e.g. collagen [Citation18,Citation19]. Hypo- and hyperfunction of this system have known clinical consequences [Citation18,Citation19].
The cystatin C concentration in plasma or serum, measured by immunochemical assays, is used as a reliable marker of glomerular filtration rate [Citation6,Citation7]. Since both hydroxylated and nonhydroxylated cystatin C are low molecular mass proteins, probably freely filtered through the glomerular membranes of the kidneys, the possibility of measuring hydroxylated and nonhydroxylated cystatin C separately by the standard addition and LC-MS-NPRP assays is not likely to increase the value of cystatin C as a marker of glomerular filtration rate.
The present study aimed at investigating if the standard addition and LC-MS-NPRP assays would be feasible for measuring the concentration of proteins in biological fluids without proteolytic degradation. We used the concentration of cystatin C in spinal fluid samples for testing the potential of the procedures and the results indicate, as far as we can judge, that the procedures might be of value and might increase the resolution obtainable by immunochemical procedures.
Acknowledgements
We are grateful to Bengt Längström for access to his LC-MS facilities and to Brian Green for valuable comments and suggestions. This investigation was supported by grants from the Alfred Österlund Foundation, the Greta and Johan Kock Foundation, the Medical Faculty of the University of Lund and from Region Skäne.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Grubb A, Löfberg H. Human γ-trace, a basic micro protein: Amino acid sequence and presence in the adenohypophysis. Proc Natl Acad Sci USA 1982;79:3024–7.
- Grubb A. Cystatin C. In Haeberli A (editor). Human protein data. VCH Verlagsgesellschaft mbH, Weinheim. 2. Installment 1993.
- Janowski R, Kozak M, Jankowska E, Grzonka Z, Grubb A, Abrahamson M, Jaskolski, M. Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat Struct Biol 2001;8:316–20.
- Abrahamson M, Dalboge H, Olafsson I, Carlsen S, Grubb A. Efficient production of native, biologically active human cystatin C by Escherichia coli. Febs Letters 1988;236: 14–18.
- Blirup-Jensen S, Grubb A, Lindström V, Schmidt C, Althaus H. Standardization of cystatin C: development of primary and secondary reference preparations. Scand J Clin Lab Invest 2008;68:67–70.
- Grubb A. Cystatin C as a biomarker in kidney disease. In Edelstein CL (editor). Biomarkers Oxford: Elsevier, 2011: 291–312. ISBN: 978-0-12-375672-5.
- Grubb A. Non-invasive estimation of glomerular filtration rate (GFR). The Lund model: simultaneous use of cystatin C- and creatinine-based GFR-prediction equations, clinical data and an internal quality check. Scand J Clin Lab Invest 2010;70:65–70.
- Löfberg H, Grubb A. Quantitation of γ-trace in human biological fluids: indications for production in the central nervous system. Scand J Clin Lab Invest 1979;39:619–26.
- Hall A, Håkansson K, Mason RW, Grubb A, Abrahamson M. Structural basis for the biological specificity of cystatin C – identification of leucine-9 in the N-terminal binding region as a selectivity-conferring residue in the inhibition of mammalian cysteine peptidases. J Biol Chem 1995;270: 5115–21.
- Kyhse-Andersen J, Schmidt C, Nordin G, Andersson B, Nilsson-Ehle P, Lindström V, Grubb A. Serum cystatin C, determined by a rapid, automated particle-enhanced turbidimetric method, is a better marker than serum creatinine for glomerular filtration rate. Clin Chem 1994; 40:1921–6.
- Boys BL, Kuprowski MC, Noel JJ, Konermann L. Protein oxidative modifications during electrospray ionization: solution phase electrochemistry or corona discharge-induced radical attack? Anal Chem 2009;81:4027–34.
- Jeppsson JO, Kobold U, Barr J, Finke A, Hoelzel W, Hoshino T, Miedema K, Mosca A, Mauri P, Paroni R, Thienpont L, Umemoto M, Weykamp C, on behalf of the International Federation of Clinical Chemistry Laboratory Medicine. Approved IFCC reference method for the measurement of HbA(1c) in human blood. Clin Chem Lab Med 2002;40:78–89.
- Stokvis E, Rosing H, Beijnen JH. Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: necessity or not? Rapid Commun Mass Sp 2005;19:401–7.
- Swinkels DW, Girelli D, Laarakkers C, Kroot J, Campostrini N, Kemna E, Tjalsma, H. Advances in quantitative hepcidin measurements by time-of-flight mass spectrometry. Plos One 2008;3:e2706.
- Grubb A, Blirup-Jensen S, Lindström V, Schmidt C, Althaus H, Zegers I on behalf of the IFCC Working Group on standardisation of cystatin C (WG-SCC): First certified reference material for cystatin C in human serum ERM-DA471/IFCC. Clin Chem Lab Med 2010;48:1619–21.
- Zegers I, Auclair G, Schimmel H, Emons H, Blirup-Jensen S, Schmidt C, Lindström V, Grubb A, Althaus H. Certification of cystatin C in the human serum reference material ERM-DA471/IFCC. EUR 24408 EN. Luxembourg: European Communities 2010:1–26. ISBN 978-92-79-07562-9.
- Hoofnagle AN, Wener MH. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J Immunol Methods 2009;347:3–11.
- Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol 2010;45:106–24.
- Hudson DM, Eyre DR. Collagen prolyl 3-hydroxylation: a major role for a minor post-translational modification? Connect Tissue Res 2013;54:245–51.